PClass: Protein Quaternary Structure Classification by Using Bootstrapping Strategy as Model Selection


 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Dataset
2.2. Feature Encoding
2.2.1. Amino Acid Composition
2.2.2. Shannon Entropy
2.2.3. Accessible Surface Area
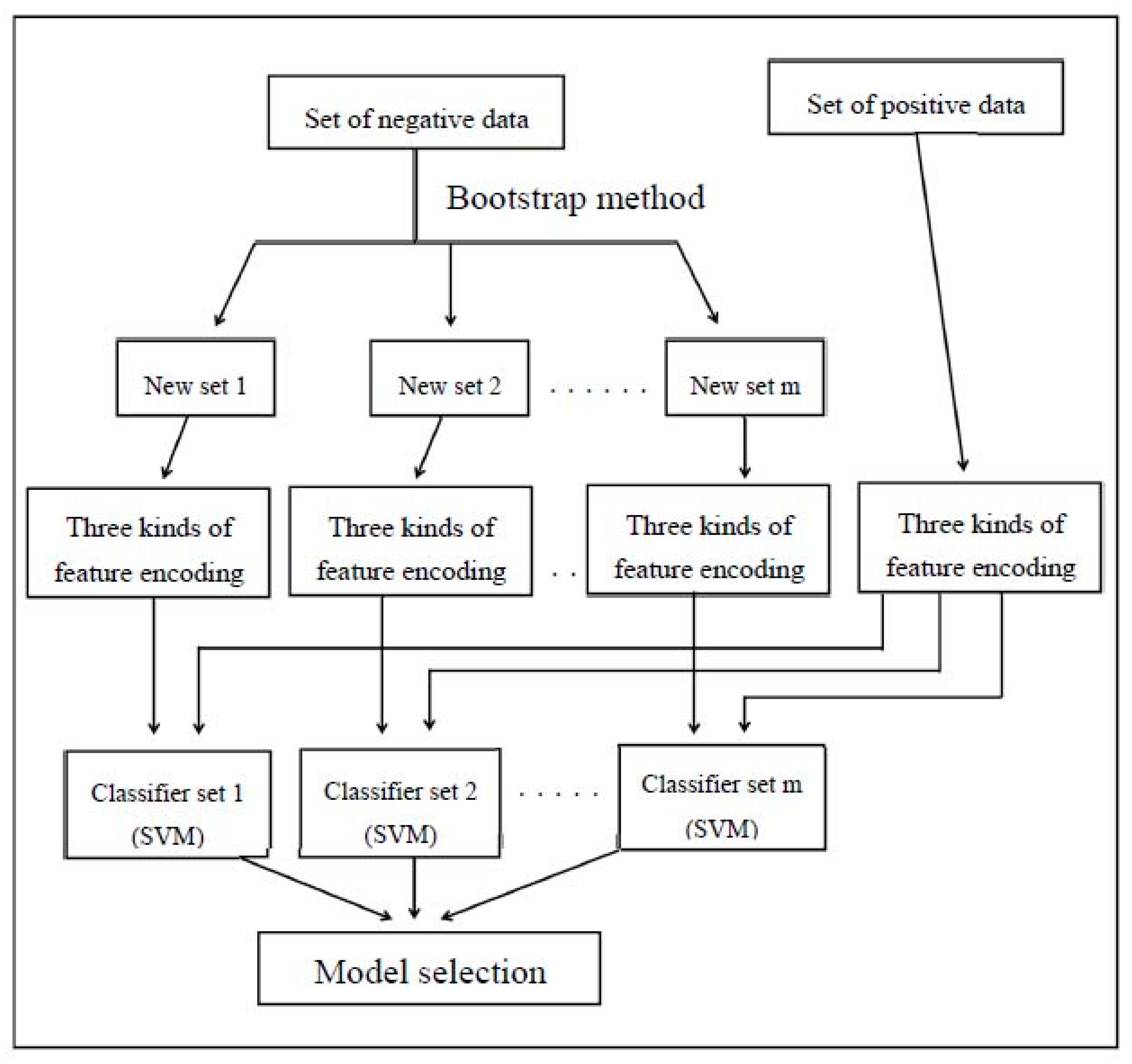
2.3. Model
3. Results and Discussion
3.1. Training of Feature Encoding in First Layer
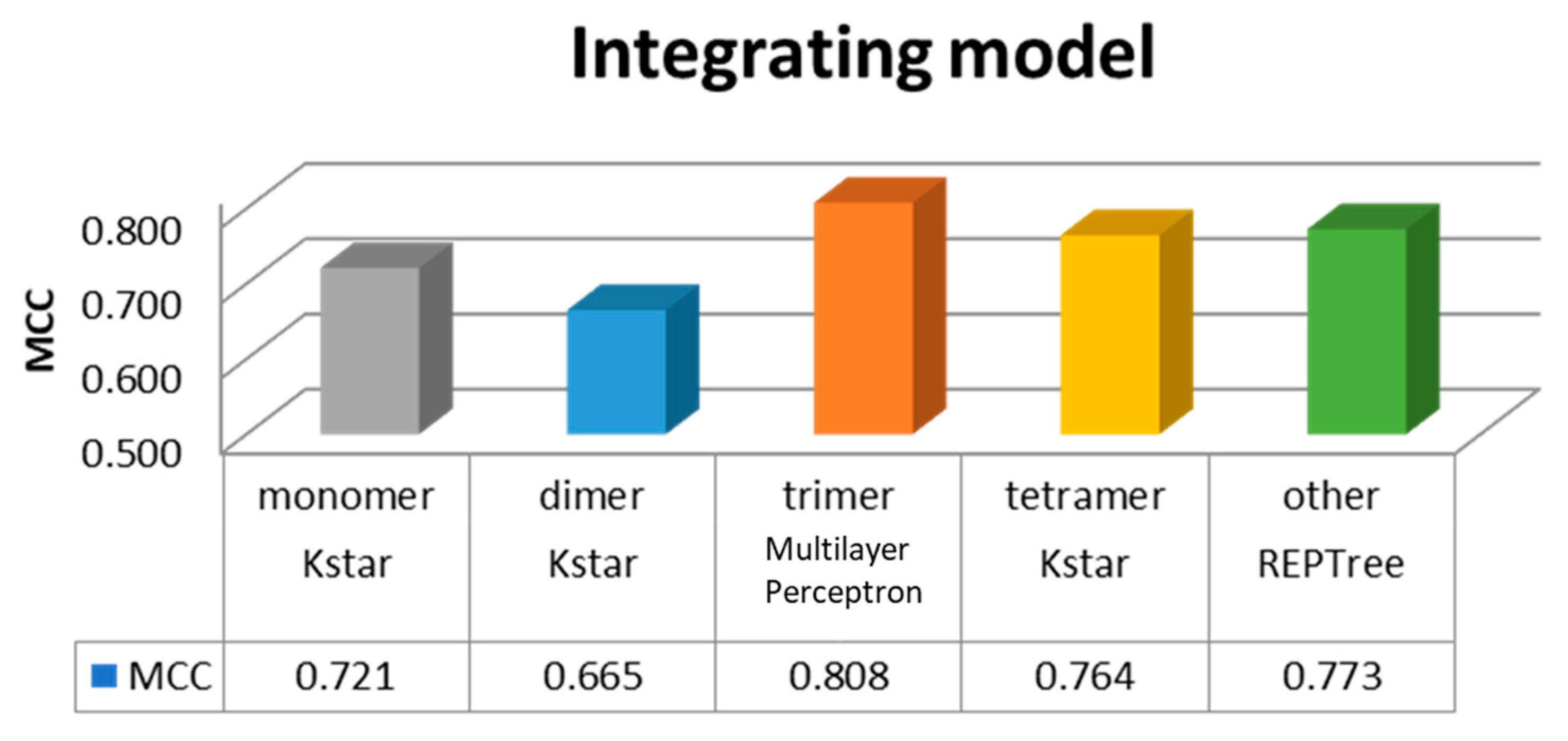
3.2. Training of Integrate Method in Two Layer
3.3. Bootstrap Method Compare with Other Method
3.4. Case Study
4. Conclusions
Supplementary Materials
Acknowledgements
Author Contributions
Conflicts of Interest
References
- Dmitriev, O.Y.; Jones, P.C.; Fillingame, R.H. Structure of the subunit c oligomer in the F1Fo ATP synthase: Model derived from solution structure of the monomer and cross-linking in the native enzyme. Proc. Natl. Acad. Sci. USA 1999, 96, 7785–7790. [Google Scholar] [CrossRef] [PubMed]
- Toledo-Ortiz, G.; Huq, E.; Quail, P.H. The Arabidopsis basic/helix-loop-helix transcription factor family. Plant Cell Online 2003, 15, 1749–1770. [Google Scholar] [CrossRef]
- Spivak-Kroizman, T.; Lemmon, M.; Dikic, I.; Ladbury, J.; Pinchasi, D.; Huang, J.; Jaye, M.; Crumley, G.; Schlessinger, J.; Lax, I. Heparin-induced oligomerization of FGF molecules is responsible for FGF receptor dimerization, activation, and cell proliferation. Cell 1994, 79, 1015–1024. [Google Scholar] [CrossRef]
- Mège, R.M.; Gavard, J.; Lambert, M. Regulation of cell–cell junctions by the cytoskeleton. Curr. Opin. Cell Boil. 2006, 18, 541–548. [Google Scholar] [CrossRef] [PubMed]
- Bulleid, N.J.; Dalley, J.A.; Lees, J.F. The C-propeptide domain of procollagen can be replaced with a transmembrane domain without affecting trimer formation or collagen triple helix folding during biosynthesis. EMBO J. 1997, 16, 6694–6701. [Google Scholar] [CrossRef] [PubMed]
