Identification of Key Pathways and Genes in the Dynamic Progression of HCC Based on WGCNA

Abstract
:1. Introduction
2. Materials and Methods
2.1. Data Processing
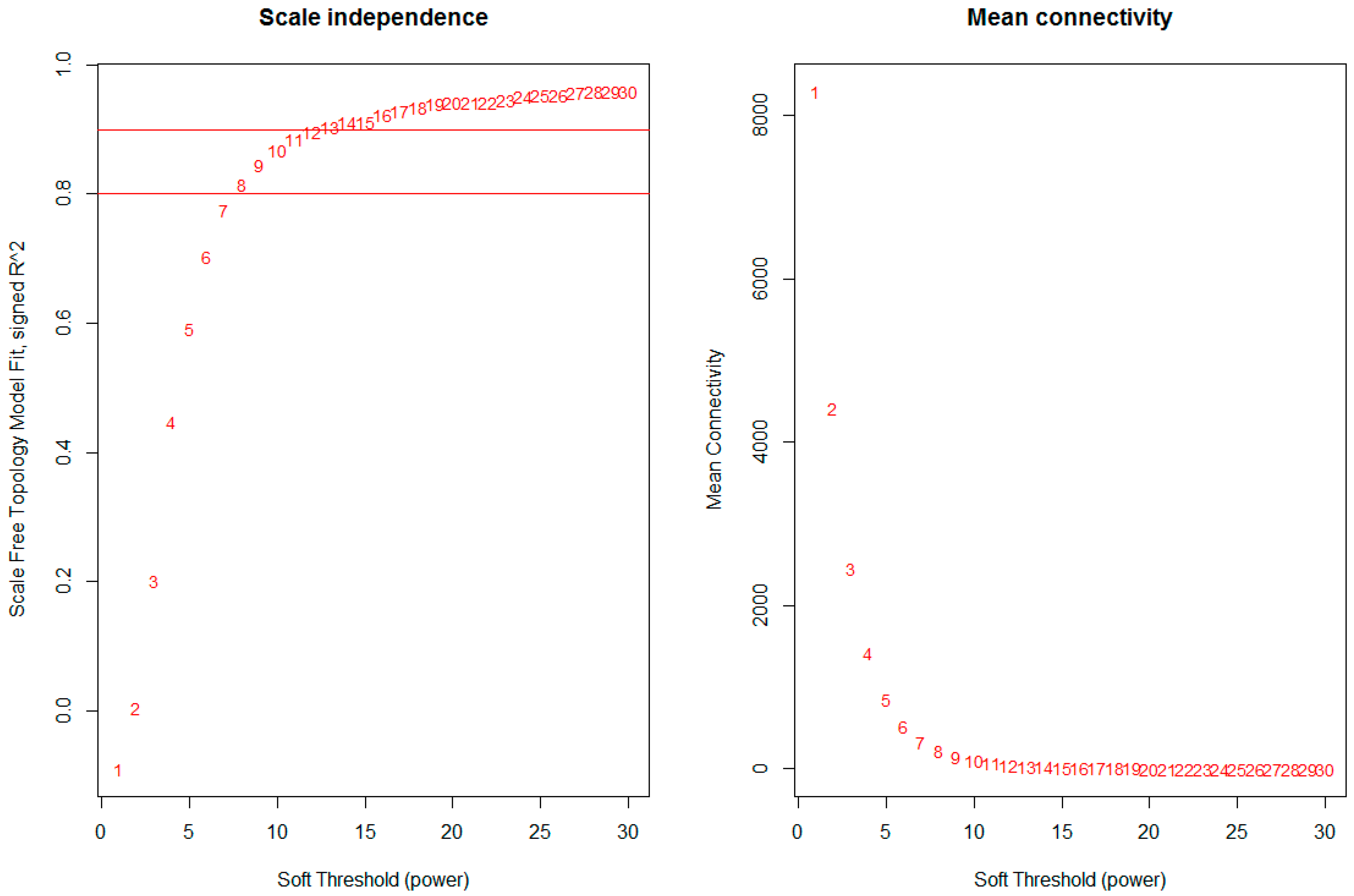
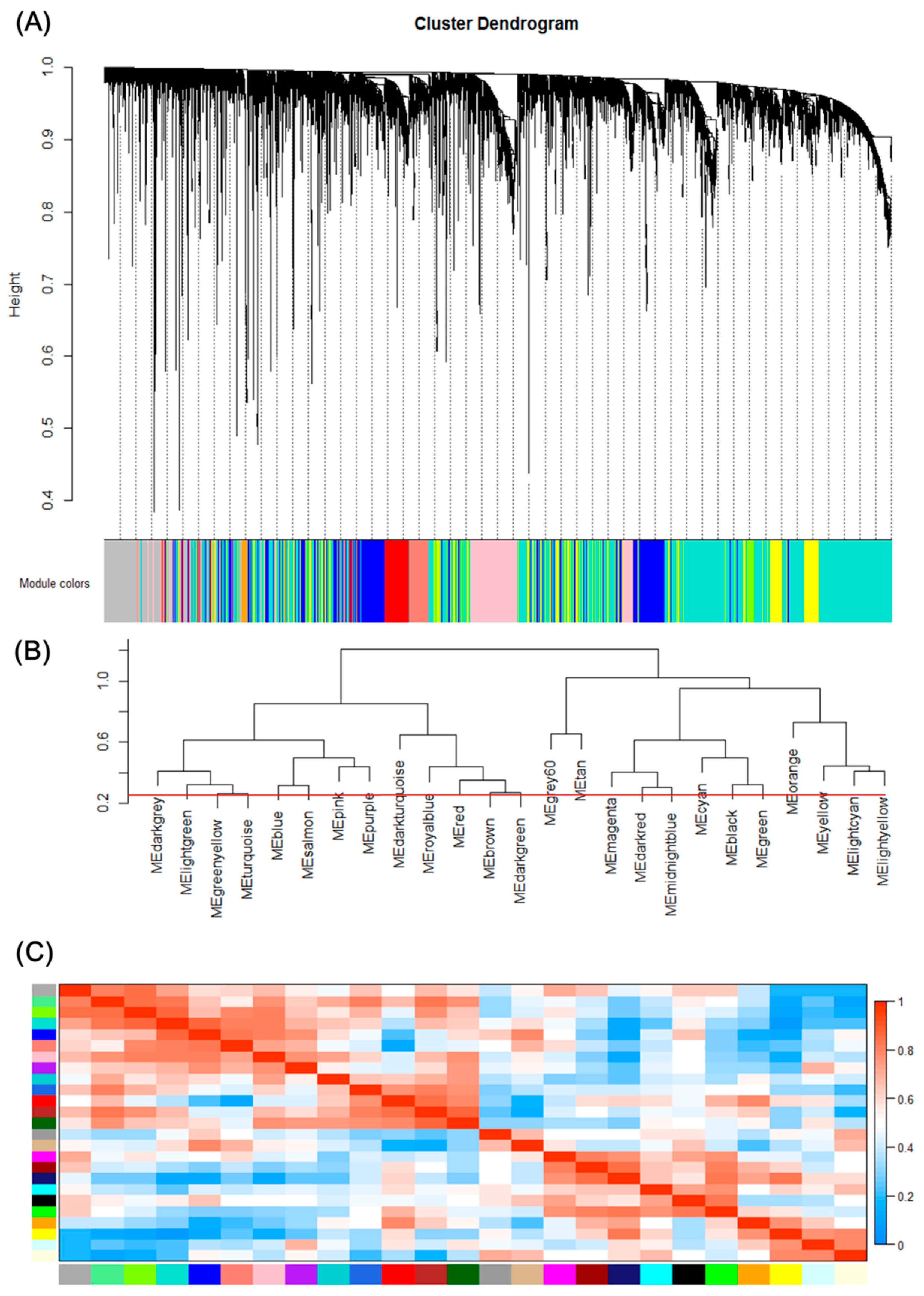
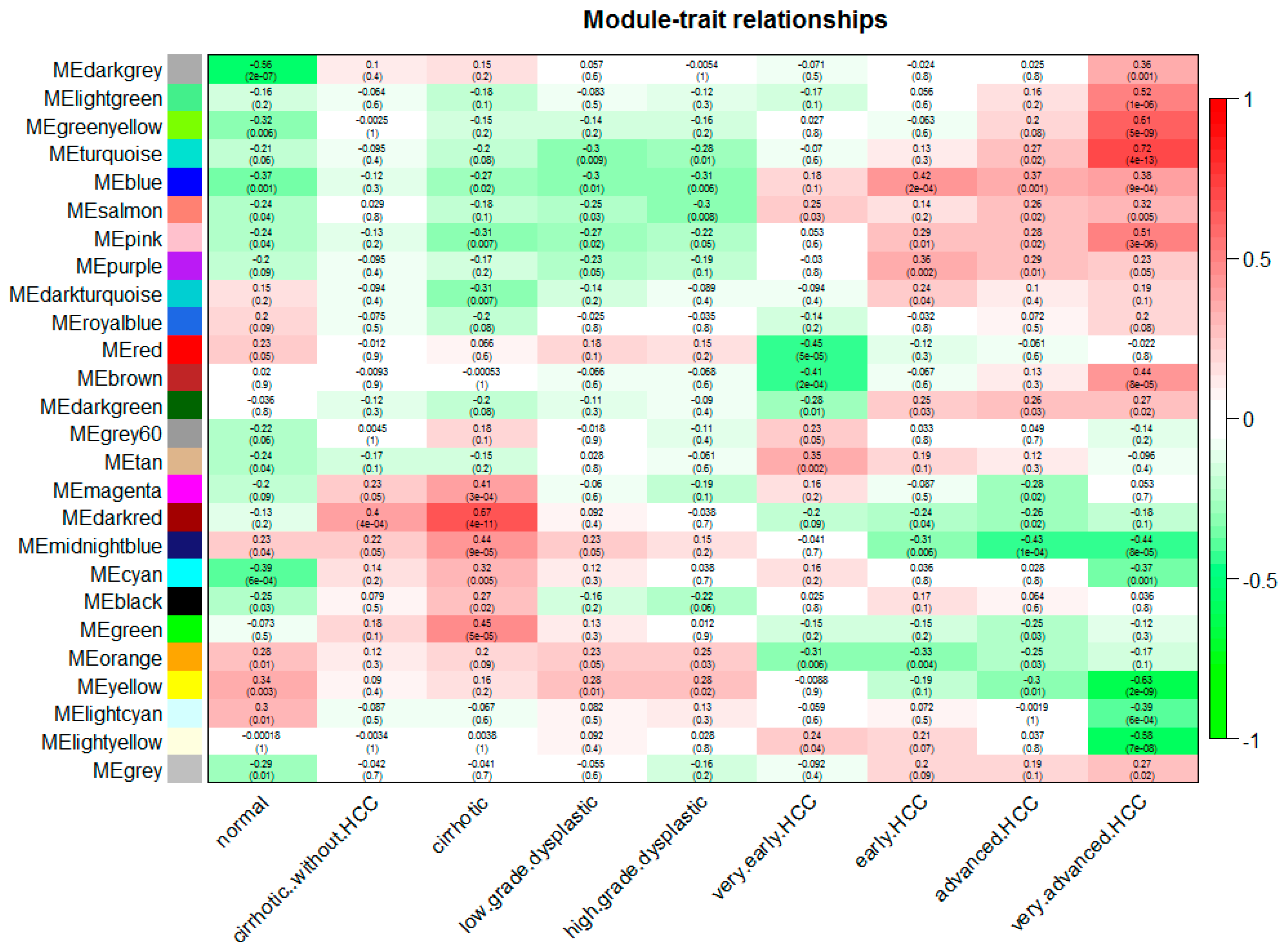
2.2. Construction of Weighted Gene Co-Expression Networks and Identification of Modules Associated with Different Stages of Hepatocellular Carcinoma
2.3. Interaction Analysis of Co-Expression Modules
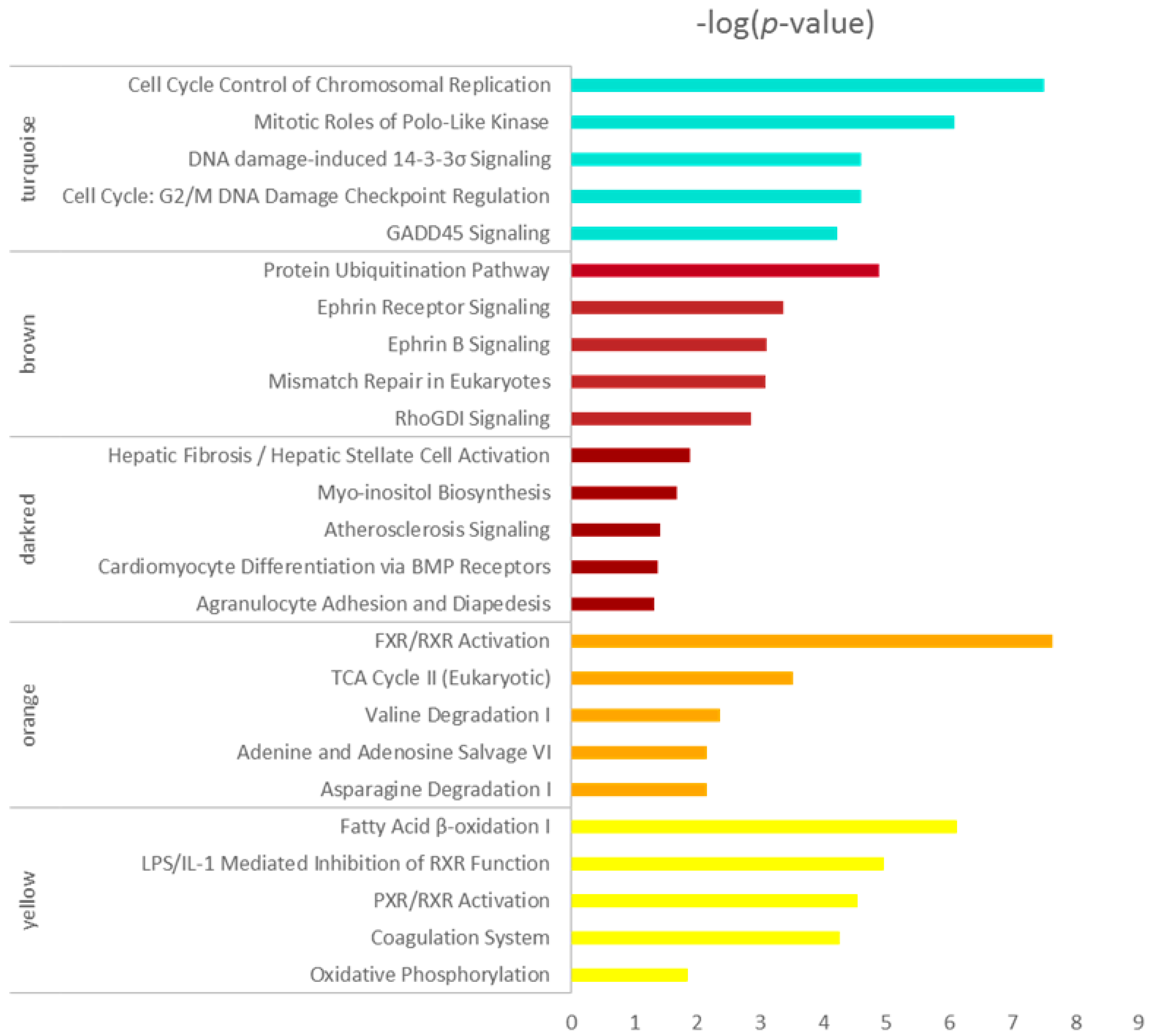
2.4. Functional Enrichment Analysis of Genes in Every Module
2.5. The Ingenuity Pathway Analysis Upstream Regulator Analysis
2.6. Protein-Protein Interaction Network Construction and Analysis for Selected Modules
2.7. Kaplan-Meier Survival Analysis
3. Results
3.1. Data Processing
3.2. Construction of Weighted Gene Co-Expression Network Identification of Modules Associated with Different Stages of HCC
3.3. Correlation between each Module
3.4. Functional Enrichment Analysis
3.5. The Ingenuity Pathway Analysis Upstream Regulator Analysis
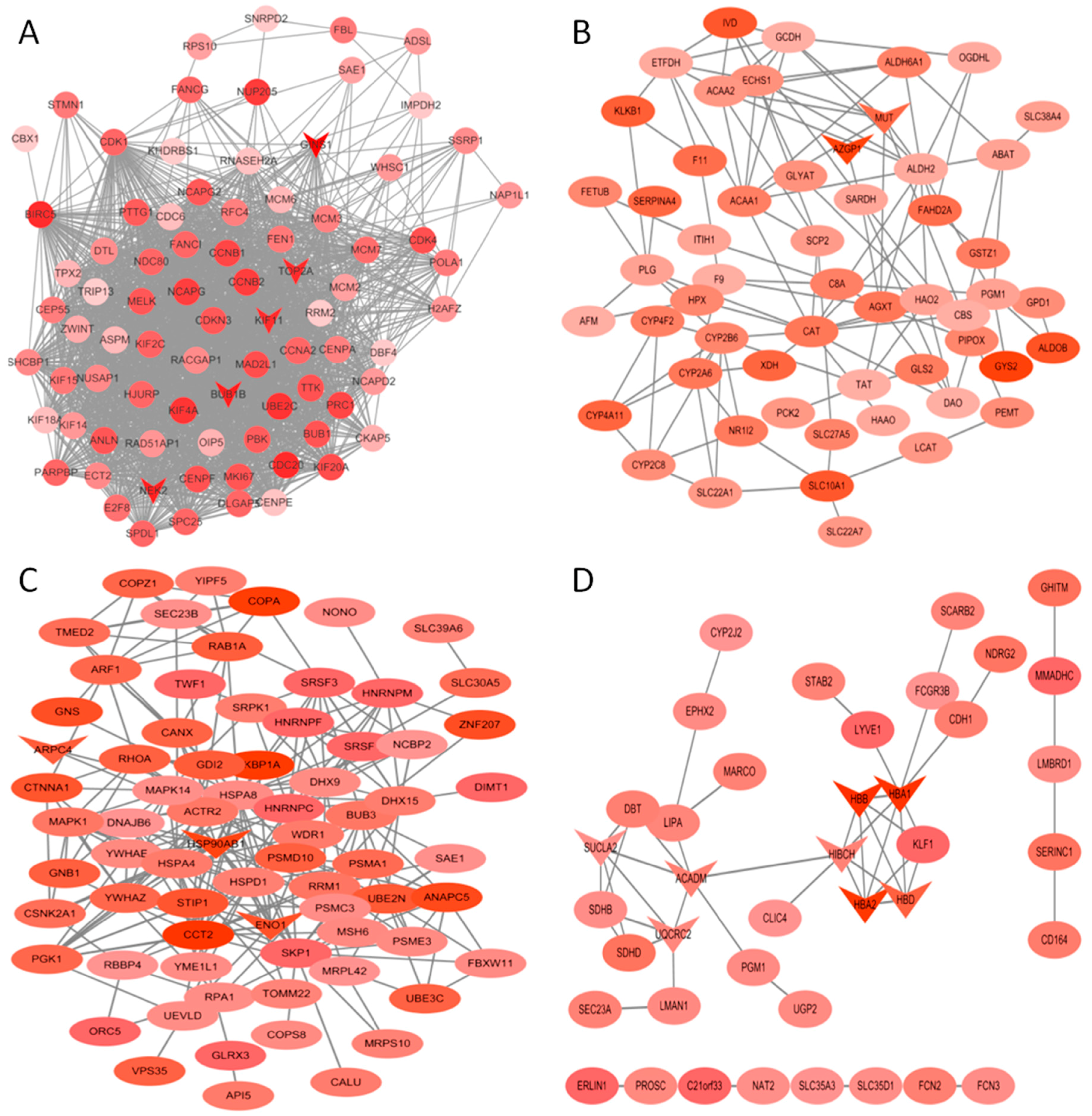
3.6. PPI Network Construction and Analysis of Selected Modules
3.7. Kaplan-Meier Survival Analysis
4. Discussion
Supplementary Materials
Conflicts of Interest
References
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. Cancer incidence and mortality worldwide: Sources, methods and major patterns in globocan 2012. Int. J. Cancer 2015, 136, E359–E386. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, K.; Saitoh, S.; Koida, I.; Arase, Y.; Tsubota, A.; Chayama, K.; Kumada, H.; Kawanishi, M. A multivariate analysis of risk factors for hepatocellular carcinogenesis: A prospective observation of 795 patients with viral and alcoholic cirrhosis. Hepatology 1993, 18, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Ringehan, M.; McKeating, J.A.; Protzer, U. Viral hepatitis and liver cancer. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 2017, 372, 1732. [Google Scholar] [CrossRef] [PubMed]
- Dhungel, B.; Jayachandran, A.; Layton, C.J.; Steel, J.C. Seek and destroy: Targeted adeno-associated viruses for gene delivery to hepatocellular carcinoma. Drug Deliv. 2017, 24, 289–299. [Google Scholar] [CrossRef] [PubMed]
- Waghray, A.; Murali, A.R.; Menon, K.N. Hepatocellular carcinoma: From diagnosis to treatment. World J. Hepatol. 2015, 7, 1020–1029. [Google Scholar] [CrossRef] [PubMed]
