Gene Therapy for Chronic HBV—Can We Eliminate cccDNA?


Abstract
:1. Introduction
2. Hepatitis B Virus Therapies Under Development
3. Gene-Based Therapies to Target Covalently Closed Circular DNA
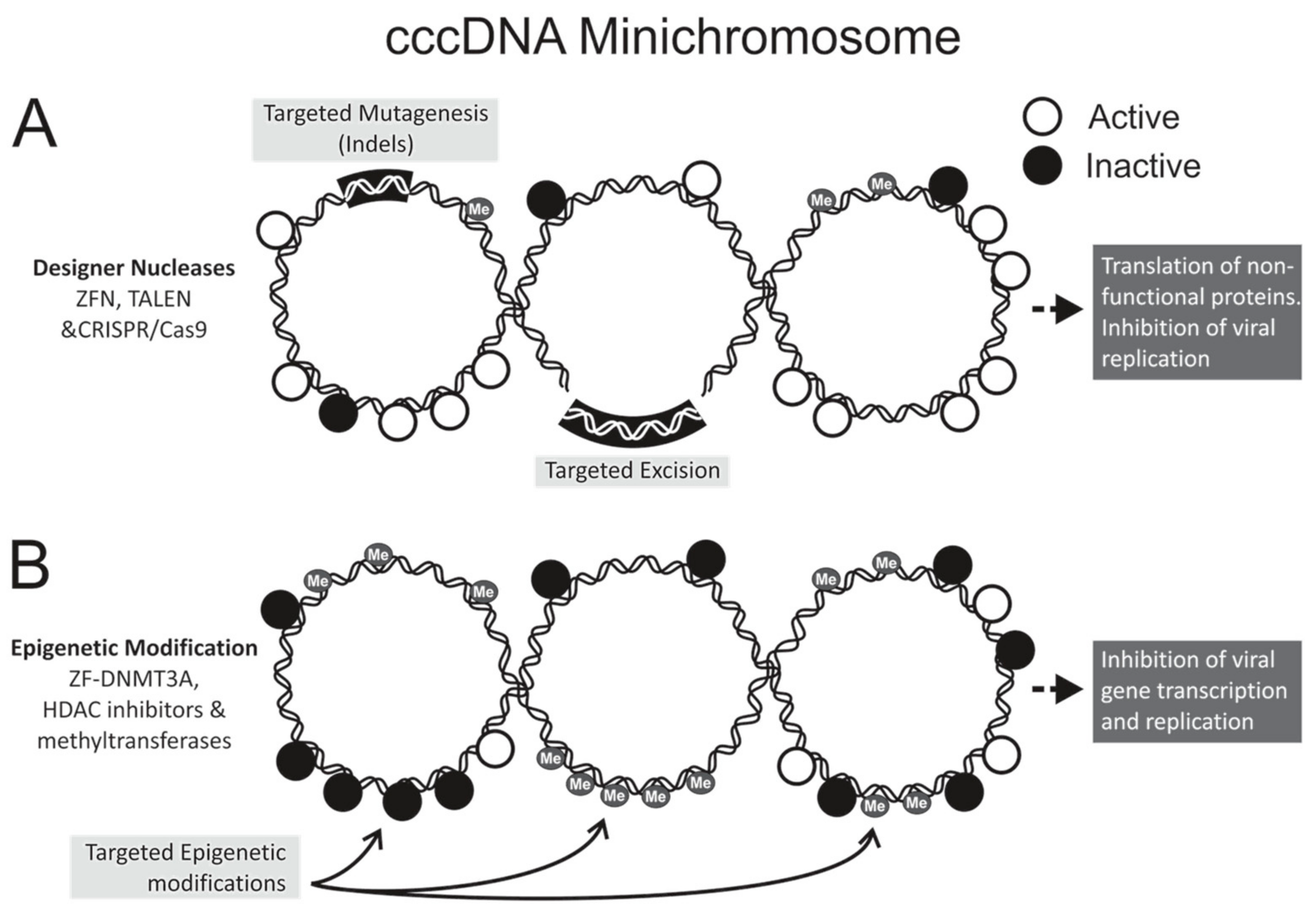
3.1. Designer Nucleases
3.2. Epigenetic Gene Silencing
3.3. Immune Modulation for Covalently Closed Circular DNA Attenuation
4. Discussion/Perspective
Acknowledgments
Author Contributions
Conflicts of Interest
References
- WHO. Global Hepatitis Report. 2017, pp. 1–83. Available online: http://www.who.int/hepatitis/ publications/global-hepatitis-report2017/en/ (accessed on 6 January 2018).