- Gustchina, E.; Li, M.; Louis, J.M.; Anderson, D.E.; Lloyd, J.; Frisch, C.; Bewley, C.A.; Gustchina, A.; Wlodawer, A.; Clore, G.M. Structural basis of HIV-1 neutralization by affinity matured Fabs directed against the internal trimeric coiled-coil of gp41. PLoS Pathog. 2010, 6, e1001182. [Google Scholar] [CrossRef] [PubMed]
- Skehel, J.J.; Wiley, D.C. Receptor binding and membrane fusion in virus entry: The influenza hemagglutinin. Annu. Rev. Biochem. 2000, 69, 531–569. [Google Scholar] [CrossRef] [PubMed]
- Gascoigne, N.; Goodnow, C.C.; Dudzik, K.I.; Oi, V.T.; Davis, M.M. Secretion of a chimeric T-cell receptor-immunoglobulin protein. Proc. Natl. Acad. Sci. USA 1987, 84, 2936–2940. [Google Scholar] [CrossRef] [PubMed]
- Ackers, G.K.; Smith, F.R. The hemoglobin tetramer: A three-state molecular switch for control of ligand affinity. Annu. Rev. Biophys. Biophys. Chem. 1987, 16, 583–609. [Google Scholar] [CrossRef] [PubMed]
- Marttila, A.T.; Airenne, K.J.; Laitinen, O.H.; Kulik, T.; Bayer, E.A.; Wilchek, M.; Kulomaa, M.S. Engineering of chicken avidin: A progressive series of reduced charge mutants. FEBS Lett. 1998, 441, 313–317. [Google Scholar] [CrossRef]
- Bailey, S.; Eliason, W.K.; Steitz, T.A. Structure of hexameric DnaB helicase and its complex with a domain of DnaG primase. Science 2007, 318, 459–463. [Google Scholar] [CrossRef] [PubMed]
- Tsao, T.S.; Tomas, E.; Murrey, H.E.; Hug, C.; Lee, D.H.; Ruderman, N.B.; Heuser, J.E.; Lodish, H.F. Role of disulfide bonds in Acrp30/adiponectin structure and signaling specificity. J. Biol. Chem. 2003, 278, 50810–50817. [Google Scholar] [CrossRef] [PubMed]
- Ciszak, E.; Smith, G.D. Crystallographic evidence for dual coordination around zinc in the T3R3 human insulin hexamer. Biochemistry 1994, 33, 1512–1517. [Google Scholar] [CrossRef] [PubMed]
- Liang, W.G.; Ren, M.; Zhao, F.; Tang, W.-J. Structures of human ccl18, ccl3, and ccl4 reveal molecular determinants for quaternary structures and sensitivity to insulin-degrading enzyme. J. Mol. Biol. 2015, 427, 1345–1358. [Google Scholar] [CrossRef] [PubMed]
- Stenkamp, R.E. Dioxygen and hemerythrin. Chem. Rev. 1994, 94, 715–726. [Google Scholar] [CrossRef]
