Identifying Patients with Atrioventricular Septal Defect in Down Syndrome Populations by Using Self-Normalizing Neural Networks and Feature Selection

 ,
,  and
and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Dataset
2.2. Feature Analysis
2.2.1. Monte Carlo Feature Selection Method
2.2.2. Incremental Feature Selection Method
2.2.3. Rule Extraction
2.3. Classification Algorithm
2.3.1. Self-Normalizing Neural Network Algorithm
2.3.2. Random Forest Algorithm
2.4. Performance Measurements
3. Results
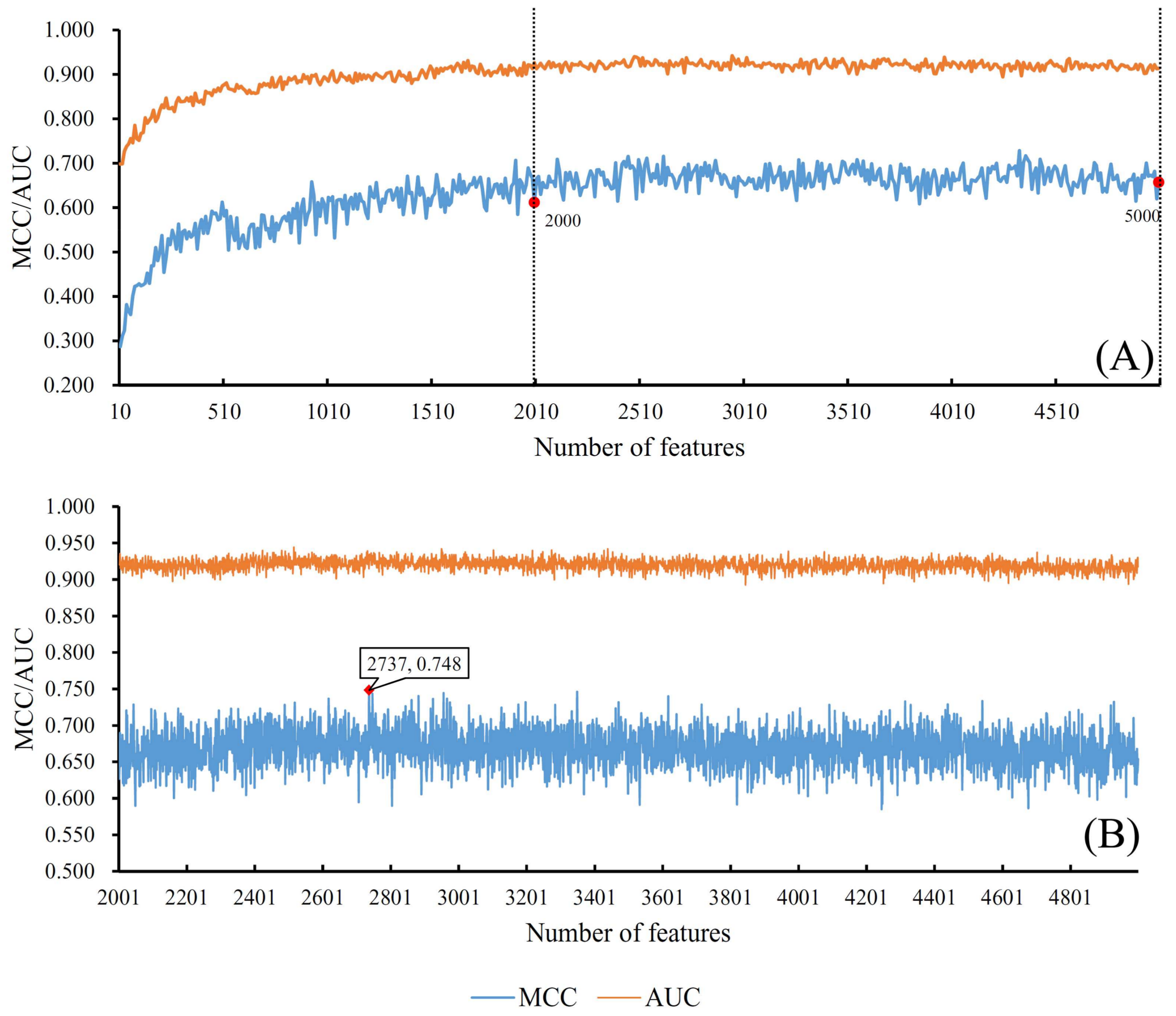
3.1. Results from Self-Normalizing Neural Network Algorithm
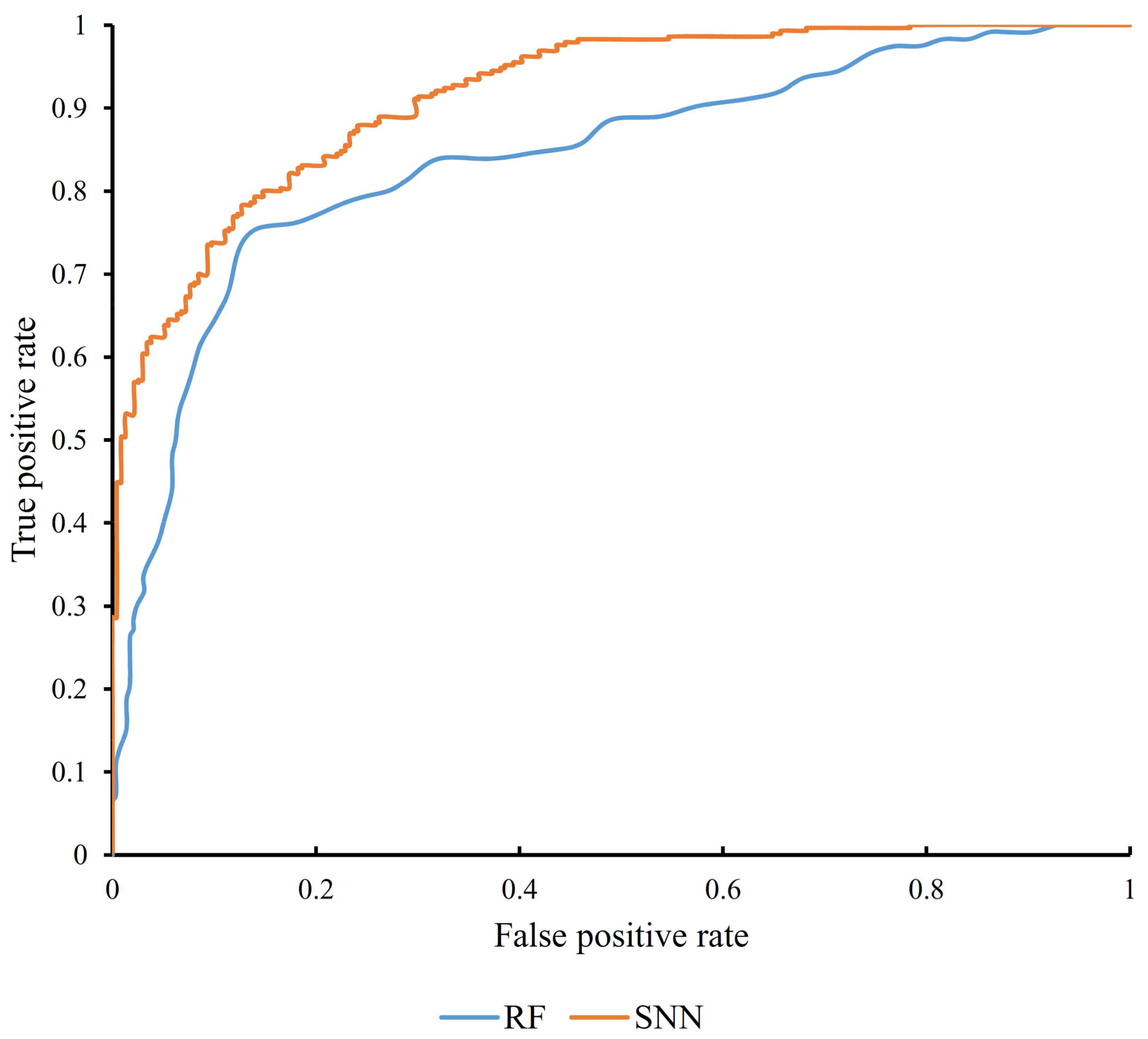
3.2. Results from Random Forest Algorithm
3.3. Decision Rules
4. Discussion
4.1. Why Use Self-Normalizing Neural Network as the Classifier
4.2. Optimal Genes Associated with Atrioventricular Septal Defect in Patients with Down Syndrome
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Lara, D.A.; Lopez, K.N. Public health research in congenital heart disease. Congenit. Heart Dis. 2014, 9, 549–558. [Google Scholar] [CrossRef] [PubMed]
- Ross, H.J.; Law, Y.; Book, W.M.; Broberg, C.S.; Burchill, L.; Cecchin, F.; Chen, J.M.; Delgado, D.; Dimopoulos, K.; Everitt, M.D.; et al. Transplantation and mechanical circulatory support in congenital heart disease: A scientific statement from the American heart association. Circulation 2016, 133, 802–820. [Google Scholar] [CrossRef] [PubMed]
- Van Hare, G.F.; Ackerman, M.J.; Evangelista, J.K.; Kovacs, R.J.; Myerburg, R.J.; Shafer, K.M.; Warnes, C.A.; Washington, R.L. Eligibility and disqualification recommendations for competitive athletes with cardiovascular abnormalities: Task force 4: Congenital heart disease: A scientific statement from the American heart association and American college of cardiology. J. Am. Coll. Cardiol. 2015, 66, 2372–2384. [Google Scholar] [CrossRef] [PubMed]
- Bermudez, B.E.; Medeiros, S.L.; Bermudez, M.B.; Novadzki, I.M.; Magdalena, N.I. Down syndrome: Prevalence and distribution of congenital heart disease in brazil. Sao Paulo Med. J. 2015, 133, 521–524. [Google Scholar] [CrossRef] [PubMed]
