Genetic Association with Subgingival Bacterial Colonization in Chronic Periodontitis

 ,
,
Abstract
:1. Introduction
2. Material and Methods
2.1. Sample Population
2.2. Genotyping
2.3. DNA-DNA Checkerboard
2.4. Statistical Analysis
3. Results
4. Discussion
Author Contributions
Conflicts of Interest
Ethical Statement
References
- Garlet, G.P. Destructive and protective roles of cytokines in periodontitis: A re-appraisal from host defense and tissue destruction viewpoints. J. Dent. Res. 2010, 89, 1349–1363. [Google Scholar] [CrossRef] [PubMed]
- Silva, N.; Abusleme, L.; Bravo, D.; Dutzan, N.; Garcia-Sesnich, J.; Vernal, R.; Hernandez, M.; Gamonal, J. Host response mechanisms in periodontal diseases. J. Appl. Oral Sci. 2015, 23, 329–355. [Google Scholar] [CrossRef] [PubMed]
- Cavalla, F.; Biguetti, C.C.; Garlet, T.P.; Trombone, A.P.F.; Garlet, G.P. Inflammatory pathways of bone resorption in periodontitis. In Pathogenesis of Periodontal Diseases Biological Concepts for Clinicians; Belibasakis, N.B.A.G., Ed.; Springer International Publishing AG: Basel, Switzerland, 2018; Volume 1, pp. 59–86. [Google Scholar]
- Graves, D.T.; Oates, T.; Garlet, G.P. Review of osteoimmunology and the host response in endodontic and periodontal lesions. J. Oral Microb. 2011, 3, 5304. [Google Scholar] [CrossRef] [PubMed]
- Meyle, J.; Chapple, I. Molecular aspects of the pathogenesis of periodontitis. Periodontology 2000 2015, 69, 7–17. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, M.; Dutzan, N.; Garcia-Sesnich, J.; Abusleme, L.; Dezerega, A.; Silva, N.; Gonzalez, F.E.; Vernal, R.; Sorsa, T.; Gamonal, J. Host-pathogen interactions in progressive chronic periodontitis. J. Dent. Res. 2011, 90, 1164–1170. [Google Scholar] [CrossRef] [PubMed]
- Hajishengallis, G. Periodontitis: From microbial immune subversion to systemic inflammation. Nat. Rev. Immunol. 2015, 15, 30–44. [Google Scholar] [CrossRef] [PubMed]
- Trombone, A.P.; Claudino, M.; Colavite, P.; de Assis, G.F.; Avila-Campos, M.J.; Silva, J.S.; Campanelli, A.P.; Ibanez, O.M.; De Franco, M.; Garlet, G.P. Periodontitis and arthritis interaction in mice involves a shared hyper-inflammatory genotype and functional immunological interferences. Genes Immun. 2010, 11, 479–489. [Google Scholar] [CrossRef] [PubMed]
- Nibali, L. Genetic influences on the periodontal microbial-host crosstalk. In Pathogenesis of Periodontal Diseases: Biological Concepts for Clinicians; Bostanci, N., Belibasakis, G.N., Eds.; Springer International Publishing: Cham, Switzerland, 2018; pp. 87–95. [Google Scholar]
- Hajishengallis, G.; Lamont, R.J. Dancing with the stars: How choreographed bacterial interactions dictate nososymbiocity and give rise to keystone pathogens, accessory pathogens, and pathobionts. Trends Microbiol. 2016, 24, 477–489. [Google Scholar] [CrossRef] [PubMed]
- Socransky, S.S.; Haffajee, A.D.; Cugini, M.A.; Smith, C.; Kent, R.L., Jr. Microbial complexes in subgingival plaque. J. Clin. Periodontol. 1998, 25, 134–144. [Google Scholar] [CrossRef] [PubMed]
- Darveau, R.P. Periodontitis: A polymicrobial disruption of host homeostasis. Nat. Rev. Microbiol. 2010, 8, 481–490. [Google Scholar] [CrossRef] [PubMed]
- Hajishengallis, G. Immunomicrobial pathogenesis of periodontitis: Keystones, pathobionts, and host response. Trends Immunol. 2014, 35, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Queiroz-Junior, C.M.; Madeira, M.F.; Coelho, F.M.; Costa, V.V.; Bessoni, R.L.; Sousa, L.F.; Garlet, G.P.; Souza Dda, G.; Teixeira, M.M.; Silva, T.A. Experimental arthritis triggers periodontal disease in mice: Involvement of TNF-α and the oral microbiota. J. Immunol. 2011, 187, 3821–3830. [Google Scholar] [CrossRef] [PubMed]
- Cavalla, F.; Araujo-Pires, A.C.; Biguetti, C.C.; Garlet, G.P. Cytokine networks regulating inflammation and immune defense in the oral cavity. Curr. Oral Health Rep. 2014, 1, 104–113. [Google Scholar] [CrossRef]