- Bo, L.; Wei, B.; Wang, Z.; Kong, D.; Gao, Z.; Miao, Z. Screening of critical genes and microRNAs in blood samples of patients with ruptured intracranial aneurysms by bioinformatic analysis of gene expression data. Med. Sci. Monit. 2017, 23, 4518–4525. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Xiao, H.; Guo, S.; Dong, L.; Chen, J. Identification of breast cancer mechanism based on weighted gene coexpression network analysis. Cancer Gene Ther. 2017, 24, 333–341. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Zhang, Q.; Ye, C.; Lv, J.M.; Liu, X.; Chen, L.; Wu, H.; Yin, L.; Cui, X.G.; Xu, D.; et al. Identification of prognostic markers of high grade prostate cancer through an integrated bioinformatics approach. J. Cancer Res. Clin. Oncol. 2017, 143, 2571–2579. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Hu, A.X.; Zhao, J.L.; Chen, F.L. Identification of key gene modules in human osteosarcoma by co-expression analysis weighted gene co-expression network analysis (WGCNA). J. Cell Biochem. 2017, 118, 3953–3959. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Lv, Q.; Teng, S.; Yu, Y.; Niu, K.; Yi, C. Identifying key genes in rheumatoid arthritis by weighted gene co-expression network analysis. Int. J. Rheum. Dis. 2017, 20, 971–979. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Ke, Z.P.; Peng, Y.G.; Cai, P.T. Coexpression analysis reveals key gene modules and pathway of human coronary heart disease. J. Cell. Biochem. 2018, 119, 2102–2109. [Google Scholar] [CrossRef] [PubMed]
- Esposti, D.D.; Hernandez-Vargas, H.; Voegele, C.; Fernandez-Jimenez, N.; Forey, N.; Bancel, B.; Le Calvez-Kelm, F.; McKay, J.; Merle, P.; Herceg, Z. Identification of novel long non-coding RNAs deregulated in hepatocellular carcinoma using RNA-sequencing. Oncotarget 2016, 7, 31862–31877. [Google Scholar] [CrossRef] [PubMed]
- Giulietti, M.; Occhipinti, G.; Principato, G.; Piva, F. Identification of candidate miRNA biomarkers for pancreatic ductal adenocarcinoma by weighted gene co-expression network analysis. Cell Oncol. 2017, 40, 181–192. [Google Scholar] [CrossRef] [PubMed]
- Liao, X.; Huang, K.; Huang, R.; Liu, X.; Han, C.; Yu, L.; Yu, T.; Yang, C.; Wang, X.; Peng, T. Genome-scale analysis to identify prognostic markers in patients with early-stage pancreatic ductal adenocarcinoma after pancreaticoduodenectomy. Oncol. Targets Ther. 2017, 10, 4493–4506. [Google Scholar] [CrossRef] [PubMed]
- Giulietti, M.; Occhipinti, G.; Principato, G.; Piva, F. Weighted gene co-expression network analysis reveals key genes involved in pancreatic ductal adenocarcinoma development. Cell Oncol. 2016, 39, 379–388. [Google Scholar] [CrossRef] [PubMed]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinformatics 2008, 9, 559. [Google Scholar] [CrossRef] [PubMed]
- Wurmbach, E.; Chen, Y.B.; Khitrov, G.; Zhang, W.; Roayaie, S.; Schwartz, M.; Fiel, I.; Thung, S.; Mazzaferro, V.; Bruix, J.; et al. Genome-wide molecular profiles of HCV-induced dysplasia and hepatocellular carcinoma. Hepatology 2007, 45, 938–947. [Google Scholar] [CrossRef] [PubMed]
