Genome Sequence of the Freshwater Yangtze Finless Porpoise




Abstract
:
{kind=link}
{kind=link}
1. Introduction
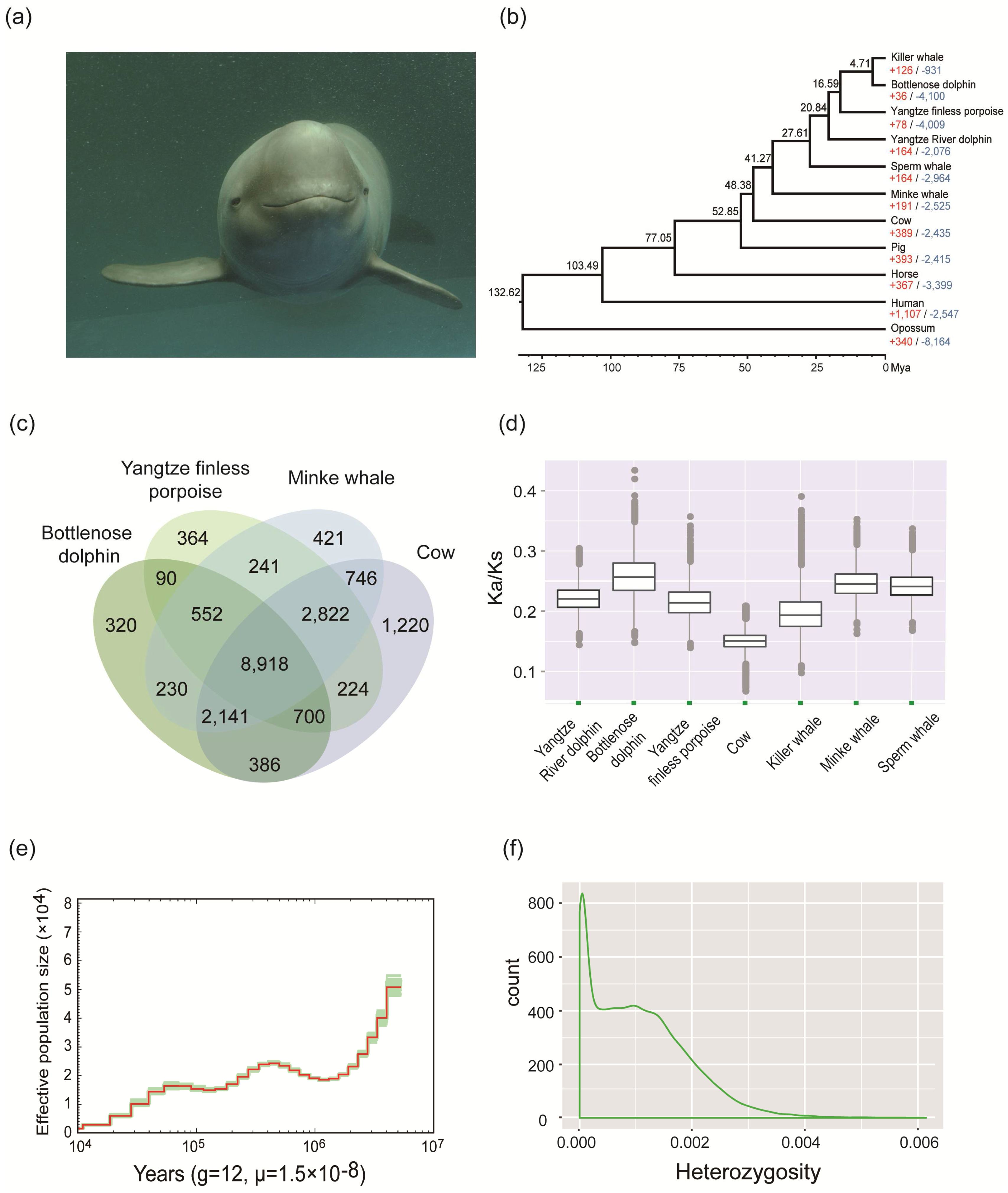
2. Materials and Methods, Results, and Discussion
Supplementary Material
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Wang, D.; Liu, R.; Zhang, X.; Yang, J.; Wei, Z.; Zhao, Q.; Wang, X. Status and conservation of the Yangtze finless porpoise. In Biology and Conservation of Freshwater Cetaceans in Asia; IUCN: Gland, Switzerland, 2000; pp. 81–85. [Google Scholar]
- Pilleri, G.; Gihr, M. On the taxonomy and ecology of the finless black porpoise, Neophocaena (Cetacea, Delphinidae). Mammalia 1975, 39, 657–674. [Google Scholar] [CrossRef] [PubMed]
- Turvey, S.T.; Pitman, R.L.; Taylor, B.L.; Barlow, J.; Akamatsu, T.; Barrett, L.A.; Zhao, X.; Reeves, R.R.; Stewart, B.S.; Wang, K.; et al. First human-caused extinction of a cetacean species? Biol. Lett. 2007, 3, 537–540. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.F.; Liu, R.J.; Zhao, Q.Z.; Zhang, G.C.; Wei, Z.; Wang, X.Q.; Yang, J. The population of finless porpoise in the middle and lower reaches of Yangtze River. Acta Theriol. Sin. 1993, 13, 260–270. [Google Scholar]
- Zhao, X.J.; Barlow, J.; Taylor, B.L.; Pitman, R.L.; Wang, K.; Wei, Z.; Stewart, B.S.; Turvey, S.T.; Akamatsu, T.; Reeves, R.R.; et al. Abundance and conservation status of the Yangtze finless porpoise in the Yangtze River, China. Biol. Conserv. 2008, 141, 3006–3018. [Google Scholar] [CrossRef]
- Mei, Z.G.; Zhang, X.Q.; Huang, S.-L.; Zhao, X.J.; Hao, Y.J.; Zhang, L.; Qian, Z.Y.; Zheng, J.S.; Wang, K.X.; Wang, D.; et al. The Yangtze finless porpoise: On an accelerating path to extinction? Biol. Conserv. 2014, 172, 117–123. [Google Scholar] [CrossRef]
- Mei, Z.G.; Huang, S.L.; Hao, Y.J.; Turvey, S.T.; Gong, W.M.; Wang, D. Accelerating population decline of Yangtze finless porpoise (Neophocaena asiaeorientalis asiaeorientalis). Biol. Conserv. 2012, 153, 192–200. [Google Scholar] [CrossRef]
- Wang, D.; Turvey, S.T.; Zhao, X.J.; Mei, Z.G. Neophocaena asiaeorientalis ssp. asiaeorientalis. IUCN Red List of Threatened Species. 2013. Version 2013.1. Available online: http://www.iucnredlist.org (accessed on 16 July 2013).
- Hayashi, K.; Yoshida, H.; Nishida, S.; Goto, M.; Pastene, L.A.; Kanda, N.; Baba, Y.; Koike, H. Genetic variation of the MHC DQB locus in the finless porpoise (Neophocaena phocaenoides). Zool. Sci. 2006, 23, 147–153. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.Y.; Frasier, T.R.; Yang, S.C.; White, B.N. Detecting recent speciation events: The case of the finless porpoise (genus Neophocaena). Heredity 2008, 101, 145–155. [Google Scholar] [CrossRef] [PubMed]
- Jefferson, T.A.; Wang, J.Y. Revision of the taxonomy of finless porpoises (genus Neophocaena): The existence of two species. J. Mar. Anim. Ecol. 2011, 4, 3–16. [Google Scholar]