- Araujo-Pires, A.C.; Francisconi, C.F.; Biguetti, C.C.; Cavalla, F.; Aranha, A.M.; Letra, A.; Trombone, A.P.; Faveri, M.; Silva, R.M.; Garlet, G.P. Simultaneous analysis of T helper subsets (Th1, Th2, Th9, Th17, Th22, Tfh, Tr1 and Tregs) markers expression in periapical lesions reveals multiple cytokine clusters accountable for lesions activity and inactivity status. J. Appl. Oral Sci. 2014, 22, 336–346. [Google Scholar] [CrossRef] [PubMed]
- Kornman, K.S. Mapping the pathogenesis of periodontitis: A new look. J. Periodontol. 2008, 79, 1560–1568. [Google Scholar] [CrossRef] [PubMed]
- Hart, T.C.; Kornman, K.S. Genetic factors in the pathogenesis of periodontitis. Periodontology 2000 1997, 14, 202–215. [Google Scholar] [CrossRef] [PubMed]
- Michalowicz, B.S.; Aeppli, D.P.; Kuba, R.K.; Bereuter, J.E.; Conry, J.P.; Segal, N.L.; Bouchard, T.J., Jr.; Pihlstrom, B.L. A twin study of genetic variation in proportional radiographic alveolar bone height. J. Dent. Res. 1991, 70, 1431–1435. [Google Scholar] [CrossRef] [PubMed]
- Michalowicz, B.S.; Diehl, S.R.; Gunsolley, J.C.; Sparks, B.S.; Brooks, C.N.; Koertge, T.E.; Califano, J.V.; Burmeister, J.A.; Schenkein, H.A. Evidence of a substantial genetic basis for risk of adult periodontitis. J. Periodontol. 2000, 71, 1699–1707. [Google Scholar] [CrossRef] [PubMed]
- Trombone, A.P.; Ferreira, S.B., Jr.; Raimundo, F.M.; de Moura, K.C.; Avila-Campos, M.J.; Silva, J.S.; Campanelli, A.P.; De Franco, M.; Garlet, G.P. Experimental periodontitis in mice selected for maximal or minimal inflammatory reactions: Increased inflammatory immune responsiveness drives increased alveolar bone loss without enhancing the control of periodontal infection. J. Periodontal Res. 2009, 44, 443–451. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Sun, X.; Xiao, L.; Xie, C.; Xuan, D.; Luo, G. Gene polymorphisms and periodontitis. Periodontology 2000 2011, 56, 102–124. [Google Scholar] [CrossRef] [PubMed]
- Razzouk, S. Regulatory elements and genetic variations in periodontal diseases. Arch. Oral Boil. 2016, 72, 106–115. [Google Scholar] [CrossRef] [PubMed]
- Garlet, G.P.; Trombone, A.P.; Menezes, R.; Letra, A.; Repeke, C.E.; Vieira, A.E.; Martins, W., Jr.; Neves, L.T.; Campanelli, A.P.; Santos, C.F.; et al. The use of chronic gingivitis as reference status increases the power and odds of periodontitis genetic studies: A proposal based in the exposure concept and clearer resistance and susceptibility phenotypes definition. J. Clin. Periodontol. 2012, 39, 323–332. [Google Scholar] [CrossRef] [PubMed]
- Loos, B.G.; Papantonopoulos, G.; Jepsen, S.; Laine, M.L. What is the contribution of genetics to periodontal risk? Dent. Clin. N. Am. 2015, 59, 761–780. [Google Scholar] [CrossRef] [PubMed]
- Vaithilingam, R.D.; Safii, S.H.; Baharuddin, N.A.; Ng, C.C.; Cheong, S.C.; Bartold, P.M.; Schaefer, A.S.; Loos, B.G. Moving into a new era of periodontal genetic studies: Relevance of large case-control samples using severe phenotypes for genome-wide association studies. J. Periodontal Res. 2014, 49, 683–695. [Google Scholar] [CrossRef] [PubMed]
- Divaris, K.; Monda, K.L.; North, K.E.; Olshan, A.F.; Lange, E.M.; Moss, K.; Barros, S.P.; Beck, J.D.; Offenbacher, S. Genome-wide association study of periodontal pathogen colonization. J. Dent. Res. 2012, 91, 21S–28S. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, A.S.; Bochenek, G.; Manke, T.; Nothnagel, M.; Graetz, C.; Thien, A.; Jockel-Schneider, Y.; Harks, I.; Staufenbiel, I.; Wijmenga, C.; et al. Validation of reported genetic risk factors for periodontitis in a large-scale replication study. J. Clin. Periodontol. 2013, 40, 563–572. [Google Scholar] [CrossRef] [PubMed]
- Shang, D.; Dong, L.; Zeng, L.; Yang, R.; Xu, J.; Wu, Y.; Xu, R.; Tao, H.; Zhang, N. Two-stage comprehensive evaluation of genetic susceptibility of common variants in FBXO38, AP3B2 and WHAMM to severe chronic periodontitis. Sci. Rep. 2015, 5, 17882. [Google Scholar] [CrossRef] [PubMed]