- Blumberg, B.S.; Alter, H.J.; Visnich, S. A “new” antigen in leukemia sera. JAMA 1965, 191, 541–546. [Google Scholar] [CrossRef] [PubMed]
- Terrault, N.A.; Bzowej, N.H.; Chang, K.M.; Hwang, J.P.; Jonas, M.M.; Murad, M.H. Aasld guidelines for treatment of chronic hepatitis B. Hepatology 2016, 63, 261–283. [Google Scholar] [CrossRef] [PubMed]
- EASL. 2017 clinical practice guidelines on the management of hepatitis B virus infection. J. Hepatol. 2017, 67, 370–398. [Google Scholar]
- Sarin, S.K.; Kumar, M.; Lau, G.K.; Abbas, Z.; Chan, H.L.; Chen, C.J.; Chen, D.S.; Chen, H.L.; Chen, P.J.; Chien, R.N.; et al. Asian-pacific clinical practice guidelines on the management of hepatitis B: A 2015 update. Hepatol. Int. 2016, 10, 1–98. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Ning, Q. Toward a cure for hepatitis B virus infection: Combination therapy involving viral suppression and immune modulation and long-term outcome. J. Infect. Dis. 2017, 216, S771–S777. [Google Scholar] [CrossRef] [PubMed]
- Hagiwara, S.; Nishida, N.; Kudo, M. Antiviral therapy for chronic hepatitis B: Combination of nucleoside analogs and interferon. World J. Hepatol. 2015, 7, 2427–2431. [Google Scholar] [CrossRef] [PubMed]
- Tuttleman, J.S.; Pourcel, C.; Summers, J. Formation of the pool of covalently closed circular viral DNA in hepadnavirus-infected cells. Cell 1986, 47, 451–460. [Google Scholar] [CrossRef]
- Newbold, J.E.; Xin, H.; Tencza, M.; Sherman, G.; Dean, J.; Bowden, S.; Locarnini, S. The covalently closed duplex form of the hepadnavirus genome exists in situ as a heterogeneous population of viral minichromosomes. J. Virol. 1995, 69, 3350–3357. [Google Scholar] [PubMed]
- Bock, C.T.; Schranz, P.; Schroder, C.H.; Zentgraf, H. Hepatitis B virus genome is organized into nucleosomes in the nucleus of the infected cell. Virus Genes 1994, 8, 215–229. [Google Scholar] [CrossRef] [PubMed]
- Pollicino, T.; Belloni, L.; Raffa, G.; Pediconi, N.; Squadrito, G.; Raimondo, G.; Levrero, M. Hepatitis B virus replication is regulated by the acetylation status of hepatitis B virus cccDNA-bound H3 and H4 histones. Gastroenterology 2006, 130, 823–837. [Google Scholar] [CrossRef] [PubMed]
- Belloni, L.; Pollicino, T.; De Nicola, F.; Guerrieri, F.; Raffa, G.; Fanciulli, M.; Raimondo, G.; Levrero, M. Nuclear HBx binds the HBV minichromosome and modifies the epigenetic regulation of cccDNA function. Proc. Natl. Acad. Sci. USA 2009, 106, 19975–19979. [Google Scholar] [CrossRef] [PubMed]
- Tropberger, P.; Mercier, A.; Robinson, M.; Zhong, W.; Ganem, D.E.; Holdorf, M. Mapping of histone modifications in episomal HBV cccDNA uncovers an unusual chromatin organization amenable to epigenetic manipulation. Proc. Natl. Acad. Sci. USA 2015, 112, E5715–5724. [Google Scholar] [CrossRef] [PubMed]
- Decorsiere, A.; Mueller, H.; van Breugel, P.C.; Abdul, F.; Gerossier, L.; Beran, R.K.; Livingston, C.M.; Niu, C.; Fletcher, S.P.; Hantz, O.; et al. Hepatitis B virus X protein identifies the Smc5/6 complex as a host restriction factor. Nature 2016, 531, 386–389. [Google Scholar] [CrossRef] [PubMed]
- Durantel, D.; Zoulim, F. New antiviral targets for innovative treatment concepts for hepatitis B virus and hepatitis delta virus. J. Hepatol. 2016, 64, S117–S131. [Google Scholar] [CrossRef] [PubMed]
- Brahmania, M.; Feld, J.; Arif, A.; Janssen, H.L. New therapeutic agents for chronic hepatitis B. Lancet. Infect. Dis. 2016, 16, e10–e21. [Google Scholar] [CrossRef]
- Cole, A.G. Modulators of HBV capsid assembly as an approach to treating hepatitis B virus infection. Curr. Opin. Pharmacol. 2016, 30, 131–137. [Google Scholar] [CrossRef] [PubMed]
- Zlotnick, A.; Venkatakrishnan, B.; Tan, Z.; Lewellyn, E.; Turner, W.; Francis, S. Core protein: A pleiotropic keystone in the HBV lifecycle. Antiviral Res. 2015, 121, 82–93. [Google Scholar] [CrossRef] [PubMed]
- Rossignol, J.F.; Keeffe, E.B. Thiazolides: A new class of drugs for the treatment of chronic hepatitis B and C. Future Microbiol. 2008, 3, 539–545. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.; Goddard, C.; Clearfield, E.; Mills, C.; Xiao, T.; Guo, H.; Morrey, J.D.; Motter, N.E.; Zhao, K.; Block, T.M.; et al. Design, synthesis, and biological evaluation of triazolo-pyrimidine derivatives as novel inhibitors of hepatitis B virus surface antigen (HBsAg) secretion. J. Med. Chem. 2011, 54, 5660–5670. [Google Scholar] [CrossRef] [PubMed]