- Luo, R.; Liu, B.; Xie, Y.; Li, Z.; Huang, W.; Yuan, J.; He, J.; Chen, Y.; Pan, Q.; Liu, Y.; et al. SOAPdenovo2: An empirically improved memory-efficient short-read de novo assembler. GigaScience 2012, 1, 18. [Google Scholar] [CrossRef] [PubMed]
- Song, L.; Florea, L.; Langmead, B. Lighter: Fast and memory-efficient sequencing error correction without counting. Genome Biol. 2014, 15, 509. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Q. Draft genome of the milu (Elaphurus davidianus). GigaScience 2017, 7, 1–6. [Google Scholar] [CrossRef]
- Zhou, X.; Sun, F.; Xu, S.; Fan, G.; Zhu, K.; Liu, X.; Yuan, C.; Shi, C.; Yang, Y.; Huang, Z.; et al. Baiji genomes reveal low genetic variability and new insights into secondary aquatic adaptations. Nat. Commun. 2013, 4, 2708. [Google Scholar] [CrossRef] [PubMed]
- Yim, H.S.; Cho, Y.S.; Guang, X.; Kang, S.G.; Jeong, J.Y.; Cha, S.S.; Oh, H.M.; Lee, J.H.; Yang, E.C.; Kwon, K.K.; et al. Minke whale genome and aquatic adaptation in cetaceans. Nat. Genet. 2014, 46, 88–92. [Google Scholar] [CrossRef] [PubMed]
- Kajitani, R.; Toshimoto, K.; Noguchi, H.; Toyoda, A.; Ogura, Y.; Okuno, M.; Yabana, M.; Harada, M.; Nagayasu, E.; Maruyama, H.; et al. Efficient de novo assembly of highly heterozygous genomes from whole-genome shotgun short reads. Genome Res. 2014, 24, 1384–1395. [Google Scholar] [CrossRef] [PubMed]
- Simao, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef] [PubMed]
- Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv, 2013; arXiv:13033997. [Google Scholar]
- Xu, Z.; Wang, H. LTR_FINDER: An efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Res. 2007, 35, W265–W268. [Google Scholar] [CrossRef] [PubMed]
- Tarailo-Graovac, M.; Chen, N. Using RepeatMasker to identify repetitive elements in genomic sequences. Curr. Protoc. Bioinform. 2009. [Google Scholar] [CrossRef]
- Foote, A.D.; Liu, Y.; Thomas, G.W.C.; Vinar, T.; Alfoldi, J.; Deng, J.; Duan, S.; van Elk, C.E.; Hunter, M.E.; Joshi, V.; et al. Convergent evolution of the genomes of marine mammals. Nat. Genet. 2015, 47, 272–275. [Google Scholar] [CrossRef] [PubMed]
- Arnason, U.; Gullberg, A.; Gretarsdottir, S.; Ursing, B.; Janke, A. The mitochondrial genome of the sperm whale and a new molecular reference for estimating eutherian divergence dates. J. Mol. Evol. 2000, 50, 569–578. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Slater, G.S.; Birney, E. Automated generation of heuristics for biological sequence comparison. BMC Bioinform. 2005, 6, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stanke, M.; Diekhans, M.; Baertsch, R.; Haussler, D. Using native and syntenically mapped cDNA alignments to improve de novo gene finding. Bioinformatics 2008, 24, 637–644. [Google Scholar] [CrossRef] [PubMed]
- Blanco, E.; Parra, G.; Guigo, R. Using geneid to identify genes. Curr. Protoc. Bioinform. 2007. [Google Scholar] [CrossRef]
- Majoros, W.H.; Pertea, M.; Salzberg, S.L. TigrScan and GlimmerHMM: Two open source ab initio eukaryotic gene-finders. Bioinformatics 2004, 20, 2878–2879. [Google Scholar] [CrossRef] [PubMed]
