The Changing Landscape in the Genetic Etiology of Human Tooth Agenesis


Abstract
:1. Introduction
2. Genetic Basis of Tooth Agenesis
2.1. Syndromes Associated with Tooth Agenesis
2.1.1. Ectodermal Dysplasia Syndromes
2.1.2. Oral-Facial Cleft Syndromes
2.1.3. Axenfeld–Rieger Syndrome
2.1.4. Familial Adenomatous Polyposis Syndrome
2.1.5. Oligodontia–Colorectal Cancer Syndrome
2.2. Isolated (Nonsyndromic) Tooth Agenesis
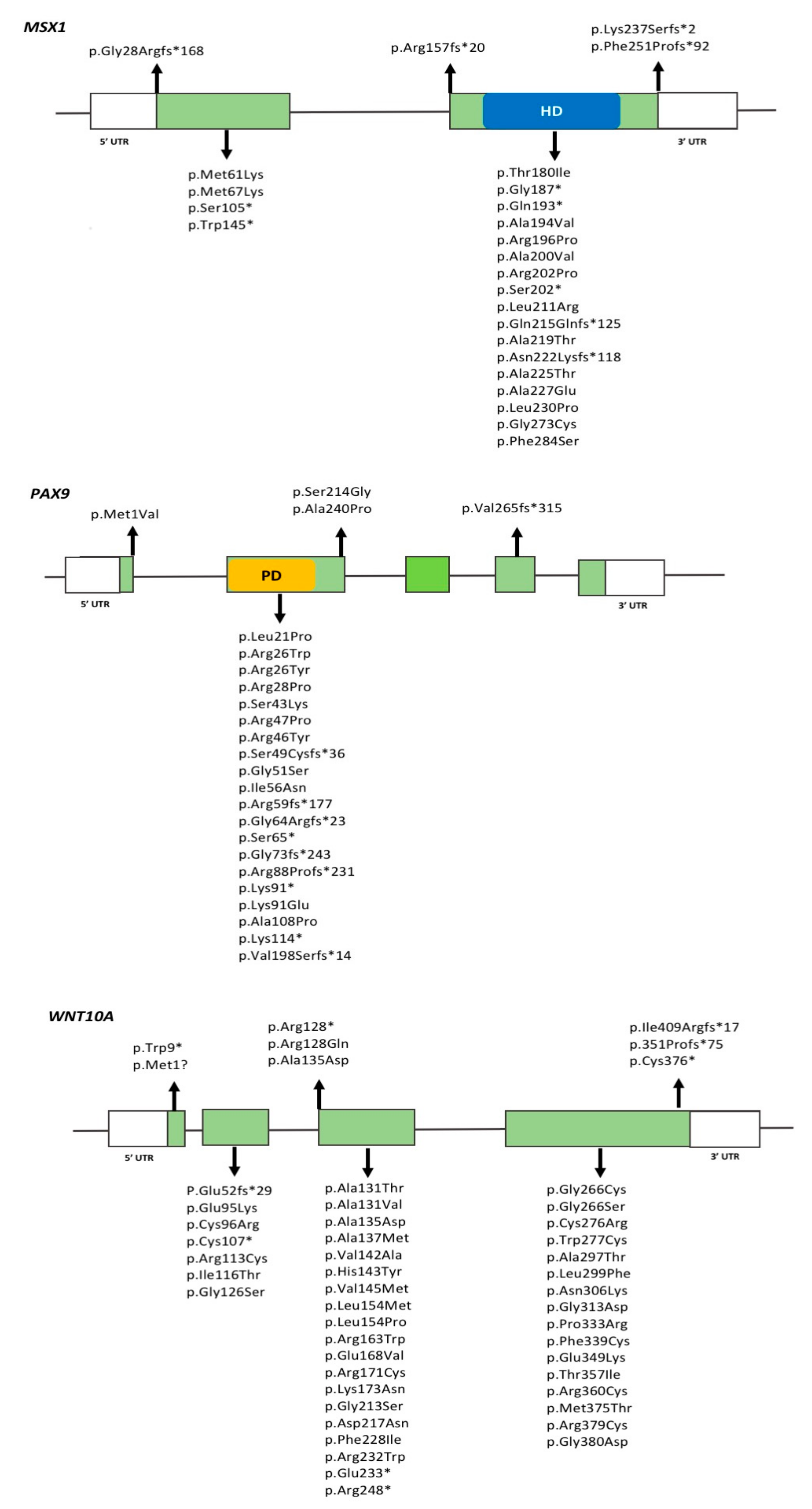
2.2.1. MSX1
2.2.2. PAX9
2.2.3. AXIN2
2.2.4. WNT10A
2.2.5. LRP6
2.2.6. Other Genes Recently Implicated in TA
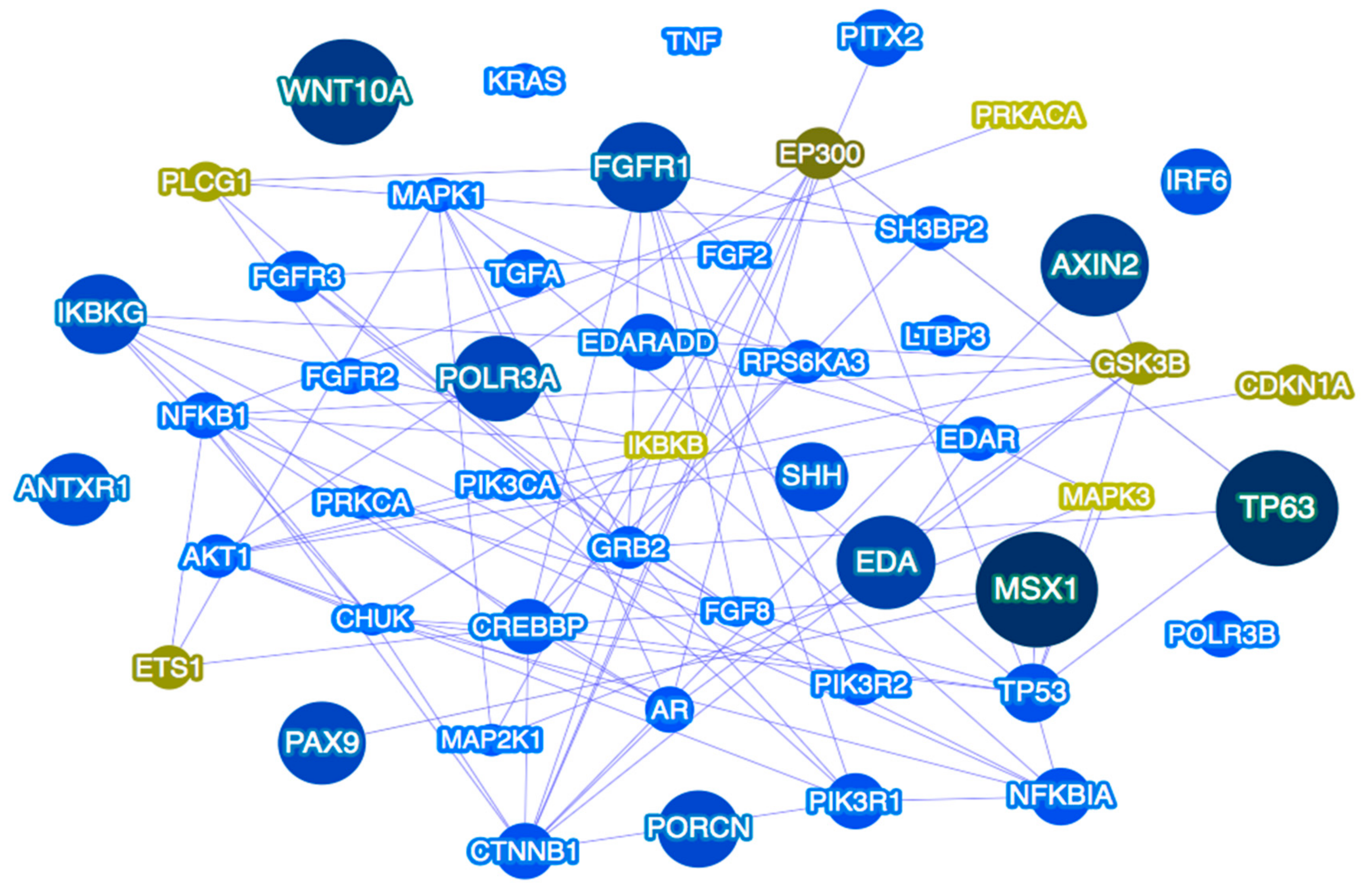
3. Monogenic vs. Oligogenic Inheritance Models
4. Genetic Pathways as the Focus of Future Studies
5. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Bei, M. Molecular genetics of tooth development. Curr. Opin. Genet. Dev. 2009, 19, 504–510. [Google Scholar] [CrossRef] [PubMed]
- Thesleff, I.; Vaahtokari, A.; Vainio, S.; Jowett, A. Molecular mechanisms of cell and tissue interactions during early tooth development. Anat. Rec. 1996, 245, 151–161. [Google Scholar] [CrossRef]
- Gorlin, R.J.; Cohen, M.M., Jr.; Levin, S.L. Syndromes of the Head and Neck, 3rd ed.; Oxford University Press: New York, NY, USA, 1990. [Google Scholar]
- Yin, W.; Bian, Z. The gene network underlying hypodontia. J. Dent. Res. 2015, 94, 878–885. [Google Scholar] [CrossRef] [PubMed]
- Polder, B.J.; Van’t Hof, M.A.; Van der Linden, F.P.; Kuijpers-Jagtman, A.M. A meta-analysis of the prevalence of dental agenesis of permanent teeth. Commun. Dent. Oral Epidemiol. 2004, 32, 217–226. [Google Scholar] [CrossRef] [PubMed]
- Carter, K.; Worthington, S. Morphologic and demographic predictors of third molar agenesis: A systematic review and meta-analysis. J. Dent. Res. 2015, 94, 886–894. [Google Scholar] [CrossRef] [PubMed]
- Ye, X.; Attaie, A.B. genetic basis of nonsyndromic and syndromic tooth agenesis. J. Pediatr. Genet. 2016, 5, 198–208. [Google Scholar] [CrossRef] [PubMed]
- Lan, Y.; Jia, S.; Jiang, R. Molecular patterning of the mammalian dentition. Semin. Cell Dev. Biol. 2014, 25, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Sun, K.; Shen, Y.; Xu, Y.; Xie, J.; Huang, R.; Zhang, Y.; Xu, C.; Zhang, X.; Wang, R.; et al. DNA methylation is critical for tooth agenesis: Implications for sporadic non-syndromic anodontia and hypodontia. Sci. Rep. 2016, 6, 19162. [Google Scholar] [CrossRef] [PubMed]
- Dinckan, N.; Du, R.; Akdemir, Z.C.; Bayram, Y.; Jhangiani, S.N.; Doddapaneni, H.; Hu, J.; Muzny, D.M.; Guven, Y.; Aktoren, O.; et al. A biallelic ANTXR1 variant expands the anthrax toxin receptor associated phenotype to tooth agenesis. Am. J. Med. Genet. A 2018, 176, 1015–1022. [Google Scholar] [CrossRef] [PubMed]
- Dinckan, N.; Du, R.; Petty, L.E.; Coban-Akdemir, Z.; Jhangiani, S.N.; Paine, I.; Baugh, E.H.; Erdem, A.P.; Kayserili, H.; Doddapaneni, H.; et al. Whole-exome sequencing identifies novel variants for tooth agenesis. J. Dent. Res. 2018, 97, 49–59. [Google Scholar] [CrossRef] [PubMed]
- Nieminen, P. Genetic basis of tooth agenesis. J. Exp. Zool. B Mol. Dev. Evol. 2009, 312, 320–342. [Google Scholar] [CrossRef] [PubMed]
- Howe, B.J.; Cooper, M.E.; Vieira, A.R.; Weinberg, S.M.; Resick, J.M.; Nidey, N.L.; Wehby, G.L.; Marazita, M.L.; Moreno Uribe, L.M. Spectrum of dental phenotypes in nonsyndromic orofacial clefting. J. Dent. Res. 2015, 94, 905–912. [Google Scholar] [CrossRef] [PubMed]
- Vieira, A.R. Oral clefts and syndromic forms of tooth agenesis as models for genetics of isolated tooth agenesis. J. Dent. Res. 2003, 82, 162–165. [Google Scholar] [CrossRef] [PubMed]
- Colige, A.; Nuytinick, L.; Hausser, I.; van Esse, A.J.; Thiry, M.; Herens, C.; Adès, L.C.; Malfait, F.; Paepe, A.D.; Franck, P.; et al. Novel types of mutation responsible for the dermatosparactic type of Ehlers-Danlos syndrome (type VIIC) and common polymorphisms in the ADAMTS2 gene. J. Invest. Dermatol. 2004, 123, 656–663. [Google Scholar] [CrossRef] [PubMed]
- Colige, A.; Sieron, A.L.; Li, S.W.; Schwarze, U.; Petty, E.; Wertelecki, W.; Wilcox, W.; Krakow, D.; Cohn, D.H.; Reardon, W.; et al. Human Ehlers-Danlos syndrome type VIIC and bovine dermatosparaxis are caused by mutations in the procollagen I N-proteinase gene. Am. J. Hum. Genet. 1999, 65, 308–317. [Google Scholar] [CrossRef] [PubMed]
- Bekhouche, M.; Colige, A. The procollagen N-proteinases ADAMTS2, 3 and 14 in pathophysiology. Matrix Biol. 2015, 44, 46–53. [Google Scholar] [CrossRef] [PubMed]
- Bayram, Y.; Pehlivan, D.; Karaca, E.; Gambin, T.; Jhangiani, S.N.; Erdin, S.; Elcioglu, N.H. Whole exome sequencing identifies three novel mutations in ANTXR1 in families with GAPO syndrome. Am. J. Med. Genet. A 2014, 164, 2328–2334. [Google Scholar] [CrossRef] [PubMed]
- Stranecky, V.; Hoischen, A.; Hartmannová, H.; Zaki, M.S.; Chaudhary, A.; Zudaie, E.; Nosková, L.; Baresová, V.; Pristoupilová, A.; Hodanová, K.; et al. Mutations in ANTXR1 cause GAPO syndrome. Am. J. Hum. Genet. 2013, 92, 792–799. [Google Scholar] [CrossRef] [PubMed]
- Salas-Alanis, J.C.; Scott, C.A.; Fajardo-Ramírez, O.R.; Duran, C.; Moreno-Treviño, M.G.; Kelsell, D.P. New ANTXR1 gene mutation for GAPO syndrome: A case report. Mol. Syndromol. 2016, 7, 160–163. [Google Scholar] [CrossRef] [PubMed]
- Marvin, M.L.; Mazzoni, S.M.; Herron, C.M.; Edwards, S.; Gruber, S.B.; Petty, E.M. AXIN2-associated autosomal dominant ectodermal dysplasia and neoplastic syndrome. Am. J. Med. Genet. A 2011, 155, 898–902. [Google Scholar] [CrossRef] [PubMed]
- Lammi, L.; Arte, S.; Somer, M.; Järvinen, H.; Lahermo, P.; Thesleff, I.; Pirinen, S.; Nieminen, P. Mutations in AXIN2 cause familial tooth agenesis and predispose to colorectal cancer. Am. J. Hum. Genet. 2004, 74, 1043–1050. [Google Scholar] [CrossRef] [PubMed]
- Malmgren, B.; Andersson, K.; Lindahl, K.; Kindmark, A.; Grigelioniene, G.; Zachariadis, V.; Dahllöf, G.; Aström, E. Tooth agenesis in osteogenesis imperfecta related to mutations in the collagen type I genes. Oral Dis. 2017, 23, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Bloch-Zupan, A.; Stachtou, J.; Emmanouil, D.; Arveiler, B.; Griffiths, D.; Lacombe, D. Oro-dental features as useful diagnostic tool in Rubinstein-Taybi syndrome. Am. J. Med. Genet. A 2007, 143, 570–573. [Google Scholar] [CrossRef] [PubMed]
- Monreal, A.W.; Zonana, J.; Ferguson, B. Identification of a new splice form of the EDA1 gene permits detection of nearly all X-linked hypohidrotic ectodermal dysplasia mutations. Am. J. Hum. Genet. 1998, 63, 380–389. [Google Scholar] [CrossRef] [PubMed]
- Han, D.; Gong, Y.; Wu, H.; Zhang, X.; Yan, M.; Wang, X.; Qu, H.; Feng, H.; Song, S. Novel EDA mutation resulting in X-linked non-syndromic hypodontia and the pattern of EDA-associated isolated tooth agenesis. Eur. J. Med. Genet. 2008, 51, 536–546. [Google Scholar] [CrossRef] [PubMed]
- Lexner, M.O.; Bardow, A.; Juncker, I.; Jensen, L.G.; Almer, L.; Kreiborg, S.; Hertz, J.M. X-linked hypohidrotic ectodermal dysplasia. Genetic and dental findings in 67 Danish patients from 19 families. Clin. Genet. 2008, 74, 252–259. [Google Scholar] [CrossRef] [PubMed]