- Vaillant, A. Nucleic acid polymers: Broad spectrum antiviral activity, antiviral mechanisms and optimization for the treatment of hepatitis B and hepatitis D infection. Antiviral Res. 2016, 133, 32–40. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Zhong, G.; Xu, G.; He, W.; Jing, Z.; Gao, Z.; Huang, Y.; Qi, Y.; Peng, B.; Wang, H.; et al. Sodium taurocholate cotransporting polypeptide is a functional receptor for human hepatitis B and D virus. Elife 2012, 3, e05570. [Google Scholar] [CrossRef] [PubMed]
- Volz, T.; Allweiss, L.; Ben, M.M.; Warlich, M.; Lohse, A.W.; Pollok, J.M.; Alexandrov, A.; Urban, S.; Petersen, J.; Lutgehetmann, M.; et al. The entry inhibitor myrcludex-B efficiently blocks intrahepatic virus spreading in humanized mice previously infected with hepatitis B virus. J. Hepatol. 2013, 58, 861–867. [Google Scholar] [CrossRef] [PubMed]
- Maepa, M.B.; Roelofse, I.; Ely, A.; Arbuthnot, P. Progress and prospects of Anti-HBV gene therapy development. Int. J. Mol. Sci 2015, 16, 17589–17610. [Google Scholar] [CrossRef] [PubMed]
- Ivacik, D.; Ely, A.; Arbuthnot, P. Countering hepatitis B virus infection using RNAi: How far are we from the clinic? Rev. Med. Virol. 2011, 21, 383–396. [Google Scholar] [CrossRef] [PubMed]
- Carmona, S.; Ely, A.; Crowther, C.; Moolla, N.; Salazar, F.H.; Marion, P.L.; Ferry, N.; Weinberg, M.S.; Arbuthnot, P. Effective inhibition of HBV replication in vivo by anti-HBx short hairpin RNAs. Mol. Ther. 2006, 13, 411–421. [Google Scholar] [CrossRef] [PubMed]
- Ely, A.; Naidoo, T.; Arbuthnot, P. Efficient silencing of gene expression with modular trimeric Pol II expression cassettes comprising microRNA shuttles. Nucleic Acids Res. 2009, 37, e91. [Google Scholar] [CrossRef] [PubMed]
- Marimani, M.D.; Ely, A.; Buff, M.C.; Bernhardt, S.; Engels, J.W.; Arbuthnot, P. Inhibition of hepatitis B virus replication in cultured cells and in vivo using 2’-O-guanidinopropyl modified siRNAs. Bioorg. Med. Chem. 2013, 21, 6145–6155. [Google Scholar] [CrossRef] [PubMed]
- Grimm, D.; Streetz, K.L.; Jopling, C.L.; Storm, T.A.; Pandey, K.; Davis, C.R.; Marion, P.; Salazar, F.; Kay, M.A. Fatality in mice due to oversaturation of cellular microRNA/short hairpin RNA pathways. Nature 2006, 441, 537–541. [Google Scholar] [CrossRef] [PubMed]
- Maepa, M.B.; Ely, A.; Grayson, W.; Arbuthnot, P. Sustained inhibition of HBV replication in vivo after systemic injection of AAVs encoding artificial antiviral primary microRNAs. Mol. Ther. Nucleic Acids 2017, 7, 190–199. [Google Scholar] [CrossRef] [PubMed]
- Koniger, C.; Wingert, I.; Marsmann, M.; Rosler, C.; Beck, J.; Nassal, M. Involvement of the host DNA-repair enzyme TDP2 in formation of the covalently closed circular DNA persistence reservoir of hepatitis B viruses. Proc. Natl. Acad. Sci. USA 2014, 111, E4244–4253. [Google Scholar] [CrossRef] [PubMed]
- Kinoshita, W.; Ogura, N.; Watashi, K.; Wakita, T. Host factor PRPF31 is involved in cccDNA production in HBV-replicating cells. Biochem. Biophys. Res. Commun. 2017, 482, 638–644. [Google Scholar] [CrossRef] [PubMed]
- Ruiz de Galarreta, M.; Lujambio, A. Therapeutic editing of hepatocyte genome in vivo. J. Hepatol. 2017, 67, 818–828. [Google Scholar] [CrossRef] [PubMed]
- Moyo, B.; Bloom, K.; Scott, T.; Ely, A.; Arbuthnot, P. Advances with using CRISPR/Cas-mediated gene editing to treat infections with hepatitis B virus and hepatitis C virus. Virus Res. 2018, 244, 311–320. [Google Scholar] [CrossRef] [PubMed]
- Locarnini, S.; Zoulim, F. Molecular genetics of HBV infection. Antivir. Ther. 2010, 15 (Suppl. S3), 3–14. [Google Scholar] [CrossRef] [PubMed]
- Beerli, R.R.; Barbas, C.F., 3rd. Engineering polydactyl zinc-finger transcription factors. Nat. Biotechnol. 2002, 20, 135–141. [Google Scholar] [CrossRef] [PubMed]
- Bitinaite, J.; Wah, D.A.; Aggarwal, A.K.; Schildkraut, I. Foki dimerization is required for DNA cleavage. Proc. Natl. Acad. Sci. USA 1998, 95, 10570–10575. [Google Scholar] [CrossRef] [PubMed]