- Zheng, S.; Tansey, W.P.; Hiebert, S.W.; Zhao, Z. Integrative network analysis identifies key genes and pathways in the progression of hepatitis C virus induced hepatocellular carcinoma. BMC Med. Genom. 2011, 4, 62. [Google Scholar] [CrossRef] [PubMed]
- Chin, C.H.; Chen, S.H.; Wu, H.H.; Ho, C.W.; Ko, M.T.; Lin, C.Y. cytoHubba: Identifying hub objects and sub-networks from complex interactome. BMC Syst. Biol. 2014, 8, S11. [Google Scholar] [CrossRef] [PubMed]
- Barrett, T.; Wilhite, S.E.; Ledoux, P.; Evangelista, C.; Kim, I.F.; Tomashevsky, M.; Marshall, K.A.; Phillippy, K.H.; Sherman, P.M.; Holko, M.; et al. Ncbi Geo: Archive for Functional Genomics Data Sets--Update. Nucleic Ac. Res. 2013, 41, D991–D995. [Google Scholar] [CrossRef] [PubMed]
- Irizarry, R.A.; Hobbs, B.; Collin, F.; Beazer-Barclay, Y.D.; Antonellis, K.J.; Scherf, U.; Speed, T.P. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics 2003, 4, 249–264. [Google Scholar] [CrossRef] [PubMed]
- R Core Team. R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing; R Core Team: Vienna, Austria, 2014; Available online: http://Www.R-Project.Org/ (accessed on 13 February 2018).
- Langfelder, P.; Horvath, S. Fast R Functions for Robust Correlations and Hierarchical Clustering. J. Stat. Softw. 2012, 46, i11. [Google Scholar] [CrossRef] [PubMed]
- Aguirre-Gamboa, R.; Gomez-Rueda, H.; Martinez-Ledesma, E.; Martinez-Torteya, A.; Chacolla-Huaringa, R.; Rodriguez-Barrientos, A.; Tamez-Pena, J.G.; Trevino, V. SurvExpress: An online biomarker validation tool and database for cancer gene expression data using survival analysis. PLoS ONE 2013, 8, e74250. [Google Scholar] [CrossRef] [PubMed]
- Aarts, M.; Linardopoulos, S.; Turner, N.C. Tumour selective targeting of cell cycle kinases for cancer treatment. Curr. Opin. Pharmacol. 2013, 13, 529–535. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Moralli, S.; Tarrado-Castellarnau, M.; Miranda, A.; Cascante, M. Targeting cell cycle regulation in cancer therapy. Pharmacol. Ther. 2013, 138, 255–271. [Google Scholar] [CrossRef] [PubMed]
- Williams, G.H.; Stoeber, K. The cell cycle and cancer. J. Pathol. 2012, 226, 352–364. [Google Scholar] [CrossRef] [PubMed]
- Liccioni, A.; Reig, M.; Bruix, J. Treatment of hepatocellular carcinoma. Dig. Dis. 2014, 32, 554–563. [Google Scholar] [CrossRef] [PubMed]
- Benada, J.; Macurek, L. Targeting the checkpoint to kill cancer cells. Biomolecules 2015, 5, 1912–1937. [Google Scholar] [CrossRef] [PubMed]
- Broustas, C.G.; Lieberman, H.B. DNA damage response genes and the development of cancer metastasis. Radiat. Res. 2014, 181, 111–130. [Google Scholar] [CrossRef] [PubMed]