- Tao, R.; Jin, B.; Guo, S.Z.; Qing, W.; Feng, G.Y.; Brooks, D.G.; Liu, L.; Xu, J.; Li, T.; Yan, Y. A novel missense mutation of the EDA gene in a Mongolian family with congenital hypodontia. J. Hum. Genet. 2006, 51, 498–502. [Google Scholar] [CrossRef] [PubMed]
- Henningsen, E.; Svendsen, M.T.; Lidballe, D.L.; Jensen, P.K. A novel mutation in the EDAR gene causes severe autosomal recessive hypohidrotic ectodermal dysplasia. Am. J. Med. Genet. A 2014, 164, 2059–2061. [Google Scholar] [CrossRef] [PubMed]
- Naeem, M.; Muhammad, D.; Ahmad, W. Novel mutations in the EDAR gene in two Pakistani consanguineous families with autosomal recessive hypohidrotic ectodermal dysplasia. Br. J. Dermatol. 2005, 153, 46–50. [Google Scholar] [CrossRef] [PubMed]
- Naqvi, S.K.; Wasif, N.; Javaid, H.; Ahmad, W. Two novel mutations in the gene EDAR causing autosomal recessive hypohidrotic ectodermal dysplasia. Orthod. Craniofac. Res. 2011, 14, 156–159. [Google Scholar] [CrossRef] [PubMed]
- Shimomura, Y.; Sato, N.; Miyashita, A.; Hashimoto, T.; Ito, M.; Kuwano, R. A rare case of hypohidrotic ectodermal dysplasia caused by compound heterozygous mutations in the EDAR gene. J. Invest. Dermatol. 2004, 123, 649–655. [Google Scholar] [CrossRef] [PubMed]
- Zeng, B.; Zhao, Q.; Li, S.; Lu, H.; Lu, J.; Ma, L.; Zhao, W.; Yu, D. Novel EDA or EDAR Mutations identified in patients with X-linked hypohidrotic ectodermal dysplasia or non-syndromic tooth agenesis. Genes 2017, 8, 259. [Google Scholar] [CrossRef] [PubMed]
- Bal, E.; Baala, L.; Cluzeau, C.; El Kerch, F.; Ouldim, K.; Hadj-Rabia, S.; Bodemer, C.; Munnich, A.; Courtois, G.; Sefiani, A. Autosomal dominant anhidrotic ectodermal dysplasias at the EDARADD locus. Hum. Mutat. 2007, 28, 703–709. [Google Scholar] [CrossRef] [PubMed]
- Headon, D.J.; Emmal, S.A.; Ferguson, B.M.; Tucker, A.S.; Justice, M.J.; Sharpe, P.T.; Zonana, J.; Overbeek, P.A. Gene defect in ectodermal dysplasia implicates a death domain adapter in development. Nature 2001, 414, 913–916. [Google Scholar] [CrossRef] [PubMed]
- Wohlfart, S.; Söder, S.; Smahi, A.; Schneider, H. A novel missense mutation in the gene EDARADD associated with an unusual phenotype of hypohidrotic ectodermal dysplasia. Am. J. Med. Genet. A 2016, 170, 249–253. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Perez, V.L.; Ide, S.E.; Strom, T.M.; Lorenz, B.; Wilson, D.; Woods, K.; King, L.; Francomano, C.; Freisinger, P.; Spranger, S.; et al. Mutations in a new gene in Ellis-van Creveld syndrome and Weyers acrodental dysostosis. Nat. Genet. 2000, 24, 283–286. [Google Scholar] [CrossRef] [PubMed]
- D’Asdia, M.C.; Torrent, I.; Consoli, F.; Ferese, R.; Magliozzi, M.; Bernardini, L.; Guida, V.; Diglio, M.C.; Marino, B.; Dallapiccola, B.; et al. Novel and recurrent EVC and EVC2 mutations in Ellis-van Creveld syndrome and Weyers acrofacial dyostosis. Eur. J. Med. Genet. 2013, 56, 80–87. [Google Scholar] [CrossRef] [PubMed]
- Ye, X.; Song, G.; Fan, M.; Shi, L.; Jabs, E.W.; Huang, S.; Guo, R.; Bian, Z. A novel heterozygous deletion in the EVC2 gene causes Weyers acrofacial dysostosis. Hum Genet 2006, 119, 199–205. [Google Scholar] [CrossRef] [PubMed]
- Shen, W.; Han, D.; Zhang, J.; Zhao, H.; Feng, H. Two novel heterozygous mutations of EVC2 cause a mild phenotype of Ellis-van Creveld syndrome in a Chinese family. Am. J. Med. Genet. A 2011, 155, 2131–2136. [Google Scholar] [CrossRef] [PubMed]
- Entesarian, M.; Matsson, H.; Klar, J.; Bergendal, B.; Olson, L.; Arakaki, R.; Hayashi, Y.; Ohuchi, H.; Falahat, B.; Bolstad, A.I.; et al. Mutations in the gene encoding fibroblast growth factor 10 are associated with aplasia of lacrimal and salivary glands. Nat Genet 2005, 37, 125–127. [Google Scholar] [CrossRef] [PubMed]
- Milunsky, J.M.; Zhao, G.; Maher, T.A.; Colby, R.; Everman, D.B. LADD syndrome is caused by FGF10 mutations. Clin. Genet. 2006, 69, 349–354. [Google Scholar] [CrossRef] [PubMed]
- Rohmann, E.; Brunner, H.G.; Kayserili, H.; Uyguner, O.; Nürnberg, G.; Lew, E.D.; Dobbie, A.; Eswarakumar, V.P.; Uzumcu, A.; Ulubil-Emeroglu, A.; et al. Mutations in different components of FGF signaling in LADD syndrome. Nat. Genet. 2006, 38, 414–417. [Google Scholar] [CrossRef] [PubMed]
- Bailleul-Forestier, I.; Gros, C.; Zenaty, D.; Bennaceur, S.; Leger, J.; de Roux, N. Dental agenesis in Kallmann syndrome individuals with FGFR1 mutations. Int. J. Paediatr. Dent. 2010, 20, 305–312. [Google Scholar] [CrossRef] [PubMed]
- Letra, A.; de Almeida, A.L.; Kaizer, R.; Esper, L.A.; Sgarbosa, S.; Granjeiro, J.M. Intraoral features of Apert’s syndrome. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. Endod. 2007, 103, e38–e41. [Google Scholar] [CrossRef] [PubMed]
- Stavropoulos, D.; Bartzela, T.; Bronkhorst, E.; Mohlin, B.; Hagberg, C. Dental agenesis patterns of permanent teeth in Apert syndrome. Eur. J. Oral Sci. 2011, 119, 198–203. [Google Scholar] [CrossRef] [PubMed]
- Ibrahimi, O.A.; Chiu, E.S.; McCarthy, J.G.; Mohammadi, M. Understanding the molecular basis of Apert syndrome. Plast. Reconstr. Surg. 2005, 115, 264–270. [Google Scholar] [PubMed]
- Meyers, G.A.; Orlow, S.J.; Munro, I.R.; Przylepa, K.A.; Jabs, E.W. Fibroblast growth factor receptor 3 (FGFR3) transmembrane mutation in Crouzon syndrome with acanthosis nigricans. Nat. Genet. 1995, 11, 462–464. [Google Scholar] [CrossRef] [PubMed]
- Reardon, W.; Wilkes, D.; Rutland, P.; Pulleyn, L.J.; Malcolm, S.; Dean, J.C.; Evans, R.D.; Jones, B.M.; Hayward, R.; Hall, C.M. Craniosynostosis associated with FGFR3 pro250arg mutation results in a range of clinical presentations including unisutural sporadic craniosynostosis. J. Med. Genet. 1997, 34, 632–636. [Google Scholar] [CrossRef] [PubMed]
- Wilkes, D.; Rutland, P.; Pulleyn, L.J.; Reardon, W.; Moss, C.; Ellis, J.P.; Winter, R.M.; Malcolm, S. A recurrent mutation, ala391glu, in the transmembrane region of FGFR3 causes Crouzon syndrome and acanthosis nigricans. J. Med. Genet. 1996, 33, 744–748. [Google Scholar] [CrossRef] [PubMed]
- Tsang, M.C.; Ling, J.Y.; King, N.M.; Chow, S.K. Oral and craniofacial morphology of a patient with Larsen syndrome. J. Craniofac. Genet. Dev. Biol. 1986, 6, 357–362. [Google Scholar] [PubMed]
- Berenstein-Aizman, G.; Hazan-Molina, H.; Drori, D.; Aizenbud, D. Axenfeld-Rieger syndrome: Dentofacial manifestation and oral rehabilitation considerations. Pediatr. Dent. 2011, 33, 440–444. [Google Scholar] [PubMed]
- O’Dwyer, E.M.; Jones, D.C. Dental anomalies in Axenfeld-Rieger syndrome. Int. J. Paediatr. Dent. 2005, 15, 459–463. [Google Scholar] [CrossRef] [PubMed]
- Kjaer, K.W.; Hansen, L.; Eiberg, H.; Leicht, P.; Opitz, J.N.; Tommerup, N. Novel Connexin 43 (GJA1) mutation causes oculo-dento-digital dysplasia with curly hair. Am. J. Med. Genet. A 2004, 127, 152–157. [Google Scholar] [CrossRef] [PubMed]
- Paznekas, W.A.; Karczeski, B.; Vermeer, S.; Lowry, R.B.; Delatycki, M.; Laurence, F.; Koivisto, P.A.; Van Maldergem, L.; Boyadjiev, S.A.; Bodurtha, J.N. GJA1 mutations, variants, and connexin 43 dysfunction as it relates to the oculodentodigital dysplasia phenotype. Hum. Mutat. 2009, 30, 724–733. [Google Scholar] [CrossRef] [PubMed]