- Cradick, T.J.; Keck, K.; Bradshaw, S.; Jamieson, A.C.; McCaffrey, A.P. Zinc-finger nucleases as a novel therapeutic strategy for targeting hepatitis B virus DNAs. Mol. Ther. 2010, 18, 947–954. [Google Scholar] [CrossRef] [PubMed]
- Weber, N.D.; Stone, D.; Sedlak, R.H.; De Silva Feelixge, H.S.; Roychoudhury, P.; Schiffer, J.T.; Aubert, M.; Jerome, K.R. AAV-mediated delivery of zinc finger nucleases targeting hepatitis B virus inhibits active replication. PLoS ONE 2014, 9, e97579. [Google Scholar] [CrossRef] [PubMed]
- Ladner, S.K.; Otto, M.J.; Barker, C.S.; Zaifert, K.; Wang, G.H.; Guo, J.T.; Seeger, C.; King, R.W. Inducible expression of human hepatitis B virus (HBV) in stably transfected hepatoblastoma cells: A novel system for screening potential inhibitors of HBV replication. Antimicrob. Agents Chemother. 1997, 41, 1715–1720. [Google Scholar] [PubMed]
- Boch, J.; Scholze, H.; Schornack, S.; Landgraf, A.; Hahn, S.; Kay, S.; Lahaye, T.; Nickstadt, A.; Bonas, U. Breaking the code of DNA binding specificity of TAL-type III effectors. Science 2009, 326, 1509–1512. [Google Scholar] [CrossRef] [PubMed]
- Moscou, M.J.; Bogdanove, A.J. A simple cipher governs DNA recognition by TAL effectors. Science 2009, 326, 1501. [Google Scholar] [CrossRef] [PubMed]
- Deng, D.; Yan, C.; Pan, X.; Mahfouz, M.; Wang, J.; Zhu, J.K.; Shi, Y.; Yan, N. Structural basis for sequence-specific recognition of DNA by TAL effectors. Science 2012, 335, 720–723. [Google Scholar] [CrossRef] [PubMed]
- Mak, A.N.; Bradley, P.; Cernadas, R.A.; Bogdanove, A.J.; Stoddard, B.L. The crystal structure of TAL effector PthXo1 bound to its DNA target. Science 2012, 335, 716–719. [Google Scholar] [CrossRef] [PubMed]
- Bloom, K.; Ely, A.; Mussolino, C.; Cathomen, T.; Arbuthnot, P. Inactivation of hepatitis B virus replication in cultured cells and in vivo with engineered transcription activator-like effector nucleases. Mol. Ther. 2013, 21, 1889–1897. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Zhang, W.; Lin, J.; Wang, F.; Wu, M.; Chen, C.; Zheng, Y.; Peng, X.; Li, J.; Yuan, Z. An efficient antiviral strategy for targeting hepatitis B virus genome using transcription activator-like effector nucleases. Mol. Ther. 2014, 22, 303–311. [Google Scholar] [CrossRef] [PubMed]
- Dreyer, T.; Nicholson, S.; Ely, A.; Arbuthnot, P.; Bloom, K. Improved antiviral efficacy using TALEN-mediated homology directed recombination to introduce artificial primary mirnas into DNA of hepatitis B virus. Biochem. Biophys. Res. Commun. 2016, 478, 1563–1568. [Google Scholar] [CrossRef] [PubMed]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef] [PubMed]
- Seeger, C.; Sohn, J.A. Targeting hepatitis B virus with CRISPR/Cas9. Mol. Ther. Nucleic Acids 2014, 3, e216. [Google Scholar] [CrossRef] [PubMed]
- Dong, C.; Qu, L.; Wang, H.; Wei, L.; Dong, Y.; Xiong, S. Targeting hepatitis B virus cccDNA by CRISPR/Cas9 nuclease efficiently inhibits viral replication. Antiviral Res. 2015, 118, 110–117. [Google Scholar] [CrossRef] [PubMed]
- Karimova, M.; Beschorner, N.; Dammermann, W.; Chemnitz, J.; Indenbirken, D.; Bockmann, J.H.; Grundhoff, A.; Luth, S.; Buchholz, F.; Schulze zur Wiesch, J.; et al. Crispr/cas9 nickase-mediated disruption of hepatitis B virus open reading frame S and X. Sci. Rep. 2015, 5, 13734. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, E.M.; Bassit, L.C.; Mueller, H.; Kornepati, A.V.; Bogerd, H.P.; Nie, T.; Chatterjee, P.; Javanbakht, H.; Schinazi, R.F.; Cullen, B.R. Suppression of hepatitis B virus DNA accumulation in chronically infected cells using a bacterial CRISP/Cas RNA-guided DNA endonuclease. Virology 2015, 476, 196–205. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Hao, R.; Chen, S.; Guo, D.; Chen, Y. Inhibition of hepatitis B virus by the CRISPR/Cas9 system via targeting the conserved regions of the viral genome. J. Gen. Virol. 2015, 96, 2252–2261. [Google Scholar] [CrossRef] [PubMed]