- Marques, S.; Fonseca, J.; Silva, P.M.; Bousbaa, H. Targeting the spindle assembly checkpoint for breast cancer treatment. Curr. Cancer Drug Targets 2015, 15, 272–281. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.H.; Guo, L.; Wu, X.H. Checkpoint inhibitors in immunotherapy of ovarian cancer. Tumour Biol. 2015, 36, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Zhang, X.; Teng, L.; Legerski, R.J. DNA damage checkpoint recovery and cancer development. Exp. Cell. Res. 2015, 334, 350–358. [Google Scholar] [CrossRef] [PubMed]
- Amann, T.; Hellerbrand, C. GLUT1 as a therapeutic target in hepatocellular carcinoma. Expert Opin. Ther. Targets 2009, 13, 1411–1427. [Google Scholar] [CrossRef] [PubMed]
- Amann, T.; Maegdefrau, U.; Hartmann, A.; Agaimy, A.; Marienhagen, J.; Weiss, T.S.; Stoeltzing, O.; Warnecke, C.; Scholmerich, J.; Oefner, P.J.; et al. GLUT1 expression is increased in hepatocellular carcinoma and promotes tumorigenesis. Am. J. Pathol. 2009, 174, 1544–1552. [Google Scholar] [CrossRef] [PubMed]
- Beyoglu, D.; Idle, J.R. The metabolomic window into hepatobiliary disease. J. Hepatol. 2013, 59, 842–858. [Google Scholar] [CrossRef] [PubMed]
- Dong, T.; Yan, Y.; Chai, H.; Chen, S.; Xiong, X.; Sun, D.; Yu, Y.; Deng, L.; Cheng, F. Pyruvate kinase M2 affects liver cancer cell behavior through up-regulation of HIF-1alpha and BCL-xL in culture. Biomed. Pharmacother. 2015, 69, 277–284. [Google Scholar] [CrossRef] [PubMed]
- Kimhofer, T.; Fye, H.; Taylor-Robinson, S.; Thursz, M.; Holmes, E. Proteomic and metabonomic biomarkers for hepatocellular carcinoma: A comprehensive review. Br. J. Cancer 2015, 112, 1141–1156. [Google Scholar] [CrossRef] [PubMed]
- Liu, A.M.; Xu, Z.; Shek, F.H.; Wong, K.F.; Lee, N.P.; Poon, R.T.; Chen, J.; Luk, J.M. miR-122 targets pyruvate kinase M2 and affects metabolism of hepatocellular carcinoma. PLoS ONE 2014, 9, e86872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oronsky, B.T.; Oronsky, N.; Fanger, G.R.; Parker, C.W.; Caroen, S.Z.; Lybeck, M.; Scicinski, J.J. Follow the ATP: Tumor energy production: A perspective. Anticancer Agents Med. Chem. 2014, 14, 1187–1198. [Google Scholar] [CrossRef] [PubMed]
- Kanzaki, R.; Naito, H.; Kise, K.; Takara, K.; Eino, D.; Minami, M.; Shintani, Y.; Funaki, S.; Kawamura, T.; Kimura, T.; Okumura, M.; Takakura, N. PSF1 (partner of SLD five 1) is a prognostic biomarker in patients with non-small cell lung cancer treated with surgery following preoperative chemotherapy or chemoradiotherapy. Ann. Surg. Oncol. 2016, 23, 4093–4100. [Google Scholar] [CrossRef] [PubMed]