- Petrof, G.; Nanda, A.; Howden, J.; Takeichi, T.; McMillan, J.R.; Aristodemou, S.; Ozoemena, L.; Liu, L.; South, A.P.; Purreyron, C.; et al. Mutations in GRHL2 result in an autosomal-recessive ectodermal Dysplasia syndrome. Am. J. Hum. Genet. 2014, 95, 308–314. [Google Scholar] [CrossRef] [PubMed]
- Kondo, S.; Schutte, B.C.; Richardsson, R.J.; Bjork, B.C.; Knight, A.S.; Watanabe, Y.; Howard, E.; de Lima, R.L.; Daack-Hirsch, S.; Sander, A.; et al. Mutations in IRF6 cause Van der Woude and popliteal pterygium syndromes. Nat. Genet. 2002, 32, 285–289. [Google Scholar] [CrossRef] [PubMed]
- Zucchero, T.M.; Cooper, M.E.; Maher, B.S.; Daak-Hirsch, S.; Nepomuceno, B.; Ribeiro, L.; Caprau, D.; Christensen, K.; Suzuki, Y.; Machida, J.; et al. Interferon regulatory factor 6 (IRF6) gene variants and the risk of isolated cleft lip or palate. N. Engl. J. Med. 2004, 351, 769–780. [Google Scholar] [CrossRef] [PubMed]
- Ho, N.C.; Lacbawan, F.; Francomano, C.A.; Ho, V. Severe hypodontia and oral xanthomas in Alagille syndrome. Am. J. Med. Genet. 2000, 93, 250–252. [Google Scholar] [CrossRef]
- Lederer, D.; Shears, D.; Benoit, V.; Verellen-Dumoulin, C.; Maystadt, I. A three generation X-linked family with Kabuki syndrome phenotype and a frameshift mutation in KDM6A. Am. J. Med. Genet. A 2014, 164, 1289–1292. [Google Scholar] [CrossRef] [PubMed]
- Van Laarhoven, P.M.; Neitzel, L.R.; Quintana, A.M.; Geiger, E.A.; Zackai, E.H.; Clouthier, D.E.; Artinger, K.B.; Ming, J.E.; Shaik, T.H. Kabuki syndrome genes KMT2D and KDM6A: Functional analyses demonstrate critical roles in craniofacial, heart and brain development. Hum. Mol. Genet. 2015, 24, 4443–4453. [Google Scholar] [CrossRef] [PubMed]
- Lerone, M.; Priolo, M.; Naselli, A.; Vignolo, A.; Romeo, G.; Silengo, M.C. Ectodermal abnormalities in Kabuki syndrome. Am. J. Med. Genet. 1997, 73, 263–266. [Google Scholar] [CrossRef]
- Matsune, K.; Matsune, K.; Shimizu, T.; Tohma, T.; Asada, Y.; Ohashi, H.; Maeda, T. Craniofacial and dental characteristics of Kabuki syndrome. Am. J. Med. Genet. 2001, 98, 185–190. [Google Scholar] [CrossRef]
- Issa, Y.A.; Kamal, L.; Rayyan, A.A.; Dweik, D.; Pierce, S.; Lee, M.K.; King, M.C.; Walsh, T.; Kanaan, M. Mutation of KREMEN1, a modulator of Wnt signaling, is responsible for ectodermal dysplasia including oligodontia in Palestinian families. Eur. J. Hum. Genet. 2016, 24, 1430–1435. [Google Scholar] [CrossRef] [PubMed]
- Forsythe, E.; Kenny, J.; Bacchelli, C.; Beales, P.L. Managing Bardet-Biedl syndrome-now and in the future. Front Pediatr. 2018, 6, 23. [Google Scholar] [CrossRef] [PubMed]
- Jumlongras, D.; Bei, M.; Stimson, J.M.; Wen-Fang, W.; DePalma, S.R.; Seidman, C.E.; Felbor, U.; Maas, R.; Seidman, J.G. A nonsense mutation in MSX1 causes Witkop syndrome. Am. J. Hum. Genet. 2001, 69, 67–74. [Google Scholar] [CrossRef] [PubMed]
- Aradhya, S.; Woffendin, H.; Jakins, T.; Bardaro, T.; Esposito, T.; Smahi, A.; Shaw, C.; Levy, M.; Munnich, A.; D’Urso, M. A recurrent deletion in the ubiquitously expressed NEMO (IKK-gamma) gene accounts for the vast majority of incontinentia pigmenti mutations. Hum. Mol. Genet. 2001, 10, 2171–2179. [Google Scholar] [CrossRef] [PubMed]
- Ku, C.L.; Dupuis-Girod, S.; Dittrich, A.M.; Bustamante, J.; Santos, O.F.; Schulze, I.; Bertrand, Y.; Couly, G.; Bodemer, C.; Bossuy, X. NEMO mutations in 2 unrelated boys with severe infections and conical teeth. Pediatrics 2005, 115, e615–e619. [Google Scholar] [CrossRef] [PubMed]
- Hirai, N.; Matsune, K.; Ohashi, H. Craniofacial and oral features of Sotos syndrome: Differences in patients with submicroscopic deletion and mutation of NSD1 gene. Am. J. Med. Genet. A 2011, 155, 2933–2939. [Google Scholar] [CrossRef] [PubMed]
- Kotilainen, J.; Pohjola, P.; Pirinen, S.; Arte, S.; Niminen, P. Premolar hypodontia is a common feature in Sotos syndrome with a mutation in the NSD1 gene. Am. J. Med. Genet. A 2009, 149, 2409–2414. [Google Scholar] [CrossRef] [PubMed]
- Shotelersuk, V.; Tifft, C.J.; Vacha, S.; Peters, K.F.; Biesecker, L.G. Discordance of oral-facial-digital syndrome type 1 in monozygotic twin girls. Am. J. Med. Genet. 1999, 86, 269–273. [Google Scholar] [CrossRef]
- Larralde de Luna, M.; Raspa, M.L.; Ibargoyen, J. Oral-facial-digital type 1 syndrome of Papillon-Leage and Psaume. Pediatr. Dermatol. 1992, 9, 52–56. [Google Scholar] [CrossRef] [PubMed]
- Brunner, H.G.; Hamel, B.C.; Bokhoven, H.V. P63 gene mutations and human developmental syndromes. Am. J. Med. Genet. 2002, 112, 284–290. [Google Scholar] [CrossRef] [PubMed]
- Rodini, E.O.; Freitas, J.A.; Richieri-Costa, A. Rapp-Hodgkin syndrome: Report of a Brazilian family. Am. J. Med. Genet. 1990, 36, 463–466. [Google Scholar] [CrossRef] [PubMed]
- Sripathomsawat, W.; Tanpaiboon, P.; Heering, J.; Dötsch, V.; Hennekam, R.C.; Kantaputra, P. Phenotypic analysis of Arg227 mutations of TP63 with emphasis on dental phenotype and micturition difficulties in EEC syndrome. Am. J. Med. Genet. A 2011, 155, 228–232. [Google Scholar] [CrossRef] [PubMed]
- Brooks, B.P.; Moroi, S.E.; Downs, C.A.; Wiltse, S.; Othman, M.I.; Semina, E.V.; Richards, J.E. A novel mutation in the PITX2 gene in a family with Axenfeld-Rieger syndrome. Ophthalmic Genet. 2004, 25, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.F.; Pressman, C.; Dyer, R.; Johnson, R.L.; Martin, J.F. Function of Rieger syndrome gene in left-right asymmetry and craniofacial development. Nature 1999, 401, 276–278. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Venugopalan, S.R.; Cao, H.; Pinho, F.O.; Paine, M.L.; Snead, M.L.; Semina, E.V.; Amendt, B.A. A model for the molecular underpinnings of tooth defects in Axenfeld-Rieger syndrome. Hum. Mol. Genet. 2014, 23, 194–208. [Google Scholar] [CrossRef] [PubMed]
- Bustos, T.; Simosa, V.; Pinto-Cisternas, J.; Abramovits, W.; Jolay, L.; Rodriguez, L.; Fernandez, L.; Ramela, M. Autosomal recessive ectodermal dysplasia: I. An undescribed dysplasia/malformation syndrome. Am. J. Med. Genet. 1991, 41, 398–404. [Google Scholar] [CrossRef] [PubMed]
- Sozen, M.A.; Suzuki, K.; Tolarova, M.M.; Bustos, T.; Fernandez Iglesias, J.E.; Spritz, R.A. Mutation of PVRL1 is associated with sporadic, non-syndromic cleft lip/palate in northern Venezuela. Nat. Genet. 2001, 29, 141–142. [Google Scholar] [CrossRef] [PubMed]
- Starr, D.G.; McClure, J.P.; Connor, J.M. Non-dermatological complications and genetic aspects of the Rothmund-Thomson syndrome. Clin. Genet. 1985, 27, 102–104. [Google Scholar] [CrossRef] [PubMed]
- Hunter, A.G. Coffin-Lowry syndrome: A 20-year follow-up and review of long-term outcomes. Am. J. Med. Genet. 2002, 111, 345–355. [Google Scholar] [CrossRef] [PubMed]
- Nanni, L.; Ming, J.E.; Du, Y.; Hall, R.K.; Aldred, M.; Bankier, A.; Muenke, M. SHH mutation is associated with solitary median maxillary central incisor: A study of 13 patients and review of the literature. Am. J. Med. Genet. 2001, 102, 1–10. [Google Scholar] [CrossRef]
- Petryk, A.; Graf, D.; Marcucio, R. Holoprosencephaly: Signaling interactions between the brain and the face, the environment and the genes, and the phenotypic variability in animal models and humans. Wiley Interdiscip. Rev. Dev. Biol. 2015, 4, 17–32. [Google Scholar] [CrossRef] [PubMed]