- Divaris, K.; Monda, K.L.; North, K.E.; Olshan, A.F.; Reynolds, L.M.; Hsueh, W.C.; Lange, E.M.; Moss, K.; Barros, S.P.; Weyant, R.J.; et al. Exploring the genetic basis of chronic periodontitis: A genome-wide association study. Hum. Mol. Genet. 2013, 22, 2312–2324. [Google Scholar] [CrossRef] [PubMed]
- Rhodin, K.; Divaris, K.; North, K.E.; Barros, S.P.; Moss, K.; Beck, J.D.; Offenbacher, S. Chronic periodontitis genome-wide association studies: Gene-centric and gene set enrichment analyses. J. Dent. Res. 2014, 93, 882–890. [Google Scholar] [CrossRef] [PubMed]
- Munz, M.; Willenborg, C.; Richter, G.M.; Jockel-Schneider, Y.; Graetz, C.; Staufenbiel, I.; Wellmann, J.; Berger, K.; Krone, B.; Hoffmann, P.; et al. A genome-wide association study identifies nucleotide variants at SIGLEC5 and DEFA1A3 as risk loci for periodontitis. Hum. Mol. Genet. 2017, 26, 2577–2588. [Google Scholar] [CrossRef] [PubMed]
- Offenbacher, S.; Divaris, K.; Barros, S.P.; Moss, K.L.; Marchesan, J.T.; Morelli, T.; Zhang, S.; Kim, S.; Sun, L.; Beck, J.D.; et al. Genome-wide association study of biologically informed periodontal complex traits offers novel insights into the genetic basis of periodontal disease. Hum. Mol. Genet. 2016, 25, 2113–2129. [Google Scholar] [CrossRef] [PubMed]
- Cavalla, F.; Biguetti, C.C.; Colavite, P.M.; Silveira, E.V.; Martins, W., Jr.; Letra, A.; Trombone, A.P.; Silva, R.M.; Garlet, G.P. TBX21-1993T/C (RS4794067) polymorphism is associated with increased risk of chronic periodontitis and increased T-bet expression in periodontal lesions, but does not significantly impact the IFN-g transcriptional level or the pattern of periodontophatic bacterial infection. Virulence 2015, 6, 293–304. [Google Scholar] [PubMed]
- Claudino, M.; Trombone, A.P.; Cardoso, C.R.; Ferreira, S.B., Jr.; Martins, W., Jr.; Assis, G.F.; Santos, C.F.; Trevilatto, P.C.; Campanelli, A.P.; Silva, J.S.; et al. The broad effects of the functional Il-10 promoter-592 polymorphism: Modulation of Il-10, Timp-3, and OPG expression and their association with periodontal disease outcome. J. Leukoc. Biol. 2008, 84, 1565–1573. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, S.B., Jr.; Trombone, A.P.; Repeke, C.E.; Cardoso, C.R.; Martins, W., Jr.; Santos, C.F.; Trevilatto, P.C.; Avila-Campos, M.J.; Campanelli, A.P.; Silva, J.S.; et al. An interleukin-1β (IL-1β) single-nucleotide polymorphism at position 3954 and red complex periodontopathogens independently and additively modulate the levels of IL-1β in diseased periodontal tissues. Infect. Immun. 2008, 76, 3725–3734. [Google Scholar] [CrossRef] [PubMed]
- Repeke, C.E.; Trombone, A.P.; Ferreira, S.B., Jr.; Cardoso, C.R.; Silveira, E.M.; Martins, W., Jr.; Trevilatto, P.C.; Silva, J.S.; Campanelli, A.P.; Garlet, G.P. Strong and persistent microbial and inflammatory stimuli overcome the genetic predisposition to higher matrix metalloproteinase-1 (MMP-1) expression: A mechanistic explanation for the lack of association of MMP1-1607 single-nucleotide polymorphism genotypes with MMP-1 expression in chronic periodontitis lesions. J. Clin. Periodontol. 2009, 36, 726–738. [Google Scholar] [PubMed]
- Trombone, A.P.; Cardoso, C.R.; Repeke, C.E.; Ferreira S.B., Jr.; Martins W., Jr.; Campanelli, A.P.; Avila-Campos, M.J.; Trevilatto, P.C.; Silva, J.S.; Garlet, G.P. Tumor necrosis factor-alpha-308G/A single nucleotide polymorphism and red-complex periodontopathogens are independently associated with increased levels of tumor necrosis factor-alpha in diseased periodontal tissues. J. Periodontal Res. 2008, 44, 598–608. [Google Scholar] [CrossRef] [PubMed]
- Schulz, S.; Zissler, N.; Altermann, W.; Klapproth, J.; Zimmermann, U.; Glaser, C.; Schaller, H.G.; Reichert, S. Impact of genetic variants of CD14 and TLR4 on subgingival periodontopathogens. Int. J. Immunogenet. 2008, 35, 457–464. [Google Scholar] [CrossRef] [PubMed]