- Camahort, R.; Shivaraju, M.; Mattingly, M.; Li, B.; Nakanishi, S.; Zhu, D.; Shilatifard, A.; Workman, J.L.; Gerton, J.L. Cse4 is part of an octameric nucleosome in budding yeast. Mol. Cell 2009, 35, 794–805. [Google Scholar] [CrossRef] [PubMed]
- Darnell, J.E. Transcription factors as targets for cancer therapy. Nat. Rev. Cancer 2002, 2, 740–749. [Google Scholar] [CrossRef] [PubMed]
- Dowierciał, A.; Wilk, P.; Rypniewski, W.; Rode, W.; Jarmuła, A. Crystal structure of mouse thymidylate synthase in tertiary complex with dUMP and raltitrexed reveals N-terminus architecture and two different active site conformations. BioMed Res. Int. 2014, 2014, 945803. [Google Scholar] [CrossRef] [PubMed]
- Wibmer, C.K.; Gorman, J.; Ozorowski, G.; Bhiman, J.N.; Sheward, D.J.; Elliott, D.H.; Rouelle, J.; Smira, A.; Joyce, M.G.; Ndabambi, N. Structure and recognition of a novel HIV-1 gp120-gp41 interface antibody that caused MPER exposure through viral escape. PLoS Pathog. 2017, 13, e1006074. [Google Scholar] [CrossRef] [PubMed]
- Kovacs, J.M.; Nkolola, J.P.; Peng, H.; Cheung, A.; Perry, J.; Miller, C.A.; Seaman, M.S.; Barouch, D.H.; Chen, B. HIV-1 envelope trimer elicits more potent neutralizing antibody responses than monomeric gp120. Proc. Natl. Acad. Sci. USA 2012, 109, 12111–12116. [Google Scholar] [CrossRef] [PubMed]
- Katen, S.P.; Tan, Z.; Chirapu, S.R.; Finn, M.; Zlotnick, A. Assembly-directed antivirals differentially bind quasiequivalent pockets to modify hepatitis B virus capsid tertiary and quaternary structure. Structure 2013, 21, 1406–1416. [Google Scholar] [CrossRef] [PubMed]
- Ogura, T.; Tong, K.I.; Mio, K.; Maruyama, Y.; Kurokawa, H.; Sato, C.; Yamamoto, M. Keap1 is a forked-stem dimer structure with two large spheres enclosing the intervening, double glycine repeat, and c-terminal domains. Proc. Natl. Acad. Sci. USA 2010, 107, 2842–2847. [Google Scholar] [CrossRef] [PubMed]