- Perez-Navero, J.L.; de la Torre-Aguilar, M.J.; Ibarra de la Rosa, I.; Gil-Campos, M.; Gomez-Guzman, E.; Merino-Cejas, C.; Munoz-Villanueva, M.C.; Llorente-Cantarero, F.J. Cardiac biomarkers of low cardiac output syndrome in the postoperative period after congenital heart disease surgery in children. Rev. Esp. Cardiol. 2017, 70, 267–274. [Google Scholar] [CrossRef] [PubMed]
- Scansen, B.A.; Schneider, M.; Bonagura, J.D. Sequential segmental classification of feline congenital heart disease. J. Vet. Cardiol. 2015, 17, S10–S52. [Google Scholar] [CrossRef] [PubMed]
- Cohen, M.S.; Jegatheeswaran, A.; Baffa, J.M.; Gremmels, D.B.; Overman, D.M.; Caldarone, C.A.; McCrindle, B.W.; Mertens, L. Echocardiographic features defining right dominant unbalanced atrioventricular septal defect: A multi-institutional congenital heart surgeons’ society study. Circ. Cardiovasc. Imaging 2013, 6, 508–513. [Google Scholar] [CrossRef] [PubMed]
- Korten, M.A.; Helm, P.C.; Abdul-Khaliq, H.; Baumgartner, H.; Kececioglu, D.; Schlensak, C.; Bauer, U.M.; Diller, G.P.; Competence network for congenital heart defects, I. Eisenmenger syndrome and long-term survival in patients with down syndrome and congenital heart disease. Heart 2016, 102, 1552–1557. [Google Scholar] [PubMed]
- Mourato, F.A.; Villachan, L.R.; Mattos Sda, S. Prevalence and profile of congenital heart disease and pulmonary hypertension in down syndrome in a pediatric cardiology service. Rev. Paul. Pediatr. 2014, 32, 159–163. [Google Scholar] [CrossRef] [PubMed]
- Ko, J.M. Genetic syndromes associated with congenital heart disease. Korean Circ. J. 2015, 45, 357–361. [Google Scholar] [CrossRef] [PubMed]
- Gelb, B.D. Genetic basis of syndromes associated with congenital heart disease. Curr. Opin. Cardiol. 2001, 16, 188–194. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, P.; Bhaumik, P.; Ghosh, S.; Ozbek, U.; Feingold, E.; Maslen, C.; Sarkar, B.; Pramanik, V.; Biswas, P.; Bandyopadhyay, B.; et al. Polymorphic haplotypes of CRELD1 differentially predispose Down syndrome and euploids individuals to atrioventricular septal defect. Am. J. Med. Genet. A 2012, 158A, 2843–2848. [Google Scholar] [CrossRef] [PubMed]
- Alcantara-Ortigoza, M.A.; De Rubens-Figueroa, J.; Reyna-Fabian, M.E.; Estandia-Ortega, B.; Gonzalez-del Angel, A.; Molina-Alvarez, B.; Velazquez-Aragon, J.A.; Villagomez-Martinez, S.; Pereira-Lopez, G.I.; Martinez-Cruz, V.; et al. Germline mutations in NKX2-5, GATA4, and CRELD1 are rare in a Mexican sample of down syndrome patients with endocardial cushion and septal heart defects. Pediatr. Cardiol. 2015, 36, 802–808. [Google Scholar] [CrossRef] [PubMed]