- Agerbaek, M.R.; Lang, N.P.; Persson, G.R. Microbiological composition associated with interleukin-1 gene polymorphism in subjects undergoing supportive periodontal therapy. J. Periodontol. 2006, 77, 1397–1402. [Google Scholar] [CrossRef] [PubMed]
- Nibali, L.; Donos, N.; Henderson, B. Periodontal infectogenomics. J. Med. Microb. 2009, 58, 1269–1274. [Google Scholar] [CrossRef] [PubMed]
- Nibali, L.; Tonetti, M.S.; Ready, D.; Parkar, M.; Brett, P.M.; Donos, N.; D’Aiuto, F. Interleukin-6 polymorphisms are associated with pathogenic bacteria in subjects with periodontitis. J. Periodontol. 2008, 79, 677–683. [Google Scholar] [CrossRef] [PubMed]
- Nibali, L.; Di Iorio, A.; Onabolu, O.; Lin, G.H. Periodontal infectogenomics: Systematic review of associations between host genetic variants and subgingival microbial detection. J. Clin. Periodontol. 2016, 43, 889–900. [Google Scholar] [CrossRef] [PubMed]
- Palmer, R.M.; Wilson, R.F.; Hasan, A.S.; Scott, D.A. Mechanisms of action of environmental factors—Tobacco smoking. J. Clin. Periodontol. 2005, 32 (Suppl. 6), 180–195. [Google Scholar] [CrossRef] [PubMed]
- Genco, R.J.; Borgnakke, W.S. Risk factors for periodontal disease. Periodontology 2000 2013, 62, 59–94. [Google Scholar] [CrossRef] [PubMed]
- Melcher, A.H.; Holowka, S.; Pharoah, M.; Lewin, P.K. Non-invasive computed tomography and three-dimensional reconstruction of the dentition of a 2800-year-old Egyptian mummy exhibiting extensive dental disease. Am. J. Phys. Anthropol. 1997, 103, 329–340. [Google Scholar] [CrossRef]
- Forshaw, R.J. Dental health and disease in ancient Egypt. Br. Dent. J. 2009, 206, 421–424. [Google Scholar] [CrossRef] [PubMed]
- Hajishengallis, G. The inflammophilic character of the periodontitis-associated microbiota. Mol. Oral Microb. 2014, 29, 248–257. [Google Scholar] [CrossRef] [PubMed]
- Armitage, G.C. Development of a classification system for periodontal diseases and conditions. Ann. Periodontol. 1999, 4, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Feres, M.; Soares, G.M.; Mendes, J.A.; Silva, M.P.; Faveri, M.; Teles, R.; Socransky, S.S.; Figueiredo, L.C. Metronidazole alone or with amoxicillin as adjuncts to non-surgical treatment of chronic periodontitis: A 1-year double-blinded, placebo-controlled, randomized clinical trial. J. Clin. Periodontol. 2012, 39, 1149–1158. [Google Scholar] [CrossRef] [PubMed]
- Araujo, M.W.; Hovey, K.M.; Benedek, J.R.; Grossi, S.G.; Dorn, J.; Wactawski-Wende, J.; Genco, R.J.; Trevisan, M. Reproducibility of probing depth measurement using a constant-force electronic probe: Analysis of inter- and intraexaminer variability. J. Periodontol. 2003, 74, 1736–1740. [Google Scholar] [CrossRef] [PubMed]
- McHugh, M.L. Interrater reliability: The kappa statistic. Biochem. Med. 2012, 22, 276–282. [Google Scholar] [CrossRef]
- Parra, F.C.; Amado, R.C.; Lambertucci, J.R.; Rocha, J.; Antunes, C.M.; Pena, S.D. Color and genomic ancestry in Brazilians. Proc. Natl. Acad. Sci. USA 2003, 100, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Pena, S.D.; Bastos-Rodrigues, L.; Pimenta, J.R.; Bydlowski, S.P. DNA tests probe the genomic ancestry of Brazilians. Braz. J. Med. Biol. Res. 2009, 42, 870–876. [Google Scholar] [CrossRef] [PubMed]
- Pena, S.D.; Di Pietro, G.; Fuchshuber-Moraes, M.; Genro, J.P.; Hutz, M.H.; Kehdy Fde, S.; Kohlrausch, F.; Magno, L.A.; Montenegro, R.C.; Moraes, M.O.; et al. The genomic ancestry of individuals from different geographical regions of Brazil is more uniform than expected. PLoS ONE 2011, 6, e17063. [Google Scholar] [CrossRef] [PubMed]
- Haffajee, A.D.; Cugini, M.A.; Tanner, A.; Pollack, R.P.; Smith, C.; Kent, R.L., Jr.; Socransky, S.S. Subgingival microbiota in healthy, well-maintained elder and periodontitis subjects. J. Clin. Periodontol. 1998, 25, 346–353. [Google Scholar] [CrossRef] [PubMed]