- Junninen, H.; Ehn, M.; Petäjä, T.; Luosujärvi, L.; Kotiaho, T.; Kostiainen, R.; Rohner, U.; Gonin, M.; Fuhrer, K.; Kulmala, M. A high-resolution mass spectrometer to measure atmospheric ion composition. Atmos. Meas. Tech. 2010, 3, 1039–1053. [Google Scholar] [CrossRef]
- Assink, H.A.; Blijenberg, B.G.; Boerma, G.J.; Leijnse, B. The introduction of bromocresol purple for the determination of serum albumin on SMAC and ACA, and the standardization procedure. J. Clin. Chem. Clin. Biochem. 1984, 22, 685–692. [Google Scholar] [CrossRef] [PubMed]
- Chou, C.Y.; Lin, Y.L.; Huang, Y.C.; Sheu, S.Y.; Lin, T.H.; Tsay, H.J.; Chang, G.G.; Shiao, M.S. Structural variation in human apolipoprotein E3 and E4: Secondary structure, tertiary structure, and size distribution. Biophys. J. 2005, 88, 455–466. [Google Scholar] [CrossRef] [PubMed]
- Oxford, J.T.; DeScala, J.; Morris, N.; Gregory, K.; Medeck, R.; Irwin, K.; Oxford, R.; Brown, R.; Mercer, L.; Cusack, S. Interaction between amino propeptides of type xi procollagen α1 chains. J. Biol. Chem. 2004, 279, 10939–10945. [Google Scholar] [CrossRef] [PubMed]
- Wolf, E.; Kim, P.S.; Berger, B. Multicoil: A program for predicting two- and three-stranded coiled coils. Protein Sci. 1997, 6, 1179–1189. [Google Scholar] [CrossRef] [PubMed]
- Woolfson, D.N.; Alber, T. Predicting oligomerization states of coiled coils. Protein Sci. 1995, 4, 1596–1607. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, C.T.; Vincent, T.L.; Green, P.J.; Woolfson, D.N. SCORER 2.0: An algorithm for distinguishing parallel dimeric and trimeric coiled-coil sequences. Bioinformatics 2011, 27, 1908–1914. [Google Scholar] [CrossRef] [PubMed]
- Testa, O.D.; Moutevelis, E.; Woolfson, D.N. Cc+: A relational database of coiled-coil structures. Nucleic Acids Res. 2009, 37, D315–D322. [Google Scholar] [CrossRef] [PubMed]
- Levy, E.D.; Pereira-Leal, J.B.; Chothia, C.; Teichmann, S.A. 3D complex: A structural classification of protein complexes. PLoS Comput. Biol. 2006, 2, e155. [Google Scholar] [CrossRef] [PubMed]