- Bamshad, M.; Le, T.; Watjkins, W.S.; Dixon, M.E.; Kramer, B.E.; Roeder, A.D.; Carey, J.C.; Root, S.; Schinzel, A.; Van Maldergem, L. The spectrum of mutations in TBX3: Genotype/Phenotype relationship in ulnar-mammary syndrome. Am. J. Hum. Genet. 1999, 64, 1550–1562. [Google Scholar] [CrossRef] [PubMed]
- Lowry, R.B.; Morgan, K.; Holmes, T.M.; Metcalf, P.J.; Stauffer, G.F. Mandibulofacial dysostosis in Hutterite sibs: A possible recessive trait. Am. J. Med. Genet. 1985, 22, 501–512. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Mernagh, J.; Bourgeois, J. Novel craniofacial and extracraniofacial findings in a case of Treacher Collins syndrome with a pathogenic mutation and a missense variant in the TCOF1 gene. Clin. Dysmorphol. 2009, 18, 63–66. [Google Scholar] [CrossRef] [PubMed]
- Mani, A.; Radhakrishnan, J.; Farhi, A.; Carew, K.S.; Waarnes, C.A.; Nelson-Williams, C.; Day, R.W.; Pober, B.; State, M.W.; Lifton, R.P. Syndromic patent ductus arteriosus: Evidence for haploinsufficient TFAP2B mutations and identification of a linked sleep disorder. Proc. Natl. Acad. Sci. USA 2005, 102, 2975–2979. [Google Scholar] [CrossRef] [PubMed]
- Korenberg, J.R. Toward a molecular understanding of Down syndrome. Prog. Clin. Biol. Res. 1993, 384, 87–115. [Google Scholar] [PubMed]
- Palaska, P.K.; Antonarakis, G.S. Prevalence and patterns of permanent tooth agenesis in individuals with Down syndrome: A meta-analysis. Eur. J. Oral Sci. 2016, 124, 317–328. [Google Scholar] [CrossRef] [PubMed]
- Peled, A.; Sarig, O.; Samuelov, L.; Bertolini, M.; Ziv, L.; Weissglas-Volkov, D.; Eskin-Schwartz, M.; Adase, C.A.; Malchin, N.; Bochner, R.; et al. Mutations in TSPEAR, encoding a regulator of notch signaling, affect tooth and hair follicle morphogenesis. PLoS Genet. 2016, 12, e1006369. [Google Scholar] [CrossRef] [PubMed]
- Almashraki, N.; Abdulnabee, M.Z.; Sukalo, M.; Alrajoudi, A.; Sharafadeen, I.; Zenker, M. Johanson-Blizzard syndrome. World J. Gastroenterol. 2011, 17, 4247–4250. [Google Scholar] [CrossRef] [PubMed]
- Adaimy, L.; Chouery, E.; Megarbane, H.; Mroueh, S.; Delague, V.; Nicolas, E.; Belguith, H.; de Mazancourt, P.; Megarbane, A. Mutation in WNT10A is associated with an autosomal recessive ectodermal dysplasia: The odonto-onycho-dermal dysplasia. Am. J. Hum. Genet. 2007, 81, 821–828. [Google Scholar] [CrossRef] [PubMed]
- Bohring, A.; Stamm, T.; Spaich, C.; Haase, C.; Spree, K.; Hehr, U.; Hoffmann, M.; Ledig, S.; Sel, S.; Wieacker, P.; et al. WNT10A mutations are a frequent cause of a broad spectrum of ectodermal dysplasias with sex-biased manifestation pattern in heterozygotes. Am. J. Hum. Genet. 2009, 85, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Nawaz, S.; Klar, J.; Wajid, M.; Aslam, M.; Tariq, M.; Schuster, J.; Baig, S.M.; Dahl, N. WNT10A missense mutation associated with a complete odonto-onycho-dermal dysplasia syndrome. Eur. J. Hum. Genet. 2009, 17, 1600–1605. [Google Scholar] [CrossRef] [PubMed]
- Monk, B.E.; Pieris, S.; Soni, V. Schopf-Schulz-Passarge syndrome. Br. J. Dermatol. 1992, 127, 33–35. [Google Scholar] [CrossRef] [PubMed]
- Schopf, E.; Schulz, H.J.; Passarge, E. Syndrome of cystic eyelids, palmo-plantar keratosis, hypodontia and hypotrichosis as a possible autosomal recessive trait. Birth Defects Orig. Artic. Ser. 1971, 7, 219–221. [Google Scholar] [PubMed]
- Bergendal, B.; Klar, J.; Stecksén-Blicks, C.; Norderyd, J.; Dahl, N. Isolated oligodontia associated with mutations in EDARADD, AXIN2, MSX1, and PAX9 genes. Am. J. Med. Genet. A 2011, 155, 1616–1622. [Google Scholar] [CrossRef] [PubMed]
- Hudson, C.D.; Witkop, C.J. Autosomal dominant hypodontia with nail dysgenesis. Report of twenty-nine cases in six families. Oral Surg. Oral Med. Oral Pathol. 1975, 39, 409–423. [Google Scholar] [CrossRef]
- Satokata, I.; Maas, R. Msx1 deficient mice exhibit cleft palate and abnormalities of craniofacial and tooth development. Nat. Genet. 1994, 6, 348–356. [Google Scholar] [CrossRef] [PubMed]
- Jarvinen, E.; Salazar-Ciudad, I.; Birchmeier, W.; Taketo, M.M.; Jernvall, J.; Thesleff, I. Continuous tooth generation in mouse is induced by activated epithelial Wnt/beta-catenin signaling. Proc. Natl. Acad. Sci. USA 2006, 103, 18627–18632. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Millar, S.E. Wnt/beta-catenin signaling in oral tissue development and disease. J. Dent. Res. 2010, 89, 318–330. [Google Scholar] [CrossRef] [PubMed]
- Yamashiro, T.; Zheng, L.; Shitaku, Y.; Saito, M.; Tsubakimoto, T.; Takada, K.; Takano-Yamamoto, T.; Thesleff, I. Wnt10a regulates dentin sialophosphoprotein mRNA expression and possibly links odontoblast differentiation and tooth morphogenesis. Differentiation 2007, 75, 452–462. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Lany, Y.; Baek, J.A.; Gao, Y.; Jiang, R. Wnt/beta-catenin signaling plays an essential role in activation of odontogenic mesenchyme during early tooth development. Dev. Biol. 2009, 334, 174–185. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Wang, S.K.; Choi, M.; Reid, B.M.; Hu, Y.; Lee, Y.L.; Herzog, C.R.; Kim-Berman, H.; Lee, M.; Benke, P.J.; et al. Taurodontism, variations in tooth number, and misshapened crowns in Wnt10a null mice and human kindreds. Mol. Genet. Genomic Med. 2015, 3, 40–58. [Google Scholar] [CrossRef] [PubMed]
- Cluzeau, C.; Hadj-Rabia, S.; Jambou, M.; Mansour, S.; Guigue, P.; Masmoudi, S.; Bal, E.; Chassaing, N.; Vincent, M.C.; Viot, G.; et al. Only four genes (EDA1, EDAR, EDARADD, and WNT10A) account for 90% of hypohidrotic/anhidrotic ectodermal dysplasia cases. Hum. Mutat. 2011, 32, 70–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plaisancie, J.; Bailleul-Forestier, I.; Gaston, V.; Vaysse, F.; Lacombe, D.; Holder-Espinasse, M.; Abramowicz, M.; Coubes, C.; Plessis, G.; Faivre, L.; et al. Mutations in WNT10A are frequently involved in oligodontia associated with minor signs of ectodermal dysplasia. Am. J. Med. Genet. A 2013, 161, 671–678. [Google Scholar] [CrossRef] [PubMed]
- Song, S.; Zhao, R.; He, H.; Zhang, J.; Feng, H.; Lin, L. WNT10A variants are associated with non-syndromic tooth agenesis in the general population. Hum. Genet. 2014, 133, 117–124. [Google Scholar] [CrossRef] [PubMed]
- van den Boogaard, M.J.; Créton, M.; Bronkhorst, Y.; van der Hout, A.; Hennekam, E.; Lindhout, D.; Cune, M.; van Amstel, H.K.P. Mutations in WNT10A are present in more than half of isolated hypodontia cases. J. Med. Genet. 2012, 49, 327–331. [Google Scholar] [CrossRef] [PubMed]
- Kantaputra, P.; Sripathomsawat, W. WNT10A and isolated hypodontia. Am. J. Med. Genet. A 2011, 155, 1119–1122. [Google Scholar] [CrossRef] [PubMed]
- Ellwanger, K.; Saito, H.; Clément-Lacroix, P.; Maltry, N.; Niedermeyer, J.; Lee, W.K.; Baron, R.; Rawadi, G.; Westphal, H.; Niehrs, C. Targeted disruption of the Wnt regulator Kremen induces limb defects and high bone density. Mol. Cell Biol. 2008, 28, 4875–4882. [Google Scholar] [CrossRef] [PubMed]
- Ingraham, C.R.; Kinoshita, A.; Kondo, S.; Yang, B.; Sajan, S.; Trout, K.J.; Malik, M.I.; Dunnwald, M.; Goudy, S.L.; Lovett, M.; et al. Abnormal skin, limb and craniofacial morphogenesis in mice deficient for interferon regulatory factor 6 (Irf6). Nat. Genet. 2006, 38, 1335–1340. [Google Scholar] [CrossRef] [PubMed]