- Haas, B.J.; Salzberg, S.L.; Zhu, W.; Pertea, M.; Allen, J.E.; Orvis, J.; White, O.; Buell, C.R.; Wortman, J.R. Automated eukaryotic gene structure annotation using EVidenceModeler and the program to assemble spliced alignments. Genome Biol. 2008, 9, R7. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, S.; Kakuta, M.; Ishida, T.; Akiyama, Y. Faster sequence homology searches by clustering subsequences. Bioinformatics 2015, 31, 1183–1190. [Google Scholar] [CrossRef] [PubMed]
- Yates, A.; Akanni, W.; Amode, M.R.; Barrell, D.; Billis, K.; Carvalho-Silva, D.; Cummins, C.; Clapham, P.; Fitzgerald, S.; Gil, L.; et al. Ensembl 2016. Nucleic Acids Res. 2016, 44, D710–D716. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Stoeckert, C.J., Jr.; Roos, D.S. OrthoMCL: Identification of ortholog groups for eukaryotic genomes. Genome Res. 2003, 13, 2178–2189. [Google Scholar] [CrossRef] [PubMed]
- De Bie, T.; Cristianini, N.; Demuth, J.P.; Hahn, M.W. CAFE: A computational tool for the study of gene family evolution. Bioinformatics 2006, 22, 1269–1271. [Google Scholar] [CrossRef] [PubMed]
- Loytynoja, A.; Goldman, N. An algorithm for progressive multiple alignment of sequences with insertions. Proc. Natl. Acad. Sci. USA 2005, 102, 10557–10562. [Google Scholar] [CrossRef] [PubMed]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [PubMed]
- Drummond, A.J.; Suchard, M.A.; Xie, D.; Rambaut, A. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol. Biol. Evol. 2012, 29, 1969–1973. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Wang, Y.; Zhao, Y.; Zhang, X.; Li, R.; Chen, L.; Zhang, G.; Jiang, Y.; Qiu, Q.; Wang, W.; et al. Draft genome of the Marco Polo Sheep (Ovis ammon polii). GigaScience 2017, 6, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Talavera, G.; Castresana, J. Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst. Biol. 2007, 56, 564–577. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z. PAML 4: Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef] [PubMed]
- Alberga, D.; Trisciuzzi, D.; Lattanzi, G.; Bennett, J.L.; Verkman, A.S.; Felice Mangiatordi, G.; Nicolotti, G. Comparative molecular dynamics study of neuromyelitis optica-immunoglobulin G binding to aquaporin-4 extracellular domains. Biochim. Biophys. Acta (BBA) Biomembr. 2017, 1859, 1326–1334. [Google Scholar] [CrossRef] [PubMed]
- Yoshikawa, Y.; Nakayama, T.; Saito, K.; Hui, P.; Morita, A.; Sato, N.; Takahashi, T.; Tamura, M.; Sato, I.; Aoi, N.; et al. Haplotype-based case-control study of the association between the guanylate cyclase activator 2B (GUCA2B, Uroguanylin) gene and essential hypertension. Hypertens. Res. 2017, 30, 789–796. [Google Scholar] [CrossRef] [PubMed]
- Ramjeesingh, M.; Li, C.; Kogan, I.; Wang, Y.; Huan, L.J.; Bear, C.E. A monomer is the minimum functional unit required for channel and ATPase activity of the cystic fibrosis transmembrane conductance regulator. Biochemistry 2001, 40, 10700–10706. [Google Scholar] [CrossRef] [PubMed]