- Mahadevaiah, G.; Gupta, M.; Ashwath, R. Down syndrome with complete atrioventricular septal defect, hypertrophic cardiomyopathy, and pulmonary vein stenosis. Tex. Heart Inst. J. 2015, 42, 458–461. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Li, Z.; Song, X.; Liu, L.; Su, G.; Cui, Y. Bioinformatic analysis of genes and microRNAs associated with atrioventricular septal defect in down syndrome patients. Int. Heart J. 2016, 57, 490–495. [Google Scholar] [CrossRef] [PubMed]
- Rambomartin, B.L.; Mulle, J.G.; Cutler, D.J.; Bean, L.J.H.; Rosser, T.C.; Dooley, K.J.; Cua, C.; Capone, G.; Maslen, C.L.; Reeves, R.H. Analysis of copy number variants on chromosome 21 in down syndrome-associated congenital heart defects. G3 2018, 8, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Clough, E.; Barrett, T. The gene expression omnibus database. Methods Mol. Biol. 2016, 1418, 93–110. [Google Scholar] [PubMed]
- Ostrovnaya, I.; Nanjangud, G.; Olshen, A.B. A classification model for distinguishing copy number variants from cancer-related alterations. BMC Bioinform. 2010, 11, 297. [Google Scholar] [CrossRef] [PubMed]
- Ding, X.; Tsang, S.Y.; Ng, S.K.; Xue, H. Application of machine learning to development of copy number variation-based prediction of cancer risk. Genom. Insights 2014, 7, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Angermueller, C.; Pärnamaa, T.; Parts, L.; Stegle, O. Deep learning for computational biology. Mol. Syst. Biol. 2016, 12, 878. [Google Scholar] [CrossRef] [PubMed]
- LeCun, Y.; Bengio, Y.; Hinton, G. Deep learning. Nature 2015, 521, 436–444. [Google Scholar] [CrossRef] [PubMed]
- Mohan, R.; Sree, P.K. An extensive survey on deep learning applications. IJIACS 2017, 6, 1Y3V4C. [Google Scholar]
- Najafabadi, M.M.; Villanustre, F.; Khoshgoftaar, T.M.; Seliya, N.; Wald, R.; Muharemagic, E. Deep learning applications and challenges in big data analytics. J. Big Data 2015, 2, 1. [Google Scholar] [CrossRef]