- Riera-Fernández, P.; Munteanu, C.R.; Escobar, M.; Prado-Prado, F.; Martín-Romalde, R.; Pereira, D.; Villalba, K.; Duardo-Sánchez, A.; González-Díaz, H. New Markov-Shannon Entropy models to assess connectivity quality in complex networks: From molecular to cellular pathway, Parasite-Host, Neural, Industry, and Legal-Social networks. J. Theor. Biol. 2012, 293, 174–188. [Google Scholar] [CrossRef] [PubMed]
- Peek, A.S. Improving model predictions for RNA interference activities that use support vector machine regression by combining and filtering features. BMC Bioinform. 2007, 8, 182. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.; Richards, F.M. The interpretation of protein structures: Estimation of static accessibility. J. Mol. Biol. 1971, 55, 379–400. [Google Scholar] [CrossRef]
- Lin, Y.-S.; Hsu, W.-L.; Hwang, J.-K.; Li, W.-H. Proportion of solvent-exposed amino acids in a protein and rate of protein evolution. Mol. Biol. Evol. 2007, 24, 1005–1011. [Google Scholar] [CrossRef] [PubMed]
- Deng, L.; Guan, J.; Dong, Q.; Zhou, S. Prediction of protein-protein interaction sites using an ensemble method. BMC Bioinform. 2009, 10, 426. [Google Scholar] [CrossRef] [PubMed]
- Frank, E.; Hall, M.; Trigg, L.; Holmes, G.; Witten, I.H. Data mining in bioinformatics using Weka. Bioinformatics 2004, 20, 2479–2481. [Google Scholar] [CrossRef] [PubMed]
- Manavalan, B.; Lee, J.; Lee, J. Random forest-based protein model quality assessment (rfmqa) using structural features and potential energy terms. PLoS ONE 2014, 9, e106542. [Google Scholar] [CrossRef] [PubMed]
- Cao, R.; Adhikari, B.; Bhattacharya, D.; Sun, M.; Hou, J.; Cheng, J. Qacon: Single model quality assessment using protein structural and contact information with machine learning techniques. Bioinformatics 2017, 33, 586–588. [Google Scholar] [CrossRef] [PubMed]
- Manavalan, B.; Lee, J. Svmqa: Support–vector-machine-based protein single-model quality assessment. Bioinformatics 2017, 33, 2496–2503. [Google Scholar] [CrossRef] [PubMed]