- Fagerberg, L.; Hallström, B.M.; Oksvold, P.; Kampf, C.; Djureinovic, D.; Odeberg, J.; Habuka, M.; Tahmasebpoor, S.; Danielsson, A.; Edlund, K.; et al. Analysis of the human tissue-specific expression by genome-wide integration of transcriptomics and antibody-based proteomics. Mol. Cell. Proteom. 2014, 13, 397–406. [Google Scholar] [CrossRef] [PubMed]
- Post, S.M.; Tomkinson, A.E.; Lee, E.Y. The human checkpoint Rad protein Rad17 is chromatin-associated throughout the cell cycle, localizes to DNA replication sites, and interacts with DNA polymerase ε. Nucleic Acids Res. 2003, 31, 5568–5575. [Google Scholar] [CrossRef] [PubMed]
- Rautio, M.; Tartarotti, B. UV radiation and freshwater zooplankton: Damage, protection and recovery. Freshw. Rev. J. Freshw. Biol. Assoc. 2010, 3, 105–131. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Wang, L.; Han, J.; Tang, X.; Ma, M.; Wang, K.; Zhang, X.; Ren, Q.; Chen, Q.; Qiu, Q. Comparative transcriptomic analysis revealed adaptation mechanism of Phrynocephalus erythrurus, the highest altitude lizard living in the Qinghai-Tibet Plateau. BMC Evol. Biol. 2015, 15, 101. [Google Scholar] [CrossRef] [PubMed]
- Shen, T.; Xu, S.; Wang, X.; Yu, W.; Zhou, K.; Yang, G. Adaptive evolution and functional constraint at TLR4 during the secondary aquatic adaptation and diversification of cetaceans. BMC Evol. Biol. 2012, 12, 39. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Yang, Y.; Zhou, X.; Xu, J.; Zhou, K.; Yang, G. Adaptive evolution of the osmoregulation-related genes in cetaceans during secondary aquatic adaptation. BMC Evol. Boil. 2013, 13, 189. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; 1000 Genome Project Data Processing Subgroup. The sequence alignment/map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Inference of human population history from individual whole-genome sequences. Nature 2011, 475, 493–496. [Google Scholar] [CrossRef] [PubMed]
- Cingolani, P.; Platts, A.; Wang, L.L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef] [PubMed]

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yuan, Y.; Zhang, P.; Wang, K.; Liu, M.; Li, J.; Zheng, J.; Wang, D.; Xu, W.; Lin, M.; Dong, L.; et al. Genome Sequence of the Freshwater Yangtze Finless Porpoise. Genes 2018, 9, 213. https://doi.org/10.3390/genes9040213
Yuan Y, Zhang P, Wang K, Liu M, Li J, Zheng J, Wang D, Xu W, Lin M, Dong L, et al. Genome Sequence of the Freshwater Yangtze Finless Porpoise. Genes. 2018; 9(4):213. https://doi.org/10.3390/genes9040213
Chicago/Turabian StyleYuan, Yuan, Peijun Zhang, Kun Wang, Mingzhong Liu, Jing Li, Jinsong Zheng, Ding Wang, Wenjie Xu, Mingli Lin, Lijun Dong, and et al. 2018. "Genome Sequence of the Freshwater Yangtze Finless Porpoise" Genes 9, no. 4: 213. https://doi.org/10.3390/genes9040213
APA StyleYuan, Y., Zhang, P., Wang, K., Liu, M., Li, J., Zheng, J., Wang, D., Xu, W., Lin, M., Dong, L., Zhu, C., Qiu, Q., & Li, S. (2018). Genome Sequence of the Freshwater Yangtze Finless Porpoise. Genes, 9(4), 213. https://doi.org/10.3390/genes9040213