- Pan, X.Y.; Shen, H.B. RNA-protein binding motifs mining with a new hybrid deep learning based cross-domain knowledge integration approach. BMC Bioinform. 2017, 18, 136. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.Y.; Fan, Y.X.; Yan, J.C.; Shen, H.B. IPMiner: Hidden ncRNA-protein interaction sequential pattern mining with stacked autoencoder for accurate computational prediction. BMC Genom. 2016, 17, 582. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Yan, J.; Li, C.; Rui, X.; Liu, L.; Bie, F. On Estimating Air Pollution from Photos Using Convolutional Neural Network. In Proceedings of the 2016 ACM on Multimedia Conference, Amsterdam, The Netherlands, 15–19 October 2016. [Google Scholar]
- Ren, Z.; Yan, J.; Ni, B.; Zha, H.; Yang, X. Unsupervised Deep Learning for Optical Flow Estimation. In Proceedings of the Thirty-First AAAI Conference on Artificial Intelligence, San Francisco, CA, USA, 4–9 February 2017. [Google Scholar]
- Ioffe, S.; Szegedy, C. Batch normalization: Accelerating deep network training by reducing internal covariate shift. In Proceedings of the International Conference on Machine Learning, Lille, France, 6–11 July 2015; pp. 448–456. [Google Scholar]
- Bengio, Y.; Simard, P.; Frasconi, P. Learning long-term dependencies with gradient descent is difficult. IEEE Trans. Neural Netw. 1994, 5, 157–166. [Google Scholar] [CrossRef] [PubMed]
- Pascanu, R.; Mikolov, T.; Bengio, Y. Understanding the exploding gradient problem. CoRR, abs/1211.5063 2012.
- Pascanu, R.; Mikolov, T.; Bengio, Y. On the Difficulty of Training Recurrent Neural Networks. In Proceedings of the International Conference on Machine Learning, Atlanta, GA, USA, 16–21 June 2013; pp. 1310–1318. [Google Scholar]
- Klambauer, G.; Unterthiner, T.; Mayr, A.; Hochreiter, S. Self-normalizing neural networks. Adv. Neural Inf. Process. Syst. 2017, 972–981. [Google Scholar]
- He, K.; Zhang, X.; Ren, S.; Sun, J. Deep Residual Learning for Image Recognition. In Proceedings of the 2016 IEEE Conference on Computer Vision and Pattern Recognition (CVPR), Las Vegas, NV, USA, 27–30 June 2016; pp. 770–778. [Google Scholar]
- Breiman, L. Random forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef]
- Rambo-Martin, B.L.; Zwick, M.E. Analysis of Copy Number Variants on Chromosome 21 in Down Syndrome-Associated Congenital Heart Defects. Available online: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE93004 (accessed on 30 January 2017).