- Manavalan, B.; Basith, S.; Shin, T.H.; Choi, S.; Kim, M.O.; Lee, G. Mlacp: Machine-learning-based prediction of anticancer peptides. Oncotarget 2017, 8, 77121–77136. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Yao, S.; Li, X.; Chen, C.; Hu, X. Genome-wide prediction of DNA methylation using DNA composition and sequence complexity in human. Int. J. Mol. Sci. 2017, 18, 420. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Training Set | Independent Test Set | |
|---|---|---|
| Monomer | 11,638 | 1513 |
| Dimer | 8570 | 1005 |
| Trimer | 1231 | 119 |
| Tetramer | 2764 | 282 |
| Other | 1527 | 176 |
| Training Set | Independent Test Set | |||
|---|---|---|---|---|
| Positive | Negative | Positive | Negative | |
| Monomer | 11,638 | 14,092 | 1535 | 1582 |
| Dimer | 8570 | 17,160 | 1005 | 2112 |
| Trimer | 1231 | 24,499 | 119 | 2998 |
| Tetramer | 2764 | 22,966 | 282 | 2835 |
| Other | 1527 | 24,203 | 176 | 2941 |
| Trimer | Tetramer | Other | |
|---|---|---|---|
| Bootstrap | 0.696 | 0.741 | 0.757 |
| Random | 0.676 | 0.727 | 0.738 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, C.-C.; Chang, C.-C.; Chen, C.-W.; Ho, S.-y.; Chang, H.-P.; Chu, Y.-W. PClass: Protein Quaternary Structure Classification by Using Bootstrapping Strategy as Model Selection. Genes 2018, 9, 91. https://doi.org/10.3390/genes9020091
Huang C-C, Chang C-C, Chen C-W, Ho S-y, Chang H-P, Chu Y-W. PClass: Protein Quaternary Structure Classification by Using Bootstrapping Strategy as Model Selection. Genes. 2018; 9(2):91. https://doi.org/10.3390/genes9020091
Chicago/Turabian StyleHuang, Chi-Chou, Chi-Chang Chang, Chi-Wei Chen, Shao-yu Ho, Hsung-Pin Chang, and Yen-Wei Chu. 2018. "PClass: Protein Quaternary Structure Classification by Using Bootstrapping Strategy as Model Selection" Genes 9, no. 2: 91. https://doi.org/10.3390/genes9020091
APA StyleHuang, C. -C., Chang, C. -C., Chen, C. -W., Ho, S. -y., Chang, H. -P., & Chu, Y. -W. (2018). PClass: Protein Quaternary Structure Classification by Using Bootstrapping Strategy as Model Selection. Genes, 9(2), 91. https://doi.org/10.3390/genes9020091