- Leslie, E.J.; Mancuso, J.L.; Schutte, B.C.; Cooper, M.E.; Durda, K.M.; L’heureux, J.; Zucchero, T.M.; Marazita, M.L.; Murray, J.C. Search for genetic modifiers of IRF6 and genotype-phenotype correlations in Van der Woude and popliteal pterygium syndromes. Am. J. Med. Genet. A 2013, 161, 2535–2544. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, K.U.; Mangold, E.; Herms, S.; Nowak, S.; Reutter, H.; Paul, A.; Becker, J.; Herberz, R.; AlChawa, T.; Nasser, E.; Böhmer, A.C. Genome-wide meta-analyses of nonsyndromic cleft lip with or without cleft palate identify six new risk loci. Nat. Genet. 2012, 44, 968–971. [Google Scholar] [CrossRef] [PubMed]
- Fitch, N.; Kaback, M. The Axenfeld syndrome and the Rieger syndrome. J. Med. Genet. 1978, 15, 30–34. [Google Scholar] [CrossRef] [PubMed]
- Chisholm, I.A.; Chudley, A.E. Autosomal dominant iridogoniodysgenesis with associated somatic anomalies: Four-generation family with Rieger’s syndrome. Br. J. Ophthalmol. 1983, 67, 529–534. [Google Scholar] [CrossRef] [PubMed]
- Semina, E.V.; Reiter, R.; Leysens, N.J.; Alward, W.L.M.; Small, K.W.; Datson, N.A.; Siegel-Bartelt, J.; Bierke-Nelson, D.; Bitoun, P.; Zabel, B.U.; et al. Cloning and characterization of a novel bicoid-related homeobox transcription factor gene, RIEG, involved in Rieger syndrome. Nat. Genet. 1996, 14, 392–399. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Selever, J.; Lu, M.F.; Martin, J.F. Genetic dissection of Pitx2 in craniofacial development uncovers new functions in branchial arch morphogenesis, late aspects of tooth morphogenesis and cell migration. Development 2003, 130, 6375–6385. [Google Scholar] [CrossRef] [PubMed]
- Hjalt, T.A.; Semina, E.V.; Amendt, B.A.; Murray, J.C. The Pitx2 protein in mouse development. Dev. Dyn. 2000, 218, 195–200. [Google Scholar] [CrossRef]
- Gardner, E.J. Follow-up study of a family group exhibiting dominant inheritance for a syndrome including intestinal polyps, osteomas, fibromas and epidermal cysts. Am. J. Hum. Genet. 1962, 14, 376–390. [Google Scholar] [PubMed]
- Letra, A.; Bjork, B.; Cooper, M.E.; Szabo-Rogers, H.; Deleyiannis, F.W.B.; Field, L.L.; Czeizel, A.E.; Ma, L.; Garlet, G.P.; Poletta, F.A.; et al. Association of AXIN2 with non-syndromic oral clefts in multiple populations. J. Dent. Res. 2012, 91, 473–478. [Google Scholar] [CrossRef] [PubMed]
- Lindor, N.M.; Win, A.K.; Gallinger, S.; Daftary, D.; Thibodeau, S.N.; Silva, R.; Letra, A. Colorectal cancer and self-reported tooth agenesis. Hered. Cancer Clin. Pract. 2014, 12, 7. [Google Scholar] [CrossRef] [PubMed]
- Williams, M.A.; Biguetti, C.; Romero-Bustillos, M.; Maheshwari, K.; Dinckan, N.; Cavalla, F.; Vieira, A.R. Colorectal cancer-associated genes are associated with tooth agenesis and may have a role in tooth development. Sci. Rep. 2018, 8, 2979. [Google Scholar] [CrossRef] [PubMed]
- Callahan, N.; Modesto, A.; Meira, R.; Seymen, F.; Patir, A.; Vieira, A.R. Axis inhibition protein 2 (AXIN2) polymorphisms and tooth agenesis. Arch. Oral Biol. 2009, 54, 45–49. [Google Scholar] [CrossRef] [PubMed]
- Mostowska, A.; Biedziak, B.; Jagodzinski, P.P. Axis inhibition protein 2 (AXIN2) polymorphisms may be a risk factor for selective tooth agenesis. J. Hum. Genet. 2006, 51, 262–266. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.C.; Zhang, J.; Wong, S.; Han, D.; Zhao, H.S.; Feng, H.L. Association between rs11001553 of DKK1 and non-syndromic tooth agenesis in the Chinese Han population. Genet. Mol. Res. 2014, 13, 7133–7139. [Google Scholar] [CrossRef] [PubMed]
- Mues, G.; Tardivel, A.; Willen, L.; Kapadia, H.; Seaman, R.; Frazier-Bowers, S.; Schneider, P.; D’Souza, R.N. Functional analysis of Ectodysplasin-A mutations causing selective tooth agenesis. Eur. J. Hum. Genet. 2010, 18, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Shen, W.; Wang, Y.; Liu, Y.; Liu, H.; Zhao, H.; Zhang, G.; Snead, M.L.; Han, D.; Feng, H. Functional study of ectodysplasin-a mutations causing non-syndromic tooth agenesis. PLoS ONE 2016, 11, e0154884. [Google Scholar] [CrossRef] [PubMed]
- Song, S.; Han, D.; Qu, H.; Gong, Y.; Wu, H.; Zhang, X.; Zhong, N.; Feng, H. EDA gene mutations underlie non-syndromic oligodontia. J. Dent. Res. 2009, 88, 126–131. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, T.; Bansal, R.; Das, P. Whole genome sequencing reveals novel non-synonymous mutation in ectodysplasin A (EDA) associated with non-syndromic X-linked dominant congenital tooth agenesis. PLoS ONE 2014, 9, e106811. [Google Scholar] [CrossRef] [PubMed]
- Vieira, A.R.; Modesto, A.; Meira, R.; Barbosa, A.R.; Lidral, A.C.; Murray, J.C. Interferon regulatory factor 6 (IRF6) and fibroblast growth factor receptor 1 (FGFR1) contribute to human tooth agenesis. Am. J. Med. Genet. A 2007, 143, 538–545. [Google Scholar] [CrossRef] [PubMed]
- Kantaputra, P.N.; Kaewgahya, M.; Hatsadaloi, A.; Vogel, P.; Kawasaki, K.; Ohazama, A.; Ketudat Cairns, J.R. GREMLIN 2 mutations and dental anomalies. J. Dent. Res. 2015, 94, 1646–1652. [Google Scholar] [CrossRef] [PubMed]
- Vieira, A.R.; Seymen, F.; Patir, A.; Menezes, R. Evidence of linkage disequilibrium between polymorphisms at the IRF6 locus and isolate tooth agenesis, in a Turkish population. Arch. Oral Biol. 2008, 53, 780–784. [Google Scholar] [CrossRef] [PubMed]
- Lidral, A.C.; Reising, B.C. The role of MSX1 in human tooth agenesis. J. Dent. Res. 2002, 81, 274–278. [Google Scholar] [CrossRef] [PubMed]
- Vastardis, H.; Karimbux, N.; Guthua, S.W.; Seidman, J.G.; Seidman, C.E. A human MSX1 homeodomain missense mutation causes selective tooth agenesis. Nat. Genet. 1996, 13, 417–421. [Google Scholar] [CrossRef] [PubMed]
- Massink, M.P.; Créton, M.A.; Spanevello, F.; Fennis, W.M.; Cune, M.S.; Savelberg, S.M.; Nijman, I.J.; Maurice, M.M.; van den Boogaard, M.J.H.; van Haaften, G. Loss-of-function mutations in the WNT co-receptor LRP6 cause autosomal-dominant oligodontia. Am. J. Hum. Genet. 2015, 97, 621–626. [Google Scholar] [CrossRef] [PubMed]
- Ockeloen, C.W.; Khandelwal, K.D.; Dreesen, K.; Ludwig, K.U.; Sullivan, R.; Van Rooij, I.A.; Thonissen, M.; Swinnen, S.; Phan, M.; Conte, F.; et al. Novel mutations in LRP6 highlight the role of WNT signaling in tooth agenesis. Genet. Med. 2016, 18, 1158–1162. [Google Scholar] [CrossRef] [PubMed]
- Dugan, S.L.; Temme, R.T.; Olson, R.A.; Mikhailov, A.; Law, R.; Mahmood, H.; Noor, A.; Vincent, J.B. New recessive truncating mutation in LTBP3 in a family with oligodontia, short stature, and mitral valve prolapse. Am. J. Med. Genet. A 2015, 167, 1396–1399. [Google Scholar] [CrossRef] [PubMed]
- Noor, A.; Windpassinger, C.; Vitcu, I.; Orlic, M.; Rafiq, M.A.; Khalid, M.; Malik, M.N.; Ayub, M.; Alman, B.; Vincent, J.B. Oligodontia is caused by mutation in LTBP3, the gene encoding latent TGF-beta binding protein 3. Am. J. Hum. Genet. 2009, 84, 519–523. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, T.; Bansal, R.; Das, P. A novel G to A transition at initiation codon and exon-intron boundary of PAX9 identified in association with familial isolated oligodontia. Gene 2017, 635, 69–76. [Google Scholar] [CrossRef] [PubMed]