- Ramanan, V.; Shlomai, A.; Cox, D.B.; Schwartz, R.E.; Michailidis, E.; Bhatta, A.; Scott, D.A.; Zhang, F.; Rice, C.M.; Bhatia, S.N. CRISPR/Cas9 cleavage of viral DNA efficiently suppresses hepatitis B virus. Sci. Rep. 2015, 5, 10833. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Xu, Z.W.; Liu, S.; Zhang, R.Y.; Ding, S.L.; Xie, X.M.; Long, L.; Chen, X.M.; Zhuang, H.; Lu, F.M. Dual gRNAs guided CRISPR/Cas9 system inhibits hepatitis B virus replication. World J. Gastroenterol. 2015, 21, 9554–9565. [Google Scholar] [CrossRef] [PubMed]
- Zhen, S.; Hua, L.; Liu, Y.H.; Gao, L.C.; Fu, J.; Wan, D.Y.; Dong, L.H.; Song, H.F.; Gao, X. Harnessing the clustered regularly interspaced short palindromic repeat (CRISPR)/CRISPR-associated Cas9 system to disrupt the hepatitis B virus. Gene Ther. 2015, 22, 404–412. [Google Scholar] [CrossRef] [PubMed]
- Sakuma, T.; Masaki, K.; Abe-Chayama, H.; Mochida, K.; Yamamoto, T.; Chayama, K. Highly multiplexed CRISPR-Cas9-nuclease and Cas9-nickase vectors for inactivation of hepatitis B virus. Genes Cells 2016, 21, 1253–1262. [Google Scholar] [CrossRef] [PubMed]
- Seeger, C.; Sohn, J.A. Complete spectrum of CRISPR/Cas9-induced mutations on HBV cccDNA. Mol. Ther. 2016, 24, 1258–1266. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Xie, K.; Xu, Y.; Wang, L.; Chen, K.; Zhang, L.; Fang, J. CRISPR/Cas9 produces anti-hepatitis B virus effect in hepatoma cells and transgenic mouse. Virus Res. 2016, 217, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Sheng, C.; Liu, H.; Liu, G.; Du, X.; Du, J.; Zhan, L.; Li, P.; Yang, C.; Qi, L.; et al. An effective molecular target site in hepatitis B virus S gene for Cas9 cleavage and mutational inactivation. Int. J. Biol. Sci. 2016, 12, 1104–1113. [Google Scholar] [CrossRef] [PubMed]
- Jiang, C.; Mei, M.; Li, B.; Zhu, X.; Zu, W.; Tian, Y.; Wang, Q.; Guo, Y.; Dong, Y.; Tan, X. A non-viral CRISPR/Cas9 delivery system for therapeutically targeting HBV DNA and pcsk9 in vivo. Cell Res. 2017, 27, 440–443. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Sheng, C.; Wang, S.; Yang, L.; Liang, Y.; Huang, Y.; Liu, H.; Li, P.; Yang, C.; Yang, X.; et al. Removal of integrated hepatitis B virus DNA using CRISPR-Cas9. Front. Cell. Infect. Microbiol. 2017, 7, 91. [Google Scholar] [CrossRef] [PubMed]
- Scott, T.; Moyo, B.; Nicholson, S.; Maepa, M.B.; Watashi, K.; Ely, A.; Weinberg, M.S.; Arbuthnot, P. ssAAVs containing cassettes encoding SaCas9 and guides targeting hepatitis B virus inactivate replication of the virus in cultured cells. Sci. Rep. 2017, 7, 7401. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Chen, R.; Zhang, R.; Ding, S.; Zhang, T.; Yuan, Q.; Guan, G.; Chen, X.; Zhang, T.; Zhuang, H.; et al. The gRNA-miRNA-gRNA ternary cassette combining CRISPR/Cas9 with RNAi approach strongly inhibits hepatitis B virus replication. Theranostics 2017, 7, 3090–3105. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Q.; Bai, L.; Zheng, S.; Liu, M.; Zhang, J.; Wang, T.; Xu, Z.; Chen, Y.; Li, J.; Duan, Z. Efficient inhibition of duck hepatitis B virus DNA by the CRISPR/Cas9 system. Mol Med Rep 2017, 16, 7199–7204. [Google Scholar] [CrossRef] [PubMed]
- Kuscu, C.; Arslan, S.; Singh, R.; Thorpe, J.; Adli, M. Genome-wide analysis reveals characteristics of off-target sites bound by the Cas9 endonuclease. Nat. Biotechnol. 2014, 32, 677–683. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Cradick, T.J.; Brown, M.T.; Deshmukh, H.; Ranjan, P.; Sarode, N.; Wile, B.M.; Vertino, P.M.; Stewart, F.J.; Bao, G. CRISPR/Cas9 systems have off-target activity with insertions or deletions between target DNA and guide RNA sequences. Nucleic Acids Res. 2014, 42, 7473–7485. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Foden, J.A.; Khayter, C.; Maeder, M.L.; Reyon, D.; Joung, J.K.; Sander, J.D. High frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat. Biotechnol. 2013, 31, 822–826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaissiere, T.; Sawan, C.; Herceg, Z. Epigenetic interplay between histone modifications and DNA methylation in gene silencing. Mutat. Res. 2008, 659, 40–48. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Chen, J.; Wu, M.; Zhang, X.; Zhang, M.; Yue, L.; Li, Y.; Liu, J.; Li, B.; Shen, F.; et al. PRMT5 restricts hepatitis B virus replication through epigenetic repression of covalently closed circular DNA transcription and interference with pregenomic RNA encapsidation. Hepatology 2017, 66, 398–415. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.H.; Li, Y.N.; Zhao, J.R.; Zhang, J.; Yan, Z. HBc binds to the CpG islands of HBV cccDNA and promotes an epigenetic permissive state. Epigenetics 2011, 6, 720–726. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Mao, R.; Yan, R.; Cai, D.; Zhang, Y.; Zhu, H.; Kang, Y.; Liu, H.; Wang, J.; Qin, Y.; et al. Transcription of hepatitis B virus covalently closed circular DNA is regulated by CpG methylation during chronic infection. PLoS ONE 2014, 9, e110442. [Google Scholar] [CrossRef] [PubMed]
- Koumbi, L.; Karayiannis, P. The epigenetic control of hepatitis B virus modulates the outcome of infection. Front. Microbiol. 2015, 6, 1491. [Google Scholar] [CrossRef] [PubMed]
- Hong, X.; Kim, E.S.; Guo, H. Epigenetic regulation of hepatitis B virus covalently closed circular DNA: Implications for epigenetic therapy against chronic hepatitis B. Hepatology 2017, 66, 2066–2077. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, M.; Deng, L.; Chen, M.; Gan, X.; Shinozaki, K.; Shoji, I.; Hotta, H. Interaction of the hepatitis B virus X protein with the lysine methyltransferase SET and MYND domain-containing 3 induces activator protein 1 activation. Microbiol. Immunol. 2016, 60, 17–25. [Google Scholar] [CrossRef] [PubMed]
- Lubyova, B.; Hodek, J.; Zabransky, A.; Prouzova, H.; Hubalek, M.; Hirsch, I.; Weber, J. PRMT5: A novel regulator of hepatitis B virus replication and an arginine methylase of HBV core. PLoS ONE 2017, 12, e0186982. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Hu, Y.; Shen, X.; Lao, Y.; Zhang, L.; Qiu, X.; Hu, J.; Gong, P.; Cui, H.; Lu, S.; et al. HBx represses RIZ1 expression by DNA methyltransferase 1 involvement in decreased miR-152 in hepatocellular carcinoma. Oncol. Rep. 2017, 37, 2811–2818. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.R.; Yang, H.C.; Kuo, Y.T.; Liu, C.J.; Yang, T.Y.; Sung, K.C.; Lin, Y.Y.; Wang, H.Y.; Wang, C.C.; Shen, Y.C.; et al. The CRISPR/Cas9 system facilitates clearance of the intrahepatic HBV templates in vivo. Mol. Ther. Nucleic Acids 2014, 3, e186. [Google Scholar] [CrossRef] [PubMed]
- Xirong, L.; Rui, L.; Xiaoli, Y.; Qiuyan, H.; Bikui, T.; Sibo, Z.; Naishuo, Z. Hepatitis B virus can be inhibited by DNA methyltransferase 3a via specific zinc-finger-induced methylation of the X promoter. Biochemistry 2014, 79, 111–123. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Zhao, Z.; Guo, J.; Huang, P.; Zhu, X.; Zhou, X.; Yang, Z.; Zhao, L.; Xu, L.; Xu, J.; et al. Creation of a six-fingered artificial transcription factor that represses the hepatitis B virus HBx gene integrated into a human hepatocellular carcinoma cell line. J. Biomol. Screen. 2013, 18, 378–387. [Google Scholar] [CrossRef] [PubMed]
- Moyo, B.; Nicholson, S.; Roelofse, I.; Crowther, C.; Bloom, K.; Mussolino, C.; Cathomen, T.; Watashi, K.; Ely, A.; Arbuthnot, P. 483. Epigenetic silencing of hepatitis B cccDNA in vitro and in vivo using AAV-delivered engineered repressor transcription activator-like effector. Mol. Ther. 2015, 23, S192. [Google Scholar] [CrossRef]
- Liu, F.; Campagna, M.; Qi, Y.; Zhao, X.; Guo, F.; Xu, C.; Li, S.; Li, W.; Block, T.M.; Chang, J.; et al. α-interferon suppresses hepadnavirus transcription by altering epigenetic modification of cccDNA minichromosomes. PLoS Pathog. 2013, 9, e1003613. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Zhao, X.; Zang, L.; Fang, X.; Zhao, J.; Yang, X.; Wang, Q.; Zheng, L.; Chang, J. Anti-hepatitis B virus activities of α-DDB-FNC, a novel nucleoside-biphenyldicarboxylate compound in cells and ducks, and its anti-immunological liver injury effect in mice. Antiviral Res. 2012, 96, 333–339. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Li, C.; Zhang, Y.; Zhu, H.; Kang, Y.; Liu, H.; Wang, J.; Qin, Y.; Mao, R.; Xie, Y.; et al. Comparative analysis of CpG islands among HBV genotypes. PLoS ONE 2013, 8, e56711. [Google Scholar] [CrossRef] [PubMed]