- Nagahama, Y.; Ueno, M.; Miyamoto, S.; Morii, E.; Minami, T.; Mochizuki, N.; Saya, H.; Takakura, N. PSF1, a DNA replication factor expressed widely in stem and progenitor cells, drives tumorigenic and metastatic properties. Cancer Res. 2010, 70, 1215–1224. [Google Scholar] [CrossRef] [PubMed]
- Wen, J.Z.; Han, X.Y.; Wei, B.; Zhang, S.; Wei, H.B. Expression of PSF1 in colon cancer tissues and its effect on the proliferation of colon cancer cells. Zhonghua Wei Chang Wai Ke Za Zhi 2013, 16, 70–74. [Google Scholar] [PubMed]
- Zhang, J.; Wu, Q.; Wang, Z.; Zhang, Y.; Zhang, G.; Fu, J.; Liu, C. Knockdown of PSF1 expression inhibits cell proliferation in lung cancer cells in vitro. Tumour Biol. 2015, 36, 2163–2168. [Google Scholar] [CrossRef] [PubMed]
- Nakahara, I.; Miyamoto, M.; Shibata, T.; Akashi-Tanaka, S.; Kinoshita, T.; Mogushi, K.; Oda, K.; Ueno, M.; Takakura, N.; Mizushima, H.; et al. Up-regulation of PSF1 promotes the growth of breast cancer cells. Genes Cells 2010, 15, 1015–1024. [Google Scholar] [CrossRef] [PubMed]
- Tahara, H.; Naito, H.; Kise, K.; Wakabayashi, T.; Kamoi, K.; Okihara, K.; Yanagisawa, A.; Nakai, Y.; Nonomura, N.; Morii, E.; et al. Evaluation of PSF1 as a prognostic biomarker for prostate cancer. Prostate Cancer Prostatic Dis. 2015, 18, 56–62. [Google Scholar] [CrossRef] [PubMed]
- Wei, H.B.; Wen, J.Z.; Wei, B.; Han, X.Y.; Zhang, S. Expression and clinical significance of GINS complex in colorectal cancer. Zhonghua Wei Chang Wai Ke Za Zhi 2011, 14, 443–447. [Google Scholar] [PubMed]
- Han, Y.; Ueno, M.; Nagahama, Y.; Takakura, N. Identification and characterization of stem cell-specific transcription of PSF1 in spermatogenesis. Biochem. Biophys. Res. Commun. 2009, 380, 609–613. [Google Scholar] [CrossRef] [PubMed]
- Ueno, M.; Itoh, M.; Kong, L.; Sugihara, K.; Asano, M.; Takakura, N. PSF1 is essential for early embryogenesis in mice. Mol. Cell. Biol. 2005, 25, 10528–10532. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Sun, X.J.; Liu, C.; Wu, Q.F.; Tai, M.H.; Wei, J.C.; Lei, L.; Meng, F.D.; Qu, K.; Xu, J. Overexpression of PSF1 is correlated with poor prognosis in hepatocellular carcinoma patients. Int. J. Biol. Markers 2015, 30, e56–e64. [Google Scholar] [CrossRef] [PubMed]
- Panvichian, R.; Tantiwetrueangdet, A.; Angkathunyakul, N.; Leelaudomlipi, S. TOP2A amplification and overexpression in hepatocellular carcinoma tissues. Biomed. Res. Int. 2015, 2015, 381602. [Google Scholar] [CrossRef] [PubMed]