- Vieira, A.R.; Meira, R.; Modesto, A.; Murray, J.C. MSX1, PAX9, and TGFA contribute to tooth agenesis in humans. J. Dent. Res. 2004, 83, 723–727. [Google Scholar] [CrossRef] [PubMed]
- Wong, S.W.; Han, D.; Zhang, H.; Liu, Y.; Zhang, X.; Miao, M.Z.; Wang, Y.; Zhao, N.; Zeng, L.; Bai, B.; et al. Nine novel PAX9 mutations and a distinct tooth agenesis genotype-phenotype. J. Dent. Res. 2018, 97, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Frazier-Bowers, S.A.; Guo, D.C.; Cavender, A.; Xue, L.; Evans, B.; King, T.; Milewicz, D.; D’Souza, R.N. A novel mutation in human PAX9 causes molar oligodontia. J. Dent. Res. 2002, 81, 129–133. [Google Scholar] [CrossRef] [PubMed]
- Mitsui, S.N.; Yasue, A.; Masuda, K.; Watanabe, K.; Horiuchi, S.; Imopto, I.; Tanaka, E. Novel PAX9 mutations cause non-syndromic tooth agenesis. J. Dent. Res. 2014, 93, 245–249. [Google Scholar] [CrossRef] [PubMed]
- Stockton, D.W.; Das, P.; Goldenberg, M.; D’Souza, R.N.; Patel, P.I. Mutation of PAX9 is associated with oligodontia. Nat. Genet. 2000, 24, 18–19. [Google Scholar] [CrossRef] [PubMed]
- Alfawaz, S.; Fong, F.; Plagnol, V.; Wong, F.S.; Fearne, J.; Kelsell, D.P. Recessive oligodontia linked to a homozygous loss-of-function mutation in the SMOC2 gene. Arch. Oral Biol. 2013, 58, 462–466. [Google Scholar] [CrossRef] [PubMed]
- Bloch-Zupan, A.; Jamet, X.; Etard, C.; Lauget, V.; Muller, J.; Geoffroy, V.; Straus, J.P.; Pelletier, V.; Marion, V.; Poch, O.; et al. Homozygosity mapping and candidate prioritization identify mutations, missed by whole-exome sequencing, in SMOC2, causing major dental developmental defects. Am. J. Hum. Genet. 2011, 89, 773–781. [Google Scholar] [CrossRef] [PubMed]
- Arzoo, P.S.; Klar, J.; Bergendal, B.; Norderyd, J.; Dahl, N. WNT10A mutations account for (1/4) of population-based isolated oligodontia and show phenotypic correlations. Am. J. Med. Genet. A 2014, 164, 353–359. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Q.; Zhao, M.; Tandon, B.; Maili, L.; Liu, X.; Zhang, A.; Baugh, E.H.; Tran, T.; Silva, R.M.; Hecht, J.T.; et al. Role of WNT10A in failure of tooth development in humans and zebrafish. Mol. Genet. Genomic. Med. 2017, 5, 730–741. [Google Scholar] [CrossRef] [PubMed]
- Kantaputra, P.N.; Hutsadaloi, A.; Kaewgahya, M.; Intachai, W.; German, R.; Koparal, M.; Leethanakul, C.; Tolun, A.; Ketudat Cairns, J.R. WNT10B mutations associated with isolated dental anomalies. Clin. Genet. 2018, 93, 992–999. [Google Scholar] [CrossRef] [PubMed]
- Yu, P.; Yang, W.; Han, D.; Wang, X.; Guo, S.; Li, J.; Li, F.; Zhang, X.; Wong, S.W.; Bai, B.; et al. Mutations in WNT10B are identified in individuals with oligodontia. Am. J. Hum. Genet. 2016, 99, 195–201. [Google Scholar] [CrossRef] [PubMed]
- Vastardis, H. The genetics of human tooth agenesis: New discoveries for understanding dental anomalies. Am. J. Orthod. Dentofacial Orthop. 2000, 117, 650–656. [Google Scholar] [CrossRef]
- Liang, J.; Von den Hoff, J.; Lange, J.; Ren, Y.; Bian, Z.; Carels, C.E. MSX1 mutations and associated disease phenotypes: Genotype-phenotype relations. Eur. J. Hum. Genet. 2016, 24, 1663–1670. [Google Scholar] [CrossRef] [PubMed]
- Peters, H.; Neubüser, A.; Kratochwil, K.; Balling, R. Pax9-deficient mice lack pharyngeal pouch derivatives and teeth and exhibit craniofacial and limb abnormalities. Genes Dev. 1998, 12, 2735–2747. [Google Scholar] [CrossRef] [PubMed]
- Peters, H.; Neubuser, A.; Balling, R. Pax genes and organogenesis: Pax9 meets tooth development. Eur. J. Oral. Sci. 1998, 106, 38–43. [Google Scholar] [CrossRef] [PubMed]
- Bonczek, O.; Balcar, V.J.; Sery, O. PAX9 gene mutations and tooth agenesis: A review. Clin. Genet. 2017, 92, 467–476. [Google Scholar] [CrossRef] [PubMed]
- Brook, A.H.; Elcock, C.; Aggarwal, M.; Lath, D.L.; Russell, J.M.; Patel, P.I.; Smith, R.N. Tooth dimensions in hypodontia with a known PAX9 mutation. Arch. Oral Biol. 2009, 54, S57–S62. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Jian, F.; Chen, J.; Wang, H.; Lin, Y.; Yang, Z.; Pan, X.; Lai, W. Sequence analysis of PAX9, MSX1 and AXIN2 genes in a Chinese oligodontia family. Arch. Oral Biol. 2011, 56, 1027–1034. [Google Scholar] [CrossRef] [PubMed]
- Vink, C.P.; Ockeloen, C.W.; Ten Kate, S.; Koolen, D.A.; Van Amstel, J.K.P.; Kuijpers-Jagtman, A.M.; Van Heumen, C.C.; Kleefstra, T.; Carels, C.E. Variability in dentofacial phenotypes in four families with WNT10A mutations. Eur. J. Hum. Genet. 2014, 22, 1063–1070. [Google Scholar] [CrossRef] [PubMed]
- He, H.; Han, D.; Feng, H.; Qu, H.; Song, S.; Bai, B.; Zhang, Z. Involvement of and interaction between WNT10A and EDA mutations in tooth agenesis cases in the Chinese population. PLoS ONE 2013, 8, e80393. [Google Scholar] [CrossRef] [PubMed]
- Pinson, K.I.; Brennan, J.; Monkley, S.; Avery, B.J.; Skarnes, W.C. An LDL-receptor-related protein mediates Wnt signalling in mice. Nature 2000, 407, 535–538. [Google Scholar] [PubMed]
- Vogel, P.; Liu, J.; Platt, K.A.; Read, R.W.; Thiel, M.; Vance, R.B.; Brommage, R. Malformation of incisor teeth in Grem2−/− mice. Vet. Pathol. 2015, 52, 224–229. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Han, D.; Song, S.; Wang, Y.; Zhao, H.; Pan, S.; Bai, B.; Feng, H. Correlation between the phenotypes and genotypes of X-linked hypohidrotic ectodermal dysplasia and non-syndromic hypodontia caused by ectodysplasin-A mutations. Eur. J. Med. Genet. 2011, 54, e377–e382. [Google Scholar] [CrossRef] [PubMed]
- Tipton, R.E.; Gorlin, R.J. Growth retardation, alopecia, pseudo-anodontia, and optic atrophy—The GAPO syndrome: Report of a patient and review of the literature. Am. J. Med. Genet. 1984, 19, 209–216. [Google Scholar] [CrossRef] [PubMed]
- Cullen, M.; Seaman, S.; Chaudhary, A.; Yang, M.Y.; Hilton, M.B.; Logsdon, D.; Croix, B.S. Host-derived tumor endothelial marker 8 promotes the growth of melanoma. Cancer Res. 2009, 69, 6021–6026. [Google Scholar] [CrossRef] [PubMed]
- Carson-Walter, E.B.; Watkins, D.N.; Nanda, A.; Vogelstein, B.; Kinzler, K.W.; Croix, B.S. Cell surface tumor endothelial markers are conserved in mice and humans. Cancer Res. 2001, 61, 6649–6655. [Google Scholar] [PubMed]
- Fernando, S.; Fletcher, B.S. Targeting tumor endothelial marker 8 in the tumor vasculature of colorectal carcinomas in mice. Cancer Res. 2009, 69, 5126–5132. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Xie, G.; Geng, P.; Zheng, C.; Li, J.; Pan, F.; Liang, H. Anti-tumor angiogenesis effect of genetic fusion vaccine encoding murine β-defensin 2 and tumor endothelial marker-8 in a CT-26 murine colorectal carcinoma model. Int. J. Clin. Exp. Med. 2015, 8, 4744–4752. [Google Scholar] [PubMed]
- Yue, H.; Liang, J.; Yang, K.; Hua, B.; Bian, Z. Functional analysis of a novel missense mutation in AXIN2 associated with non-syndromic tooth agenesis. Eur. J. Oral Sci. 2016, 124, 228–233. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, T.; Hosomichi, K.; Yano, K.; Kim, Y.I.; Nakaoka, H.; Kimura, R.; Shirota, T. Comprehensive genetic exploration of selective tooth agenesis of mandibular incisors by exome sequencing. Hum. Genome Var. 2017, 4, 17005. [Google Scholar] [CrossRef] [PubMed]