- Draminski, M.; Rada-Iglesias, A.; Enroth, S.; Wadelius, C.; Koronacki, J.; Komorowski, J. Monte carlo feature selection for supervised classification. Bioinformatics 2008, 24, 110–117. [Google Scholar] [CrossRef] [PubMed]
- Draminski, M. Available online:. Available online: http://www.ipipan.eu/staff/m.draminski/mcfs.html (accessed on 5 April 2018).
- Johnson, D.S. Approximation algorithms for combinatorial problems. J. Comput. Syst. Sci. 1974, 9, 256–278. [Google Scholar] [CrossRef]
- Cohen, W.W. Fast Effective Rule Induction. In Proceedings of the Twelfth International Conference on Machine Learning, Tahoe City, CA, USA, 9–12 July 1995; pp. 115–123. [Google Scholar]
- Johannes, F.; Widmer, G. Incremental Reduced Error Pruning. In Proceedings of the Eleventh Annual Conference on Machine Learning, New Brunswick, NJ, USA, 10–13 July 1994. [Google Scholar]
- Quinlan, J.R. Learning logical definitions from relations. Mach. Learn. 1990, 266, 239–266. [Google Scholar] [CrossRef]
- Brunnk, C.A.; Pazzani, M.J. An investigation of noise-tolerant relational concept learning algorithms. In Proceedings of the 8th International Workshop on Machine Learning, Evanston, IL, USA, 1991; Elsevier: Amsterdam, The Netherlands, 1991; pp. 389–393. [Google Scholar]
- Ho, T.K. Random Decision Forests. In Proceedings of the 3rd International Conference on Document Analysis and Recognition, Montreal, QC, USA, 14–16 August 1995; pp. 278–282. [Google Scholar]
- Ho, T.K. The random subspace method for constructing decision forests. IEEE Trans. Pattern Anal. Mach. Intell. 1998, 20, 832–844. [Google Scholar]
- Ting, K.M.; Witten, I.H. Stacking Bagged and Dagged Models. In Proceedings of the Fourteenth International Conference on Machine Learning, San Francisco, CA, USA, 8–12 July 1997. [Google Scholar]
- Chen, L.; Zhang, Y.-H.; Zheng, M.; Huang, T.; Cai, Y.-D. Identification of compound–protein interactions through the analysis of gene ontology, KEGG enrichment for proteins and molecular fragments of compounds. Mol. Genet. Genom. 2016, 291, 2065–2079. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Chu, C.; Huang, T.; Kong, X.; Cai, Y.D. Prediction and analysis of cell-penetrating peptides using pseudo-amino acid composition and random forest models. Amino Acids 2015, 47, 1485–1493. [Google Scholar] [CrossRef] [PubMed]
- Li, B.-Q.; Zheng, L.-L.; Hu, L.-L.; Feng, K.-Y.; Huang, G.; Chen, L. Prediction of linear B-cell epitopes with mRMR feature selection and analysis. Curr. Bioinform. 2016, 11, 22–31. [Google Scholar] [CrossRef]
- Li, B.Q.; Hu, L.L.; Chen, L.; Feng, K.Y.; Cai, Y.D.; Chou, K.C. Prediction of protein domain with mRMR feature selection and analysis. PLoS ONE 2012, 7, e39308. [Google Scholar] [CrossRef] [PubMed]
- Casanova, R.; Saldana, S.; Chew, E.Y.; Danis, R.P.; Greven, C.M.; Ambrosius, W.T. Application of random forests methods to diabetic retinopathy classification analyses. PLoS ONE 2014, 9, e98587. [Google Scholar] [CrossRef] [PubMed]
- Kandaswamy, K.K.; Chou, K.-C.; Martinetz, T.; Möller, S.; Suganthan, P.; Sridharan, S.; Pugalenthi, G. AFP-Pred: A random forest approach for predicting antifreeze proteins from sequence-derived properties. J. Theor. Biol. 2011, 270, 56–62. [Google Scholar] [CrossRef] [PubMed]
- Pugalenthi, G.; Kandaswamy, K.; Chou, K.-C.; Vivekanandan, S.; Kolatkar, P. RSARF: Prediction of residue solvent accessibility from protein sequence using random forest method. Protein Pept. Lett. 2011, 19, 50–56. [Google Scholar] [CrossRef]