- Wong, N.; Yeo, W.; Wong, W.L.; Wong, N.L.; Chan, K.Y.; Mo, F.K.; Koh, J.; Chan, S.L.; Chan, A.T.; Lai, P.B.; et al. TOP2A overexpression in hepatocellular carcinoma correlates with early age onset, shorter patients survival and chemoresistance. Int. J. Cancer 2009, 124, 644–652. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Wang, L.; Jiang, M.; Lin, H.; Qi, L.; Diao, H. PTHLH coupling upstream negative regulation of fatty acid biosynthesis and Wnt receptor signal to downstream peptidase activity-induced apoptosis network in human hepatocellular carcinoma by systems-theoretical analysis. J. Recept. Signal Transduct. Res. 2012, 32, 250–256. [Google Scholar] [CrossRef] [PubMed]
- Jin, B.; Wang, W.; Du, G.; Huang, G.Z.; Han, L.T.; Tang, Y.Z.; Fan, D.G.; Li, J.; Zhang, S.Z. Identifying hub genes and dysregulated pathways in hepatocellular carcinoma. Eur. Rev. Med. Pharmacol. Sci. 2015, 19, 592–601. [Google Scholar] [PubMed]
- Qi, L.; Wang, L.; Huang, J.; Jiang, M.; Diao, H.; Zhou, H.; Li, X.; Jiang, Z. Activated amelogenin Y-linked (AMELY) regulation and angiogenesis in human hepatocellular carcinoma by biocomputation. Oncol. Lett. 2013, 5, 1075–1079. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Huang, J.; Jiang, M.; Lin, H.; Qi, L.; Diao, H. Activated PTHLH coupling feedback phosphoinositide to G-protein receptor signal-induced cell adhesion network in human hepatocellular carcinoma by systems-theoretic analysis. Sci. World J. 2012, 2012, 428979. [Google Scholar]
- Li, G.; Zhong, Y.; Shen, Q.; Zhou, Y.; Deng, X.; Li, C.; Chen, J.; Zhou, Y.; He, M. NEk2 serves as a prognostic biomarker for hepatocellular carcinoma. Int. J. Oncol. 2017, 50, 405–413. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Zhou, S.; Jiang, S.; Liu, X.; Wang, Y.; Zheng, X.; Zhou, H.; Li, X.; Cai, X. NEK2 regulates stem-like properties and predicts poor prognosis in hepatocellular carcinoma. Oncol. Rep. 2016, 36, 853–862. [Google Scholar] [CrossRef] [PubMed]
- Wen, S.; Liu, Y.; Yang, M.; Yang, K.; Huang, J.; Feng, D. Increased NEK2 in hepatocellular carcinoma promotes cancer progression and drug resistance by promoting PP1/Akt and Wnt activation. Oncol. Rep. 2016, 36, 2193–2199. [Google Scholar] [CrossRef] [PubMed]
- Wubetu, G.Y.; Morine, Y.; Teraoku, H.; Yoshikawa, M.; Ishikawa, D.; Yamada, S.; Ikemoto, T.; Saito, Y.U.; Imura, S.; Shimada, M. High NEK2 expression is a predictor of tumor recurrence in hepatocellular carcinoma patients after hepatectomy. Anticancer Res. 2016, 36, 757–762. [Google Scholar] [PubMed]
- Tian, H.; Ge, C.; Zhao, F.; Zhu, M.; Zhang, L.; Huo, Q.; Li, H.; Chen, T.; Xie, H.; Cui, Y.; et al. Downregulation of AZGP1 by Ikaros and histone deacetylase promotes tumor progression through the PTEN/Akt and CD44s pathways in hepatocellular carcinoma. Carcinogenesis 2017, 38, 207–217. [Google Scholar] [CrossRef] [PubMed]