- Mestnik, M.J.; Feres, M.; Figueiredo, L.C.; Duarte, P.M.; Lira, E.A.; Faveri, M. Short-term benefits of the adjunctive use of metronidazole plus amoxicillin in the microbial profile and in the clinical parameters of subjects with generalized aggressive periodontitis. J. Clin. Periodontol. 2010, 37, 353–365. [Google Scholar] [CrossRef] [PubMed]
- Sampaio, E.; Rocha, M.; Figueiredo, L.C.; Faveri, M.; Duarte, P.M.; Gomes Lira, E.A.; Feres, M. Clinical and microbiological effects of azithromycin in the treatment of generalized chronic periodontitis: A randomized placebo-controlled clinical trial. J. Clin. Periodontol. 2011, 38, 838–846. [Google Scholar] [CrossRef] [PubMed]
- Benjamini, Y.; Krieger, A.M.; Yekutieli, D. Adaptive linear step-up procedures that control the false discovery rate. Biometrika 2006, 93, 491–507. [Google Scholar] [CrossRef]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.; Daly, M.J.; et al. Plink: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [PubMed]
- Velsko, I.M.; Cruz-Almeida, Y.; Huang, H.; Wallet, S.M.; Shaddox, L.M. Cytokine response patterns to complex biofilms by mononuclear cells discriminate patient disease status and biofilm dysbiosis. J. Oral Microb. 2017, 9, 1330645. [Google Scholar] [CrossRef] [PubMed]
- Boutin, S.; Hagenfeld, D.; Zimmermann, H.; El Sayed, N.; Hopker, T.; Greiser, H.K.; Becher, H.; Kim, T.S.; Dalpke, A.H. Clustering of subgingival microbiota reveals microbial disease ecotypes associated with clinical stages of periodontitis in a cross-sectional study. Front. Microbiol. 2017, 8, 340. [Google Scholar] [CrossRef] [PubMed]
- Hong, E.P.; Park, J.W. Sample size and statistical power calculation in genetic association studies. Genom. Inform. 2012, 10, 117–122. [Google Scholar] [CrossRef] [PubMed]
- Ai, D.; Huang, R.; Wen, J.; Li, C.; Zhu, J.; Xia, L.C. Integrated metagenomic data analysis demonstrates that a loss of diversity in oral microbiota is associated with periodontitis. BMC Genom. 2017, 18, 1041. [Google Scholar] [CrossRef] [PubMed]
- Koo, I.; Yao, S.; Zhang, X.; Kim, S. Comparative analysis of false discovery rate methods in constructing metabolic association networks. J. Bioinform. Comput. Biol. 2014, 12, 1450018. [Google Scholar] [CrossRef] [PubMed]
- Blainey, P.; Krzywinski, M.; Altman, N. Points of significance: Replication. Nat. Methods 2014, 11, 879–880. [Google Scholar] [CrossRef] [PubMed]
- Chapple, I.L.; Bouchard, P.; Cagetti, M.G.; Campus, G.; Carra, M.C.; Cocco, F.; Nibali, L.; Hujoel, P.; Laine, M.L.; Lingstrom, P.; et al. Interaction of lifestyle, behaviour or systemic diseases with dental caries and periodontal diseases: Consensus report of group 2 of the joint EFP/ORCA workshop on the boundaries between caries and periodontal diseases. J. Clin. Periodontol. 2017, 44 (Suppl. 18), S39–S51. [Google Scholar] [CrossRef] [PubMed]
- Warnakulasuriya, S.; Dietrich, T.; Bornstein, M.M.; Casals Peidro, E.; Preshaw, P.M.; Walter, C.; Wennstrom, J.L.; Bergstrom, J. Oral health risks of tobacco use and effects of cessation. Int. Dent. J. 2010, 60, 7–30. [Google Scholar] [PubMed]
- Lek, M.; Karczewski, K.J.; Minikel, E.V.; Samocha, K.E.; Banks, E.; Fennell, T.; O’Donnell-Luria, A.H.; Ware, J.S.; Hill, A.J.; Cummings, B.B.; et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016, 536, 285–291. [Google Scholar] [CrossRef] [PubMed]
- Hansen, C.J.; Burnell, K.K.; Brogden, K.A. Antimicrobial activity of substance P and neuropeptide Y against laboratory strains of bacteria and oral microorganisms. J. Neuroimmunol. 2006, 177, 215–218. [Google Scholar] [CrossRef] [PubMed]
- Lundy, F.T.; El Karim, I.A.; Linden, G.J. Neuropeptide Y (NPY) and NPY Y1 receptor in periodontal health and disease. Arch. Oral Boil. 2009, 54, 258–262. [Google Scholar] [CrossRef] [PubMed]
- Woods, T.A.; Du, M.; Carmody, A.; Peterson, K.E. Neuropeptide Y negatively influences monocyte recruitment to the central nervous system during retrovirus infection. J. Virol. 2015, 90, 2783–2793. [Google Scholar] [CrossRef] [PubMed]