- Pan, X.Y.; Zhang, Y.N.; Shen, H.B. Large-scale prediction of human protein-protein interactions from amino acid sequence based on latent topic features. J. Proteome Res. 2010, 9, 4992–5001. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Sun, X.; Feng, K.; Wang, S.; Zhang, Y.H.; Wang, S.; Lu, L.; Cai, Y.D. Predicting citrullination sites in protein sequences using mRMR method and random forest algorithm. Comb. Chem. High Throughput Screen 2017, 20, 164–173. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Zhang, Y.H.; Huang, T.; Cai, Y.D. Gene expression profiling gut microbiota in different races of humans. Sci. Rep. 2016, 6, 23075. [Google Scholar] [CrossRef] [PubMed]
- Witten, I.H.; Frank, E. Data Mining: Practical Machine Learning Tools and Techniques; Morgan Kaufmann: San Francisco, CA, USA, 2005. [Google Scholar]
- Kohavi, R. A Study of Cross-Validation and Bootstrap for Accuracy Estimation and Model Selection. In Proceedings of the 14th International Joint Conference on Artificial Intelligence, Montreal, QC, Canada, 20–25 August 1995; Lawrence Erlbaum Associates Ltd.: Mahwah, NJ, USA; pp. 1137–1145. [Google Scholar]
- Chen, L.; Wang, S.; Zhang, Y.-H.; Li, J.; Xing, Z.-H.; Yang, J.; Huang, T.; Cai, Y.-D. Identify key sequence features to improve CRISPR sgRNA efficacy. IEEE Access 2017, 5, 26582–26590. [Google Scholar] [CrossRef]
- Chen, L.; Li, J.; Zhang, Y.-H.; Feng, K.; Wang, S.; Zhang, Y.; Huang, T.; Kong, X.; Cai, Y.-D. Identification of gene expression signatures across different types of neural stem cells with the Monte-carlo feature selection method. J. Cell. Biochem. 2017, 119, 3394–3403. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Zhang, Y.H.; Lu, G.; Huang, T.; Cai, Y.D. Analysis of cancer-related lncRNAs using gene ontology and KEGG pathways. Artif. Intell. Med. 2017, 76, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Zeng, W.-M.; Cai, Y.-D.; Feng, K.-Y.; Chou, K.-C. Predicting anatomical therapeutic chemical (ATC) classification of drugs by integrating chemical-chemical interactions and similarities. PLoS ONE 2012, 7, e35254. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Chu, C.; Zhang, Y.-H.; Zheng, M.-Y.; Zhu, L.; Kong, X.; Huang, T. Identification of drug-drug interactions using chemical interactions. Curr. Bioinform. 2017, 12, 526–534. [Google Scholar] [CrossRef]
- Chen, L.; Feng, K.Y.; Cai, Y.D.; Chou, K.C.; Li, H.P. Predicting the network of substrate-enzyme-product triads by combining compound similarity and functional domain composition. BMC Bioinform. 2010, 11, 293. [Google Scholar] [CrossRef] [PubMed]
- Matthews, B.W. Comparison of the predicted and observed secondary structure of T4 phage lysozyme. Biochim. Biophys. Acta 1975, 405, 442–451. [Google Scholar] [CrossRef]
- Ramachandran, D.; Mulle, J.G.; Locke, A.E.; Bean, L.J.; Rosser, T.C.; Bose, P.; Dooley, K.J.; Cua, C.L.; Capone, G.T.; Reeves, R.H.; et al. Contribution of copy-number variation to down syndrome-associated atrioventricular septal defects. Genet. Med. 2015, 17, 554–560. [Google Scholar] [CrossRef] [PubMed]
- Michaud, J.; Kudoh, J.; Berry, A.; Bonne-Tamir, B.; Lalioti, M.D.; Rossier, C.; Shibuya, K.; Kawasaki, K.; Asakawa, S.; Minoshima, S.; et al. Isolation and characterization of a human chromosome 21q22.3 gene (WDR4) and its mouse homologue that code for a WD-repeat protein. Genomics 2000, 68, 71–79. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.H.; Kim, S.Y.; Park, S.Y.; Lee, S.Y.; Kim, M.J.; Han, Y.J.; Lee, S.W.; Chung, J.H.; Kim, M.Y.; Yang, J.H.; et al. Non-invasive epigenetic detection of fetal trisomy 21 in first trimester maternal plasma. PLoS ONE 2011, 6, e27709. [Google Scholar] [CrossRef] [PubMed]