- Koppenol, W.H.; Bounds, P.L.; Dang, C.V. Otto Warburg's contributions to current concepts of cancer metabolism. Nat. Rev. Cancer 2011, 11, 325–337. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Sanchez, R.; Rodriguez-Enriquez, S.; Marin-Hernandez, A.; Saavedra, E. Energy metabolism in tumor cells. FEBS J. 2007, 274, 1393–1418. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.L.; Lin, H.C.; Lin, K.H.; Lin, L.S.; Hsieh, C.E.; Ko, C.J.; Hung, Y.J.; Lin, P.Y. Low hemoglobin level is associated with the development of delirium after hepatectomy for hepatocellular carcinoma patients. PLoS ONE 2015, 10, e0119199. [Google Scholar] [CrossRef] [PubMed]
- Donadon, V.; Balbi, M.; Valent, F.; Avogaro, A. Glycated hemoglobin and antidiabetic strategies as risk factors for hepatocellular carcinoma. World J. Gastroenterol. 2010, 16, 3025–3032. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Li, L.Z.; Zhang, C.Z.; Yi, C.; Liu, L.L.; Zhou, X.; Xie, G.B.; Cai, M.Y.; Li, Y.; Yun, J.P. Decreased expression of zinc-alpha2-glycoprotein in hepatocellular carcinoma associates with poor prognosis. J. Transl. Med. 2012, 10, 106. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.Y.; Chen, R.; Yu, J.X.; Liu, T.; Qu, Y.; Lu, L.G. AZGP1 suppresses epithelial-to-mesenchymal transition and hepatic carcinogenesis by blocking TGFbeta1-ERK2 pathways. Cancer Lett. 2016, 374, 241–249. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.X.; Zhao, J.S.; Li, J.J.; Wang, T.; Cheng, S.Q.; Yuan, Y.; Wang, F.; Wang, X.F.; Xie, D. Liver cancer: EphrinA2 promotes tumorigenicity through Rac1/Akt/NF-kappaB signaling pathway. Hepatology 2010, 51, 535–544. [Google Scholar] [CrossRef] [PubMed]
- Qin, G.; Dang, M.; Gao, H.; Wang, H.; Luo, F.; Chen, R. Deciphering the protein-protein interaction network regulating hepatocellular carcinoma metastasis. Biochim. Biophys. Acta 2017, 1865, 1114–1122. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Module | Upstream Regulator | Type | p Value of Overlap | Target Molecules in Dataset |
|---|---|---|---|---|
| turquoise | MYC | transcription regulator | 3.94 × 10−10 | CCNB1, CCNB2, CDC20, CDK1, MCM6 |
| MED1 | transcription regulator | 1.06 × 10−9 | BIRC5, CCNB1, CDC20, CDK1, CDK4, CENPA | |
| FOXM1 | transcription regulator | 0.000308 | CCNB2, CDC20 | |
| ATP7B | transporter | 0.000373 | CDC20, PTTG1, TRIP13 | |
| CNR1 | g-protein coupled receptor | 0.00157 | CCNB2, CDK1 | |
| PPARA | ligand-dependent nuclear receptor | 0.00521 | CCNB1, CDK1, CDK4, H2AFZ, TOP2A | |
| TOB1 | transcription regulator | 0.00538 | HJURP, SPDL1 | |
| brown | NFE2L2 | transcription regulator | 0.00662 | ARF1, HSP90AB1, PSMA1, PSMC3, STIP1, TMED2 |
| LMF1 | other | 0.00911 | CANX | |
| mir-451 | microRNA | 0.00911 | YWHAZ | |