- Stadler, J.; Le, T.P.; Haas, P.; Nave, H. Distinct effects of NPY13-36, a specific NPY Y2 agonist, in a model of rodent endotoxemia on leukocyte subsets and cytokine levels. Ann. Anat. 2011, 193, 486–493. [Google Scholar] [CrossRef] [PubMed]
- Hargett, S.R.; Walker, N.N.; Hussain, S.S.; Hoehn, K.L.; Keller, S.R. Deletion of the Rab Gap Tbc1d1 modifies glucose, lipid, and energy homeostasis in mice. Am. J. Physiol. Endocrinol. Metab. 2015, 309, E233–E245. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Chen, Q.; Xie, B.; Quan, C.; Sheng, Y.; Zhu, S.; Rong, P.; Zhou, S.; Sakamoto, K.; MacKintosh, C.; et al. Disruption of the AMPK-TBC1D1 nexus increases lipogenic gene expression and causes obesity in mice via promoting IGF1 secretion. Proc. Natl. Acad. Sci. USA 2016, 113, 7219–7224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Demmer, R.T.; Breskin, A.; Rosenbaum, M.; Zuk, A.; LeDuc, C.; Leibel, R.; Paster, B.; Desvarieux, M.; Jacobs, D.R., Jr.; Papapanou, P.N. The subgingival microbiome, systemic inflammation and insulin resistance: The oral infections, glucose intolerance and insulin resistance study. J. Clin. Periodontol. 2017, 44, 255–265. [Google Scholar] [CrossRef] [PubMed]
- Demmer, R.T.; Jacobs, D.R., Jr.; Singh, R.; Zuk, A.; Rosenbaum, M.; Papapanou, P.N.; Desvarieux, M. Periodontal bacteria and prediabetes prevalence in origins: The oral infections, glucose intolerance, and insulin resistance study. J. Dent. Res. 2015, 94, 201S–211S. [Google Scholar] [CrossRef] [PubMed]
- Silva-Boghossian, C.M.; Cesario, P.C.; Leao, A.T.T.; Colombo, A.P.V. Subgingival microbial profile of obese women with periodontal disease. J. Periodontol. 2018, 89, 186–194. [Google Scholar] [CrossRef] [PubMed]
- Maciel, S.S.; Feres, M.; Goncalves, T.E.; Zimmermann, G.S.; da Silva, H.D.; Figueiredo, L.C.; Duarte, P.M. Does obesity influence the subgingival microbiota composition in periodontal health and disease? J. Clin. Periodontol. 2016, 43, 1003–1012. [Google Scholar] [CrossRef] [PubMed]
- Garlet, G.P.; Cardoso, C.R.; Mariano, F.S.; Claudino, M.; de Assis, G.F.; Campanelli, A.P.; Avila-Campos, M.J.; Silva, J.S. Regulatory T cells attenuate experimental periodontitis progression in mice. J. Clin. Periodontol. 2010, 37, 591–600. [Google Scholar] [CrossRef] [PubMed]
- Yu, P.; Hu, Y.; Liu, Z.; Kawai, T.; Taubman, M.A.; Li, W.; Han, X. Local induction of B cell interleukin-10 competency alleviates inflammation and bone loss in ligature-induced experimental periodontitis in mice. Infect. Immun. 2017, 85, e00645-16. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Ma, S.; Guo, J.; Jiang, J.; Luo, K.; Yan, F.; Xiao, Y. Effect of local hIL-10 gene therapy on experimental periodontitis in ovariectomized rats. Acta Odontol. Scand. 2017, 75, 268–279. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, W.; Rutz, S.; Crellin, N.K.; Valdez, P.A.; Hymowitz, S.G. Regulation and functions of the IL-10 family of cytokines in inflammation and disease. Annu. Rev. Immunol. 2011, 29, 71–109. [Google Scholar] [CrossRef] [PubMed]
- Garlet, G.P.; Cardoso, C.R.; Campanelli, A.P.; Garlet, T.P.; Avila-Campos, M.J.; Cunha, F.Q.; Silva, J.S. The essential role of IFN-γ in the control of lethal Aggregatibacter actinomycetemcomitans infection in mice. Microbes Infect. 2008, 10, 489–496. [Google Scholar] [CrossRef] [PubMed]
- Lu, K.; Mahbub, R.; Cable, P.H.; Ru, H.; Parry, N.M.; Bodnar, W.M.; Wishnok, J.S.; Styblo, M.; Swenberg, J.A.; Fox, J.G.; et al. Gut microbiome phenotypes driven by host genetics affect arsenic metabolism. Chem. Res. Toxicol. 2014, 27, 172–174. [Google Scholar] [CrossRef] [PubMed]
- Sumner, C.J.; d’Ydewalle, C.; Wooley, J.; Fawcett, K.A.; Hernandez, D.; Gardiner, A.R.; Kalmar, B.; Baloh, R.H.; Gonzalez, M.; Zuchner, S.; et al. A dominant mutation in FBXO38 causes distal spinal muscular atrophy with calf predominance. Am. J. Hum. Genet. 2013, 93, 976–983. [Google Scholar] [CrossRef] [PubMed]