- Jonsson, L.; Magnusson, T.E.; Thordarson, A.; Jonsson, T.; Geller, F.; Feenstra, B.; Rivadeneira, F. Rare and common variants conferring risk of tooth agenesis. J. Dent. Res. 2018, 97, 515–522. [Google Scholar] [CrossRef] [PubMed]
- Letra, A.; Menezes, R.; Granjeiro, J.M.; Vieira, A.R. Defining subphenotypes for oral clefts based on dental development. J. Dent. Res. 2007, 86, 986–991. [Google Scholar] [CrossRef] [PubMed]
- Posey, J.E.; Harel, T.; Liu, P.; Rosenfeld, J.A.; James, R.A.; Coban Akdemir, Z.H.; Xia, F. Resolution of disease phenotypes resulting from multilocus genomic variation. N. Engl. J. Med. 2017, 376, 21–31. [Google Scholar] [CrossRef] [PubMed]
- Arte, S.; Parmanen, S.; Pirinen, S.; Alaluusua, S.; Nieminen, P. Candidate gene analysis of tooth agenesis identifies novel mutations in six genes and suggests significant role for WNT and EDA signaling and allele combinations. PLoS ONE 2013, 8, e73705. [Google Scholar] [CrossRef] [PubMed]
- Fedi, P.; Bafico, A.; Soria, A.N.; Burgess, W.H.; Miki, T.; Bottaro, D.P.; Aaronson, S.A. Isolation and biochemical characterization of the human Dkk-1 homologue, a novel inhibitor of mammalian Wnt signaling. J. Biol. Chem. 1999, 274, 19465–19472. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Huang, X.; Xu, X.; Mayo, J.; Bringas, P. Jr.; Jiang, R.; Wang, S.; Chai, Y. SMAD4-mediated WNT signaling controls the fate of cranial neural crest cells during tooth morphogenesis. Development 2011, 138, 1977–1989. [Google Scholar] [CrossRef] [PubMed]
- McGrath, J.A.; Kivirikko, S.; Ciatti, S.; Moss, C.; Dunnill, M.G.S.; Eady, R.A.; Uitto, J. A homozygous nonsense mutation in the α3 chain gene of laminin 5 (LAMA3) in Herlitz junctional epidermolysis bullosa: Prenatal exclusion in a fetus at risk. Genomics 1995, 29, 282–284. [Google Scholar] [CrossRef] [PubMed]
- Ryan, M.C.; Lee, K.; Miyashita, Y.; Carter, W.G. Targeted disruption of the LAMA3 gene in mice reveals abnormalities in survival and late stage differentiation of epithelial cells. J. Cell Biol. 1999, 145, 1309–1323. [Google Scholar] [CrossRef] [PubMed]
- Tasanen, K.; Eble, J.A.; Aumailley, M.; Schumann, H.; Baetge, J.; Tu, H.; Bruckner-Tuderman, L. Collagen XVII is destabilized by a glycine substitution mutation in the cell adhesion domain Col15. J. Biol. Chem. 2000, 275, 3093–3099. [Google Scholar] [CrossRef] [PubMed]
- Salvi, A.; Giacopuzzi, E.; Bardellini, E.; Amadori, F.; Ferrari, L.; De Petro, G.; Majorana, A. Mutation analysis by direct and whole exome sequencing in familial and sporadic tooth agenesis. Int. J. Mol. Med. 2016, 38, 1338–1348. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Zhao, P.; Liu, Y.; Zhang, X.; Fu, J.; Yu, H.M.I.; Zhang, Z. Intra-epithelial requirement of canonical Wnt signaling for tooth morphogenesis. J. Biol. Chem. 2013, 288, 12080–12089. [Google Scholar] [CrossRef] [PubMed]
- De Coster, P.J.; Marks, L.A.; Martens, L.C.; Huysseune, A. Dental agenesis: Genetic and clinical perspectives. J. Oral Pathol. Med. 2009, 38, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Phenolyzer. Available online: http://phenolyzer.wglab.org (accessed on 3 May 2018).



{kind=link}
{kind=link}
| Gene/Locus | OMIM | Chromosome | Syndrome | Inheritance | Dental/Oral Phenotypes | Animal Model | Animal Model Phenotype | Reference |
|---|---|---|---|---|---|---|---|---|
| ADAMTS2 | 604539 | 5q35.3 | Ehlers–Danlos syndrome | AR | Hypodontia, microdontia, tooth discoloration | Yes Bovine | Dermatosparatic phenotype resembling EDS type VII C | [15,16,17] |
| ANTXR1 | 606410 | 2p13.3 | Growth retardation, alopecia, pseudoanodontia, and optic atrophy (GAPO) syndrome | AR | Hypodontia, delayed eruption | Yes | Growth delay, bone loss, shortened skulls with frontal bossing, and midfacial hypoplasia | [18,19,20] |
| AXIN2 | 604025 | 17q24.1 | Oligodontia-colorectal cancer syndrome | AD | Oligodontia | Yes | Abnormal cranium morphology | [21,22] |
| COL1A1/2 | 120150 | 17q21.33 | Osteogenesis imperfecta type 1 | AD | Hypodontia, oligodontia | Yes | Lethal, bone fractures | [23] |
| CREBBP | 600140 | 16p13.3 | Rubinstein–Taybi syndrome | AD | Hypodontia, retrognathia, micrognathia, arched/narrow palate, talon cusps, dental crowding, screwdriver incisors, cross bite, and enamel hypoplasia | Yes | Skeletal malformations | [24] |
| EDA | 300451 | Xq13.1 | Ectodermal dysplasia, hypohidrotic | XLR | Anodontia, hypodontia, misshapen teeth, microdontia | Yes Canine | Incomplete set of conically shaped teeth | [25,26,27,28] |
| EDAR | 604095 | 2q13 | Ectodermal dysplasia, hypohidrotic/hair/tooth type | AR | Anodontia, hypodontia, oligodontia | Yes Mouse | Decreased molar number, small incisor, small molars, abnormal enamel knot morphology | [29,30,31,32,33] |
| EDARADD | 606603 | 1q42-q43 | Ectodermal dysplasia, hypohidrotic/hair/tooth type | AD | Anodontia, hypodontia, taurodontism, microdontia | Yes Mouse | Abnormal tooth morphology, decreased molar number, small molars, abnormal enamel morphology | [34,35,36] |
| EVC | 604831 | 4p16.2 | Ellis–van Creveld syndrome and Weyers acrofacial dysostosis | AR/AD | Natal teeth, enamel abnormalities, hypodontia, microdontia | Yes Mouse | Enamel defects, abnormal tooth morphology | [37,38,39] |
| EVC2 | 607261 | 4p16.2 | Ellis–van Creveld syndrome and Weyers acrofacial dysostosis | AR/AD | Natal teeth, enamel abnormalities, hypodontia, oligodontia, microdontia | Yes Mouse | Microdontia, small upper incisors, small cranium | [37,38,39,40] |
| FGF10 | 602115 | 5p12 | Lacrimoauriculodentodigital syndrome | AD | Hypodontia (maxillary incisors), microdontia, delayed eruption, enamel dysplasia | Yes Mouse | Abnormal tooth morphology, short incisors, small molars, abnormal palatal development, abnormal tongue morphology | [41,42,43] |
| FGFR1 | 136350 | 8p11.23 | Kallmann syndrome | XLR | Hypodontia, cleft lip/palate | Yes Mouse | Abnormal cranium morphology, facial asymmetry, long incisors | [44] |
| FGFR2 | 176943 | 10q26.13 | Lacrimoauriculodentodigital syndrome | AD | Hypodontia (maxillary incisors), microdontia peg laterals, delayed eruption, enamel dysplasia | Yes Mouse | Arrest of tooth development, long incisors, decreased molar number, micrognathia | [43] |
| Apert syndrome | AD | Hypodontia (maxillary canines), enamel opacities, ectopic eruptions, gingival hyperplasia | Yes Mouse | Arrest of tooth development, long incisors, decreased molar number, micrognathia | [45,46,47] | |||
| FGFR3 | 134934 | 4p16.3 | Crouzon syndrome with acanthosis nigricans | AD | Hypodontia, malocclusion, cementomas, delayed eruption, midface hypoplasia | Yes Mouse | Tooth misalignment, long incisors, malocclusion, prognathia, maxillary retrognathia | [48,49,50] |
| FLNB | 603381 | 3p14.3 | Larsen syndrome | AD | Hypodontia, delayed dental development, class 3 occlusion, morphological anomalies | Yes Mouse | Abnormal cranium morphology | [51] |
| FOXC1 | 601090 | 6p25.3 | Axenfeld–Rieger syndrome type 3 | AD | Hypodontia, microdontia, taurodontism | Yes, Mouse | Short mandible | [52,53] |