- Almeida, C.B.; Traina, F.; Lanaro, C.; Canalli, A.A.; Saad, S.T.; Costa, F.F.; Conran, N. High expression of the cGMP-specific phosphodiesterase, PDE9a, in sickle cell disease (SCD) and the effects of its inhibition in erythroid cells and SCD neutrophils. Br. J. Haematol. 2008, 142, 836–844. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.I.; Zhu, G.; Sasaki, T.; Cho, G.S.; Hamdani, N.; Holewinski, R.; Jo, S.H.; Danner, T.; Zhang, M.; Rainer, P.P.; et al. Phosphodiesterase 9A controls nitric-oxide-independent cGMP and hypertrophic heart disease. Nature 2015, 519, 472–476. [Google Scholar] [CrossRef] [PubMed]
- Gorenflo, M.; Zheng, C.; Poge, A.; Bettendorf, M.; Werle, E.; Fiehn, W.; Ulmer, H.E. Metabolites of the L-arginine-NO pathway in patients with left-to-right shunt. Clin. Lab. 2001, 47, 441–447. [Google Scholar] [PubMed]
- Gorenflo, M.; Ullmann, M.V.; Eitel, K.; Gross, J.; Fiehn, W.; Hagl, S.; Dreyhaupt, J. Plasma L-arginine and metabolites of nitric oxide synthase in patients with left-to-right shunt after intracardiac repair. Chest 2005, 127, 1184–1189. [Google Scholar] [CrossRef]
- Seghaye, M.C.; Duchateau, J.; Bruniaux, J.; Demontoux, S.; Detruit, H.; Bosson, C.; Lecronier, G.; Mokhfi, E.; Serraf, A.; Planche, C. Endogenous nitric oxide production and atrial natriuretic peptide biological activity in infants undergoing cardiac operations. Crit. Care Med. 1997, 25, 1063–1070. [Google Scholar] [CrossRef] [PubMed]
- Rachidi, M.; Lopes, C.; Costantine, M.; Delabar, J.M. C21orf5, a new member of dopey family involved in morphogenesis, could participate in neurological alterations and mental retardation in down syndrome. DNA Res. 2005, 12, 203–210. [Google Scholar] [CrossRef] [PubMed]
- Rachidi, M.; Lopes, C.; Delezoide, A.L.; Delabar, J.M. C21orf5, a human candidate gene for brain abnormalities and mental retardation in down syndrome. Cytogenet. Genome Res. 2006, 112, 16–22. [Google Scholar] [CrossRef] [PubMed]
- Antonarakis, S.E. Down syndrome and the complexity of genome dosage imbalance. Nat. Rev. Genet. 2017, 18, 147–163. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Madugula, V. Mechanisms of ciliary targeting: Entering importins and rabs. Cell. Mol. Life Sci. 2018, 75, 597–606. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Meng, D.; Zhu, B.; Pan, J. Mechanism of ciliary disassembly. Cell. Mol. Life Sci. 2016, 73, 1787–1802. [Google Scholar] [CrossRef] [PubMed]
- Kondo, T.; Onitsuka, S.; Ryoji, O.; Kihara, T.; Goto, Y.; Satoh, T.; Nakazawa, H.; Toma, H. Analysis of prognostic factors related to primary superficial bladder cancer tumor recurrence in prophylactic intravesical epirubicin therapy. Int. J. Urol. 1999, 6, 178–183. [Google Scholar] [CrossRef] [PubMed]
- Slusarski, D.C.; Pelegri, F. Calcium signaling in vertebrate embryonic patterning and morphogenesis. Dev. Biol. 2007, 307, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Gu, W.; Hurles, M.E.; Lupski, J.R. Copy number variation in human health, disease, and evolution. Annu. Rev. Genom. Hum. Genet. 2009, 10, 451–481. [Google Scholar] [CrossRef] [PubMed]
- Mlynarski, E.E.; Sheridan, M.B.; Xie, M.; Guo, T.; Racedo, S.E.; McDonald-McGinn, D.M.; Gai, X.; Chow, E.W.; Vorstman, J.; Swillen, A.; et al. Copy-number variation of the glucose transporter gene SLC2A3 and congenital heart defects in the 22q11.2 deletion syndrome. Am. J. Hum. Genet. 2015, 96, 753–764. [Google Scholar] [CrossRef] [PubMed]
- Asai, S.; Yamaki, A.; Kudoh, J.; Shimizu, N.; Shimizu, Y. Analysis of the promoter region of human placenta-specific DSCR4 gene. Biochim. Biophys. Acta 2008, 1779, 40–50. [Google Scholar] [CrossRef] [PubMed]
- Al-Biltagi, M.A. Echocardiography in children with down syndrome. World J. Clin. Pediatr. 2013, 2, 36–45. [Google Scholar] [CrossRef] [PubMed]