- Saygun, I.; Nizam, N.; Keskiner, I.; Bal, V.; Kubar, A.; Acikel, C.; Serdar, M.; Slots, J. Salivary infectious agents and periodontal disease status. J. Periodontal Res. 2011, 46, 235–239. [Google Scholar] [CrossRef] [PubMed]
- Stingu, C.S.; Jentsch, H.; Eick, S.; Schaumann, R.; Knofler, G.; Rodloff, A. Microbial profile of patients with periodontitis compared with healthy subjects. Quintessence Int. 2012, 43, e23–e31. [Google Scholar] [PubMed]
- Tsuchida, S.; Satoh, M.; Takiwaki, M.; Nomura, F. Ubiquitination in periodontal disease: A review. Int. J. Mol. Sci. 2017, 18, 1476. [Google Scholar] [CrossRef] [PubMed]
- Resque, R.; Gusmao, L.; Geppert, M.; Roewer, L.; Palha, T.; Alvarez, L.; Ribeiro-dos-Santos, A.; Santos, S. Male lineages in Brazil: Intercontinental admixture and stratification of the European background. PLoS ONE 2016, 11, e0152573. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Zhao, Y.; Yang, S.; Zhang, H.; Wu, Q.; Chen, F. An integrated evolutionary analysis of miRNA-lncRNA in mammals. Mol. Biol. Rep. 2014, 41, 201–207. [Google Scholar] [CrossRef] [PubMed]
- Brinkworth, J.F. Infectious disease and the diversification of the human genome. Hum. Biol. 2017, 89, 47–65. [Google Scholar] [CrossRef] [PubMed]
- Scannapieco, F.A.; Cantos, A. Oral inflammation and infection, and chronic medical diseases: Implications for the elderly. Periodontology 2000 2016, 72, 153–175. [Google Scholar] [CrossRef] [PubMed]
- Weinstock, J.V.; Elliott, D.E. Helminths and the IBD hygiene hypothesis. Inflamm. Bowel Dis. 2009, 15, 128–133. [Google Scholar] [CrossRef] [PubMed]
- Wilson, J.N.; Rockett, K.; Keating, B.; Jallow, M.; Pinder, M.; Sisay-Joof, F.; Newport, M.; Kwiatkowski, D. A hallmark of balancing selection is present at the promoter region of interleukin 10. Genes Immun. 2006, 7, 680–683. [Google Scholar] [CrossRef] [PubMed]
- Mestnik, M.J.; Feres, M.; Figueiredo, L.C.; Soares, G.; Teles, R.P.; Fermiano, D.; Duarte, P.M.; Faveri, M. The effects of adjunctive metronidazole plus amoxicillin in the treatment of generalized aggressive periodontitis: A 1-year double-blinded, placebo-controlled, randomized clinical trial. J. Clin. Periodontol. 2012, 39, 955–961. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Healthy (n = 91) | Chronic Periodontitis (n = 76) | |
|---|---|---|
| Gender distribution | 50 f/41 m | 39 f/37 m |
| Age | 45.1 ± 5.9 | 46.8 ± 6.1 |
| Clinical parameters | value ± SD | value ± SD |
| Probing depth | 2.2 ± 0.6 | 4.5 ± 0.7 |
| Clinical Attachment Loss | 0.6 ± 0.2 | 4.2 ± 0.7 |
| % Bleeding on probing | 4.5 ± 2.7 | 66.2 ± 8.9 |
| Plaque index | 30.2 ± 6.2 | 56.9 ± 9.1 |
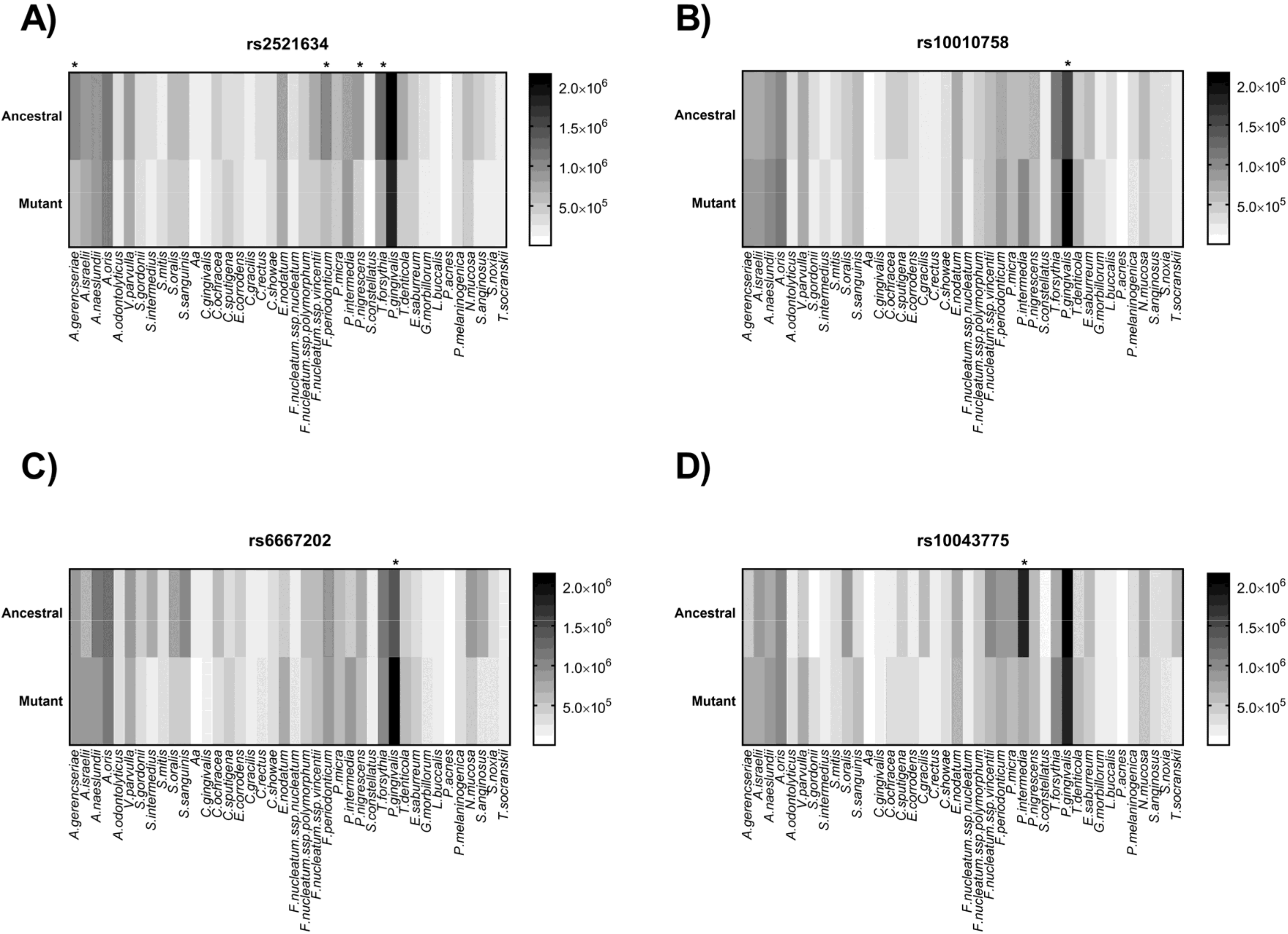
| Code | Nearby Gene | Allele | Ancestral | Chrm. | Position | Association | MAF * | Relative Position |
|---|---|---|---|---|---|---|---|---|
| rs2521634 | NPY | G/A | G | 7 | 37614913 | Severe CP | 0.24 | Intergenic variant |
| rs7762544 | NCR2 | G/A | G | 6 | 147785313 | Severe Chronic Periodontitis | 0.15 | Intergenic variant |
| rs12032672 | PKN2 | A/C | A | 1 | 74883781 | Red complex | 0.44 | Intergenic variant |
| rs10010758 | TBC1D1 | T/C | T | 4 | 1842012 | Red complex | 0.29 | Intron variant |
| rs1932040 | RUNX2 | A/G | A | 6 | 88398224 | Orange complex | 0.4 | Intergenic variant |
| rs9942773 | CSMD3 | A/C | C | 8 | 22115347 | Orange complex | 0.19 | Intergenic variant |
| rs1616122 | VAMP3 | C/T | C | 1 | 137669543 | Orange complex | 0.42 | Intron variant |
| rs11621969 | FOS | T/C | T | 14 | 7444172 | Aa | 0.15 | Intergenic variant |
| rs9287989 | WAPAL | T/C | C | 2 | 45804766 | Aa | 0.5 | Regulatory region variant |
| rs8080364 | KIAA0753 | C/T | T | 17 | 24344565 | Control | 0.27 | NCTEV |
| rs2836981 | BRWD1-IT2 | G/A | G | 21 | 39610235 | Control | 0.39 | Intron variant |
| rs11695297 | MYT1L | T/A | A | 2 | 22088619 | Control | 0.47 | Intron variant |
| rs1537415 | GTL6D1 | G/C | G | 9 | 6838736 | AP | 0.31 | Intron variant |