| orange | LEP | growth factor | 0.00714 | ACADM, EPHX2, LIPA, UGP2 |
| PLG | peptidase | 0.00787 | CLIC4, STAB2 | |
| yellow | PPARA | ligand-dependent nuclear receptor | 7.66 × 10−8 | ACAA1, ALDH2, ALDOB, C8A, CAT, CYP2B6, CYP2C8, CYP4A11, GPD1, GSTZ1, HPX, SCP2 |
| GPD1 | enzyme | 0.00000244 | C8A, CYP2B6, CYP2C8, CYP39A1, CYP4A22, F11 | |
| SLC25A13 | transporter | 0.00000244 | C8A, CYP2B6, CYP2C8, CYP39A1, CYP4A22, F11 | |
| FECH | enzyme | 0.0000416 | CYP2B6, CYP2C8, CYP4A11, SLC10A1 | |
| RORC | ligand-dependent nuclear receptor | 0.0000458 | CYP2A6 (includes others), CYP2B6, CYP2C8, CYP39A1, CYP4A11, RDH16 | |
| RORA | ligand-dependent nuclear receptor | 0.0000485 | CYP2A6 (includes others), CYP2B6, CYP2C8, CYP39A1, CYP4A11, RDH16 | |
| LEP | growth factor | 0.0000887 | ALDOB, CYP2C8, CYP4A11, PEMT, SCP2, SLC27A5 | |
| EHHADH | enzyme | 0.000106 | ACAA1, CYP4A11, SCP2 | |
| HSD17B4 | enzyme | 0.000106 | ACAA1, CYP4A11, SCP2 | |
| ACOX1 | enzyme | 0.000492 | ACAA1, CAT, CYP4A11 | |
| POR | enzyme | 0.000531 | CYP2A6 (includes others), CYP2B6, CYP2C8, CYP39A1, CYP4A11 | |
| TCF7L2 | transcription regulator | 0.00056 | ACAA1, CYP2C8, GYS2 | |
| AHR | ligand-dependent nuclear receptor | 0.000667 | ALDH2, CYP2B6, CYP2C8, RDH16, SLC22A7 | |
| NR1H4 | ligand-dependent nuclear receptor | 0.000878 | CYP2B6, LCAT, NR1I2, SLC10A1 | |
| HNF4A | transcription regulator | 0.0011 | CAT, HPX, NR1I2, SCP2, SLC10A1 | |
| ZBTB20 | transcription regulator | 0.00172 | CYP2B6, CYP2C8, GHR | |
| NR1I3 | ligand-dependent nuclear receptor | 0.00186 | CYP2A6 (includes others), CYP2B6, CYP2C8, CYP39A1 | |
| STAT1 | transcription regulator | 0.00344 | CYP2C8, CYP4A11 | |
| STAT6 | transcription regulator | 0.004 | CIDEB, CYP4A11 | |
| ABCC4 | transporter | 0.00687 | CYP2B6 | |
| IL25 | cytokine | 0.00687 | CIDEB | |
| TERC | other | 0.00894 | CYP2C8, CYP4A11 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yin, L.; Cai, Z.; Zhu, B.; Xu, C. Identification of Key Pathways and Genes in the Dynamic Progression of HCC Based on WGCNA. Genes 2018, 9, 92. https://doi.org/10.3390/genes9020092
Yin L, Cai Z, Zhu B, Xu C. Identification of Key Pathways and Genes in the Dynamic Progression of HCC Based on WGCNA. Genes. 2018; 9(2):92. https://doi.org/10.3390/genes9020092
Chicago/Turabian StyleYin, Li, Zhihui Cai, Baoan Zhu, and Cunshuan Xu. 2018. "Identification of Key Pathways and Genes in the Dynamic Progression of HCC Based on WGCNA" Genes 9, no. 2: 92. https://doi.org/10.3390/genes9020092
APA StyleYin, L., Cai, Z., Zhu, B., & Xu, C. (2018). Identification of Key Pathways and Genes in the Dynamic Progression of HCC Based on WGCNA. Genes, 9(2), 92. https://doi.org/10.3390/genes9020092