| GJA1 | 121014 | 6q22.31 | Oculodentodigital dysplasia | AD, AR | Microdontia, enamel hypoplasia, hypodontia, delayed eruption | Yes Mouse | Abnormal tooth morphology, microdontia, small mandible and maxilla, reduced enamel thickness | [54,55] |
| GRHL2 | 608576 | 8q22.3 | Ectodermal dysplasia/short stature syndrome | AR | Delayed eruption, hypodontia, enamel hypoplasia | Yes, Mouse | Abnormal cranium morphology, facial and midline clefts | [56] |
| IRF6 | 607199 | 1q32.2 | van der Woude syndrome | AD | Hypodontia, cleft lip/palate | Yes Mouse | Abnormal tooth morphology, abnormal palatal development, small mandible | [57,58] |
| JAG1 | 601920 | 20p12.2 | Alagille Syndrome | AD | Hypodontia, enamel hypoplasia and opacities, hypomineralization | Yes mouse | Short maxilla, malocclusion, abnormal palate morphology | [59] |
| KDM6A | 300128 | Xp11.3 | Kabuki syndrome 2 | XLD | High-arched palate, malocclusion, microdontia, a small dental arch, hypodontia, severe maxillary recession, conical teeth | Yes Mouse | Cranioschisis | [60,61] |
| KMT2D | 602113 | 12q13.12 | Kabuki syndrome 1 | AD | High-arched palate, malocclusion, microdontia, a small dental arch, hypodontia, severe maxillary retrognathia, conical teeth | Yes Mouse | Short maxilla, flattened snout | [62,63] |
| KREMEN1 | 609898 | 22q12.1 | Ectodermal dysplasia, hair/tooth type | AR | Oligodontia, hypodontia, alveolar ridge deficiency, increased palatal depth | Yes | No craniofacial phenotype | [64] |
| MKKS | 604896 | 20p12.2 | Bardet-Biedl syndrome | AR | Dental crowding, high-arched palate, hypodontia, malocclusion, enamel hypoplasia, retrognathia | Yes Mouse | Abnormal olfactory epithelium | [65] |
| MSX1 | 142983 | 4p16.1 | Witkop syndrome | AD | Hypodontia, oligodontia | Yes Mouse | Arrest of tooth development, nail bed and nail plates defective, cleft palate | [66] |
| NEMO | 300248 | Xq28 | Incontinentia pigmenti | XLD | Hypodontia, anodontia, microdontia | Yes Mouse | No craniofacial phenotype | [67,68] |
| NSD1 | 606681 | 5q35.3 | Sotos syndrome I | AD | Hypodontia, enamel defects, malocclusion | Yes Mouse | No craniofacial phenotypes | [69,70] |
| OFD1 | 300170 | Xp22.2 | Orofaciodigital syndrome I | XLD | Hypodontia, missing lateral incisors, canine malposition, micrognathia | Yes | Primary cilia formed then disappeared, renal cysts | [71,72] |
| P63 | 603273 | 3q28 | Orofacial cleft 8, Rapp-Hodgkin, and Ectrodactyly, ectodermal dysplasia, and cleft lip/palate syndrome 3 | AD | Hypodontia, enamel hypoplasia, extensive dental caries, hypodontia of the mandibular canines, generalized microdontia, prominent marginal ridges of permanent maxillary incisors, round-shaped permanent molars, and barrel-shaped permanent maxillary central incisors | Yes Mouse | Arrest of tooth development, small mandible and maxilla, abnormal craniofacial development, cleft palate | [73,74,75] |
| PITX2 | 601542 | 4q25 | Axenfeld–Rieger syndrome, type 1 | AD | Hypodontia, microdontia, enamel hypoplasia | Yes Mouse | Abnormal maxilla and mandible morphology, arrested tooth development | [53,76,77,78] |
| PVRL1 | 600644 | 11q23.3 | Cleft lip/palate-ectodermal dysplasia | AR | Hypodontia, cleft lip and palate, abnormal dental morphology, microdontia | Yes Mouse | Abnormal tooth morphology | [79,80] |
| RECQL4 | 603780 | 8q24.3 | Rothmund–Thomson syndrome | AR | Hypodontia, microdontia, hypoplastic teeth | Yes Mouse | Delayed tooth eruption, cleft palate | [81] |
| RSK2 | 300075 | Xp22.12 | Coffin–Lowry syndrome | XLD | High narrow palate, midline lingual furrow, hypodontia, and microdontia | Yes Mouse | Abnormal tooth morphology, supernumerary teeth | [82] |
| SHH | 600725 | 7q36.3 | Holoprosencephaly | AD | Cleft lip and palate, single central incisor, micrognathia | Yes | Abnormal tooth morphology, microdontia | [83,84] |
| TBX3 | 601621 | 12q24.21 | Ulnar-mammary syndrome | AD | Hypodontia, ectopic and hypoplastic canines | Yes | Secondary palate clefting | [85] |
| TCOF1 | 606847 | 5q32-q33 | Treacher Collins syndrome | AD | Hypodontia, micrognathia, malocclusion, spaced teeth | Yes | Short mandible and maxilla | [86,87] |
| TFAP2B | 601601 | 6p12.3 | Char syndrome | AD | Oligodontia, hypodontia, thick lips, retention of primary teeth | Yes | No craniofacial phenotype | [88] |
| Trisomy 21 | 190685 | 21q22.13 | Down syndrome | IC | Hypodontia, delayed eruption, barrel-shaped permanent maxillary central incisors | Yes | General hypoplasia and developmental delay, hydronephrosis, heart and neurologic defects | [89,90] |
| TSPEAR | 612920 | 21q22.3 | Ectodermal dysplasia | AR | Hypodontia, microdontia | Yes | No craniofacial phenotypes | [91] |
| UBR1 | 605981 | 15q15.2 | Johanson–Blizzard syndrome | AR | Oligodontia | Yes | No craniofacial phenotypes | [92] |
| WNT10A | 606268 | 2q35 | Odontoonychodermal dysplasia | AR | Oligodontia, hypodontia, microdontia | Yes | Arrested tooth development of molars, supernumerary molars, abnormal tooth morphology | [93,94,95] |
| Schopf–Schulz–Passarge syndrome | AR | Oligodontia, hypodontia, microdontia | Yes | Arrested tooth development of molars, supernumerary molars, abnormal tooth morphology | [94,96,97] |
| Gene | OMIM | Chromosome | Dental Phenotypes | Reference(s) |
|---|---|---|---|---|
| AXIN2 | 604025 | 17q24.1 | Oligodontia, hypodontia | [22,98,124,125] |
| ANTXR1 | 606410 | 2p13.3 | Oligodontia, hypodontia | [10] |
| COL17A1 | 113811 | 10q25.1 | Hypodontia | [11] |
| DKK1 | 605189 | 10q21.1 | Hypodontia | [11,126] |
| EDA | 300451 | Xq13.1 | Oligodontia, hypodontia | [33,127,128,129,130] |
| EDAR | 604095 | 2q13 | Oligodontia, hypodontia | [33] |
| EDARADD | 606603 | 1q42-q43 | Oligodontia, hypodontia | [33] |
| FGFR1 | 136350 | 8p11.23 | Hypodontia | [131] |
| GREM2 | 608832 | 1q43 | Hypodontia, microdontia, taurodontia | [132] |
| IRF6 | 607199 | 1q32.2 | Hypodontia, lip pits | [131,133] |
| MSX1 | 142983 | 4p16.2 | Oligodontia, hypodontia | [98,134,135] |
| LAMA3 | 600805 | 18q11.2 | Hypodontia | [11] |
| LRP6 | 603507 | 12p13.2 | Oligodontia | [11,136,137] |
| LTBP3 | 602090 | 11q13.1 | Oligodontia, hypodontia | [138,139] |
| PAX9 | 167416 | 14q13.3 | Oligodontia, hypodontia, microdontia | [98,140,141,142,143,144,145] |
| SMOC2 | 607223 | 6q27 | Oligodontia, microdontia, abnormal morphology | [146,147] |
| WNT10A | 606268 | 2q35 | Oligodontia, hypodontia | [11,108,109,110,148,149] |
| WNT10B | 601906 | 12q13.12 | Oligodontia, microdontia | [150,151] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Williams, M.A.; Letra, A. The Changing Landscape in the Genetic Etiology of Human Tooth Agenesis. Genes 2018, 9, 255. https://doi.org/10.3390/genes9050255
Williams MA, Letra A. The Changing Landscape in the Genetic Etiology of Human Tooth Agenesis. Genes. 2018; 9(5):255. https://doi.org/10.3390/genes9050255
Chicago/Turabian StyleWilliams, Meredith A., and Ariadne Letra. 2018. "The Changing Landscape in the Genetic Etiology of Human Tooth Agenesis" Genes 9, no. 5: 255. https://doi.org/10.3390/genes9050255
APA StyleWilliams, M. A., & Letra, A. (2018). The Changing Landscape in the Genetic Etiology of Human Tooth Agenesis. Genes, 9(5), 255. https://doi.org/10.3390/genes9050255