- Pelleri, M.C.; Gennari, E.; Locatelli, C.; Piovesan, A.; Caracausi, M.; Antonaros, F.; Rocca, A.; Donati, C.M.; Conti, L.; Strippoli, P.; et al. Genotype-phenotype correlation for congenital heart disease in down syndrome through analysis of partial trisomy 21 cases. Genomics 2017, 109, 391–400. [Google Scholar] [CrossRef] [PubMed]
- Ait Yahya-Graison, E.; Aubert, J.; Dauphinot, L.; Rivals, I.; Prieur, M.; Golfier, G.; Rossier, J.; Personnaz, L.; Creau, N.; Blehaut, H.; et al. Classification of human chromosome 21 gene-expression variations in down syndrome: Impact on disease phenotypes. Am. J. Hum. Genet. 2007, 81, 475–491. [Google Scholar] [CrossRef] [PubMed]
- Ramos, P.S.; Brown, E.E.; Kimberly, R.P.; Langefeld, C.D. Genetic factors predisposing to systemic lupus erythematosus and lupus nephritis. Semin. Nephrol. 2010, 30, 164–176. [Google Scholar] [CrossRef] [PubMed]
- Lintas, C.; Sacco, R.; Persico, A.M. Genome-wide expression studies in autism spectrum disorder, rett syndrome, and down syndrome. Neurobiol. Dis. 2012, 45, 57–68. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Morishima, M.; Yu, T.; Matsui, S.; Zhang, L.; Fu, D.; Pao, A.; Costa, A.C.; Gardiner, K.J.; Cowell, J.K.; et al. Genetic analysis of down syndrome-associated heart defects in mice. Hum. Genet. 2011, 130, 623–632. [Google Scholar] [CrossRef] [PubMed]
- Rost, I.; Fiegler, H.; Fauth, C.; Carr, P.; Bettecken, T.; Kraus, J.; Meyer, C.; Enders, A.; Wirtz, A.; Meitinger, T.; et al. Tetrasomy 21pter→q21.2 in a male infant without typical down’s syndrome dysmorphic features but moderate mental retardation. J. Med. Genet. 2004, 41, e26. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Classification Algorithm | Number of Features | MCC | AUC |
|---|---|---|---|
| SNN | 2737 | 0.748 | 0.915 |
| Random forest | 132 | 0.582 | 0.834 |
| Classification | Rules | Features | Criteria |
|---|---|---|---|
| With AVSD | Rule 1 | A_16_P41408273 | ≤−0.00593 |
| With AVSD | Rule 2 | A_16_P03593084 | ≥−0.0164 |
| A_16_P03593084 | ≤0.075 | ||
| A_16_P41408273 | ≥0.0248 | ||
| Without DS | Rule 3 | Other conditions |
| No. | Feature Name | Gene Name |
|---|---|---|
| 1 | A_16_P03593084 | PDE9A |
| 2 | A_16_P03583086 | DOPEY2 |
| 3 | A_16_P03587947 | LCA5L |
| 4 | A_16_P21251330 | DSCR4 |
| 5 | A_16_P41466725 | ITGB2 |
| 6 | A_16_P41430034 | U16296 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pan, X.; Hu, X.; Zhang, Y.H.; Feng, K.; Wang, S.P.; Chen, L.; Huang, T.; Cai, Y.D. Identifying Patients with Atrioventricular Septal Defect in Down Syndrome Populations by Using Self-Normalizing Neural Networks and Feature Selection. Genes 2018, 9, 208. https://doi.org/10.3390/genes9040208
Pan X, Hu X, Zhang YH, Feng K, Wang SP, Chen L, Huang T, Cai YD. Identifying Patients with Atrioventricular Septal Defect in Down Syndrome Populations by Using Self-Normalizing Neural Networks and Feature Selection. Genes. 2018; 9(4):208. https://doi.org/10.3390/genes9040208
Chicago/Turabian StylePan, Xiaoyong, Xiaohua Hu, Yu Hang Zhang, Kaiyan Feng, Shao Peng Wang, Lei Chen, Tao Huang, and Yu Dong Cai. 2018. "Identifying Patients with Atrioventricular Septal Defect in Down Syndrome Populations by Using Self-Normalizing Neural Networks and Feature Selection" Genes 9, no. 4: 208. https://doi.org/10.3390/genes9040208
APA StylePan, X., Hu, X., Zhang, Y. H., Feng, K., Wang, S. P., Chen, L., Huang, T., & Cai, Y. D. (2018). Identifying Patients with Atrioventricular Septal Defect in Down Syndrome Populations by Using Self-Normalizing Neural Networks and Feature Selection. Genes, 9(4), 208. https://doi.org/10.3390/genes9040208