- Vivekanandan, P.; Thomas, D.; Torbenson, M. Hepatitis B viral DNA is methylated in liver tissues. J. Viral Hepat. 2008, 15, 103–107. [Google Scholar] [CrossRef] [PubMed]
- Jain, S.; Chang, T.T.; Chen, S.; Boldbaatar, B.; Clemens, A.; Lin, S.Y.; Yan, R.; Hu, C.T.; Guo, H.; Block, T.M.; et al. Comprehensive DNA methylation analysis of hepatitis B virus genome in infected liver tissues. Sci. Rep. 2015, 5, 10478. [Google Scholar] [CrossRef] [PubMed]
- Vivekanandan, P.; Kannangai, R.; Ray, S.C.; Thomas, D.L.; Torbenson, M. Comprehensive genetic and epigenetic analysis of occult hepatitis B from liver tissue samples. Clin. Infect. Dis. 2008, 46, 1227–1236. [Google Scholar] [CrossRef] [PubMed]
- Kaur, P.; Paliwal, A.; Durantel, D.; Hainaut, P.; Scoazec, J.Y.; Zoulim, F.; Chemin, I.; Herceg, Z. DNA methylation of hepatitis B virus (HBV) genome associated with the development of hepatocellular carcinoma and occult HBV infection. J. Infect. Dis. 2010, 202, 700–704. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Li, Y.; Mu, S.; Zhang, J.; Yan, Z. Evidence that methylation of hepatitis B virus covalently closed circular DNA in liver tissues of patients with chronic hepatitis B modulates HBV replication. J. Med. Virol. 2009, 81, 1177–1183. [Google Scholar] [CrossRef] [PubMed]
- Thakore, P.I.; Black, J.B.; Hilton, I.B.; Gersbach, C.A. Editing the epigenome: Technologies for programmable transcriptional modulation and epigenetic regulation. Nature methods 2016, 13, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Archin, N.M.; Keedy, K.S.; Espeseth, A.; Dang, H.; Hazuda, D.J.; Margolis, D.M. Expression of latent human immunodeficiency type 1 is induced by novel and selective histone deacetylase inhibitors. AIDS 2009, 23, 1799–1806. [Google Scholar] [CrossRef] [PubMed]
- West, A.C.; Johnstone, R.W. New and emerging HDAC inhibitors for cancer treatment. J. Clin. Investig. 2014, 124, 30–39. [Google Scholar] [CrossRef] [PubMed]
- Aurisicchio, L.; Delmastro, P.; Salucci, V.; Paz, O.G.; Rovere, P.; Ciliberto, G.; La Monica, N.; Palombo, F. Liver-specific alpha 2 interferon gene expression results in protection from induced hepatitis. J. Virol. 2000, 74, 4816–4823. [Google Scholar] [CrossRef] [PubMed]
- Fiedler, M.; Rodicker, F.; Salucci, V.; Lu, M.; Aurisicchio, L.; Dahmen, U.; Jun, L.; Dirsch, O.; Putzer, B.M.; Palombo, F.; et al. Helper-dependent adenoviral vector-mediated delivery of woodchuck-specific genes for alpha interferon (IFN-α) and IFN-γ: IFN-α but not IFN-γ reduces woodchuck hepatitis virus replication in chronic infection in vivo. J. Virol. 2004, 78, 10111–10121. [Google Scholar] [CrossRef] [PubMed]
- Dumortier, J.; Schonig, K.; Oberwinkler, H.; Low, R.; Giese, T.; Bujard, H.; Schirmacher, P.; Protzer, U. Liver-specific expression of interferon γ following adenoviral gene transfer controls hepatitis b virus replication in mice. Gene Ther. 2005, 12, 668–677. [Google Scholar] [CrossRef] [PubMed]
- Berraondo, P.; Ochoa, L.; Crettaz, J.; Rotellar, F.; Vales, A.; Martinez-Anso, E.; Zaratiegui, M.; Ruiz, J.; Gonzalez-Aseguinolaza, G.; Prieto, J. IFN-α gene therapy for woodchuck hepatitis with adeno-associated virus: Differences in duration of gene expression and antiviral activity using intraportal or intramuscular routes. Mol. Ther. 2005, 12, 68–76. [Google Scholar] [CrossRef] [PubMed]
- Berraondo, P.; Di Scala, M.; Korolowicz, K.; Thampi, L.M.; Otano, I.; Suarez, L.; Fioravanti, J.; Aranda, F.; Ardaiz, N.; Yang, J.; et al. Liver-directed gene therapy of chronic hepadnavirus infection using interferon alpha tethered to apolipoprotein A-I. J. Hepatol. 2015, 63, 329–336. [Google Scholar] [CrossRef] [PubMed]
- Crettaz, J.; Otano, I.; Ochoa-Callejero, L.; Benito, A.; Paneda, A.; Aurrekoetxea, I.; Berraondo, P.; Rodriguez-Madoz, J.R.; Astudillo, A.; Kreppel, F.; et al. Treatment of chronic viral hepatitis in woodchucks by prolonged intrahepatic expression of interleukin-12. J. Virol. 2009, 83, 2663–2674. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Madoz, J.R.; Liu, K.H.; Quetglas, J.I.; Ruiz-Guillen, M.; Otano, I.; Crettaz, J.; Butler, S.D.; Bellezza, C.A.; Dykes, N.L.; Tennant, B.C.; et al. Semliki forest virus expressing interleukin-12 induces antiviral and antitumoral responses in woodchucks with chronic viral hepatitis and hepatocellular carcinoma. J. Virol. 2009, 83, 12266–12278. [Google Scholar] [CrossRef] [PubMed]
- Di Scala, M.; Otano, I.; Gil-Farina, I.; Vanrell, L.; Hommel, M.; Olague, C.; Vales, A.; Galarraga, M.; Guembe, L.; Ortiz de Solorzano, C.; et al. Complementary effects of interleukin-15 and alpha interferon induce immunity in hepatitis b virus transgenic mice. J. Virol. 2016, 90, 8563–8574. [Google Scholar] [CrossRef] [PubMed]
- Bohne, F.; Chmielewski, M.; Ebert, G.; Wiegmann, K.; Kurschner, T.; Schulze, A.; Urban, S.; Kronke, M.; Abken, H.; Protzer, U. T cells redirected against hepatitis B virus surface proteins eliminate infected hepatocytes. Gastroenterology 2008, 134, 239–247. [Google Scholar] [CrossRef] [PubMed]
- Gehring, A.J.; Xue, S.A.; Ho, Z.Z.; Teoh, D.; Ruedl, C.; Chia, A.; Koh, S.; Lim, S.G.; Maini, M.K.; Stauss, H.; et al. Engineering virus-specific T cells that target HBV infected hepatocytes and hepatocellular carcinoma cell lines. J. Hepatol. 2011, 55, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Kah, J.; Koh, S.; Volz, T.; Ceccarello, E.; Allweiss, L.; Lutgehetmann, M.; Bertoletti, A.; Dandri, M. Lymphocytes transiently expressing virus-specific T cell receptors reduce hepatitis B virus infection. J. Clin. Investig. 2017, 127, 3177–3188. [Google Scholar] [CrossRef] [PubMed]
- Krebs, K.; Bottinger, N.; Huang, L.R.; Chmielewski, M.; Arzberger, S.; Gasteiger, G.; Jager, C.; Schmitt, E.; Bohne, F.; Aichler, M.; et al. T cells expressing a chimeric antigen receptor that binds hepatitis B virus envelope proteins control virus replication in mice. Gastroenterology 2013, 145, 456–465. [Google Scholar] [CrossRef] [PubMed]
- Wisskirchen, K.; Metzger, K.; Schreiber, S.; Asen, T.; Weigand, L.; Dargel, C.; Witter, K.; Kieback, E.; Sprinzl, M.F.; Uckert, W.; et al. Isolation and functional characterization of hepatitis B virus-specific T-cell receptors as new tools for experimental and clinical use. PLoS ONE 2017, 12, e0182936. [Google Scholar] [CrossRef] [PubMed]
- Kosinska, A.D.; Liu, J.; Lu, M.; Roggendorf, M. Therapeutic vaccination and immunomodulation in the treatment of chronic hepatitis B: Preclinical studies in the woodchuck. Med. Microbiol. Immunol. 2015, 204, 103–114. [Google Scholar] [CrossRef] [PubMed]
- Cornu, T.I.; Mussolino, C.; Cathomen, T. Refining strategies to translate genome editing to the clinic. Nat. Med. 2017, 23, 415–423. [Google Scholar] [CrossRef] [PubMed]
- Hardee, L.C.; Arévalo-Soliz, M.L.; Hornstein, D.B.; Zechiedrich, L. Advances in non-viral DNA vectors for gene therapy. Genes 2017, 8, 65. [Google Scholar] [CrossRef] [PubMed]
- Baruteau, J.; Waddington, S.N.; Alexander, I.E.; Gissen, P. Delivering efficient liver-directed AAV-mediated gene therapy. Gene Ther. 2017, 24, 263. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.M.; Cradick, T.J.; Fine, E.J.; Bao, G. Nuclease target site selection for maximizing on-target activity and minimizing off-target effects in genome editing. Mol. Ther. 2016, 24, 475–487. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| ZFN | TALEN | CRISPR/Cas | ||
|---|---|---|---|---|
| DNA binding domain |
|
|
| |
| Nuclease domain |
|
|
| |
| Advantages |
|
|
| |
| Disadvantages |
|
|
| |
| HBV model systems |
| |||
| cccDNA | Cleavage (%) |
| ||
| Reduction (%) |
|
| ||
| Alternative effector domain |
| |||
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bloom, K.; Maepa, M.B.; Ely, A.; Arbuthnot, P. Gene Therapy for Chronic HBV—Can We Eliminate cccDNA? Genes 2018, 9, 207. https://doi.org/10.3390/genes9040207
Bloom K, Maepa MB, Ely A, Arbuthnot P. Gene Therapy for Chronic HBV—Can We Eliminate cccDNA? Genes. 2018; 9(4):207. https://doi.org/10.3390/genes9040207
Chicago/Turabian StyleBloom, Kristie, Mohube Betty Maepa, Abdullah Ely, and Patrick Arbuthnot. 2018. "Gene Therapy for Chronic HBV—Can We Eliminate cccDNA?" Genes 9, no. 4: 207. https://doi.org/10.3390/genes9040207
APA StyleBloom, K., Maepa, M. B., Ely, A., & Arbuthnot, P. (2018). Gene Therapy for Chronic HBV—Can We Eliminate cccDNA? Genes, 9(4), 207. https://doi.org/10.3390/genes9040207