| rs1333048 | CDKN2A/2B | A/C | A | 9 | 43163827 | AP; CP | 0.45 | Downstream gene variant |
| rs6667202 | IL10 | C/A | C | 1 | 205023715 | AP | 0.31 | Intergenic variant |
| rs3826782 | VAV1 | G/A | G | 19 | 41487293 | Severe Chronic Periodontitis | 0.15 | Intron variant |
| rs10043775 | FBXO38 | T/C | C | 5 | 6478800 | Red complex | 0.26 | Missense variant |
| rs4794067 | TBX21 | T/C | C | 17 | 176425987 | Chronic Periodontitis | 0.25 | Upstream gene variant |
| rs2891168 | CDKN2B-AS1 | A/G | A | 9 | 115190203 | Aggressive Periodontitis | 0.42 | Intron variant |
| Bacterial Species Assayed by DNA-DNA Hybridization Checkerboard | |||
|---|---|---|---|
| Actinomyces gerencseriae | Streptococcus sanguinis | Fusobacterium nucleatum spp. nucleatum | Treponema denticola |
| Actinomyces israelii | Aggregatibacter actinomycetemcomitans | Fusobacterium nucleatum spp. polymorphum | Eubacterium saburreum |
| Actinomyces naeslundii | Capnocytophaga gingivalis | Fusobacterium nucleatum spp. vincentii | Gemella morbillorum |
| Actinomyces oris | Capnocytophaga ochracea | Fusobacterium periodonticum | Leptotrichia buccalis |
| Actinomyces odontolyticus | Capnocytophaga sputigena | Parvimonas micra | Propionibacterium acnes |
| Veillonella parvula | Eikenella corrodens | Prevotella intermedia | Prevotella melaninogenica |
| Streptococcus gordonii | Campylobacter gracilis | Prevotella nigrescens | Neisseria mucosa |
| Streptococcus intermedius | Campylobacter rectus | Streptococcus constellatus | Streptococcus anginosus |
| Streptococcus mitis | Campylobacter showae | Tannerella forsythia | Selenomonas noxia |
| Streptococcus oralis | Eubacterium nodatum | Porphyromonas gingivalis | Treponema socranskii |
| SNP | Control [n (%)] | CP [n (%)] | Data Analysis | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Ancestral HZ | HetZ | Mutant HZ | Mutant AF | Ancestral HZ | HetZ | Mutant HZ | Mutant AF | H-W | Chi-Square | p-Value | |
| rs2521634 | 46 (51.7) | 31 (34.8) | 12 (13.4) | 36 (25) | 39 (54.1) | 30 (41.6) | 3 (4.1) | 55 (30.8) | 0.443 | 4.245 | 0.12 |
| rs7762544 | 7 (9.6) | 20 (27.3) | 46 (63) | 112 (76.7) | 0 (0) | 22 (33.3) | 44 (66.6) | 110 (83.3) | 0.048 | NA | NA |
| rs12032672 | 25 (40.3) | 30 (48.3) | 7 (11.3) | 44 (35.5) | 20 (31.2) | 35 (54.7) | 9 (14.1) | 53 (41.4) | 0.65 | 1.159 | 0.56 |
| rs10010758 | 22 (62.8) | 11 (31.4) | 2 (5.7) | 15 (21.4) | 32 (57.1) | 16 (28.6) | 8 (14.3) | 32 (28.5) | 0.33 | 1.168 | 0.44 |
| rs1932040 | 3 (3.6) | 33 (39.3) | 48 (57.1) | 129 (76.7) | 6 (8.2) | 36 (49.3) | 31 (42.5) | 98 (67.1) | 0.44 | 4.038 | 0.13 |
| rs9942773 | 4 (5) | 27 (33.7) | 49 (61.2) | 125 (78.1) | 6 (9.4) | 19 (29.7) | 39 (60.9) | 97 (75.7) | 0.6 | 1.164 | 0.55 |
| rs1616122 | 27 (31.8) | 35 (41.1) | 23 (27) | 81 (47.6) | 14 (18.4) | 43 (56.6) | 19 (25) | 81 (53.2) | 0.057 | 4.835 | 0.08 |
| rs11621969 | 60 (71.4) | 22 (26.2) | 2 (2.4) | 26 (15.4) | 49 (64.5) | 24 (31.5) | 3 (3.9) | 30 (19.7) | 0.75 | 0.999 | 0.61 |
| rs9287989 | 28 (32.5) | 47 (54.6) | 11 (12.7) | 69 (40) | 22 (29.7) | 37 (50) | 15 (20.2) | 67 (45.2) | 0.111 | 1.635 | 0.44 |
| rs8080364 | 37 (44) | 39 (46.4) | 8 (9.5) | 55 (32.7) | 46 (60.5) | 27 (35.5) | 3 (3.9) | 33 (21.7) | 0.282 | 5.043 | 0.08 |
| rs2836981 | 14 (16.1) | 42 (48.3) | 31 (35.6) | 104 (59.7) | 7 (9.2) | 40 (52.6) | 29 (38.1) | 98 (64.4) | 0.808 | 1.714 | 0.42 |
| rs11695297 | 43 (53.7) | 25 (31.2) | 12 (15) | 49 (30.6) | 20 (27.4) | 38 (52) | 15 (20.5) | 68 (46.5) | 0.029 | NA | NA |
| rs1537415 | 12 (14.1) | 37 (43.5) | 36 (42.3) | 109 (64.1) | 6 (7.9) | 39 (51.3) | 31 (40.8) | 101 (66.4) | 0.97 | 1.929 | 0.38 |
| rs1333048 | 17 (25) | 33 (48.5) | 18 (26.4) | 69 (50.7) | 16 (25.8) | 28 (45.1) | 18 (29) | 64 (51.6) | 0.993 | 0.1636 | 0.92 |
| rs6667202 | 12 (13.6) | 30 (34.1) | 46 (52.2) | 122 (69.3) | 10 (13.1) | 35 (46) | 31 (40.8) | 97 (63.8) | 0.191 | 3.365 | 0.18 |
| rs3826782 | 64 (78) | 18 (21.9) | 0 (0) | 18 (10.9) | 57 (76) | 16 (21.3) | 2 (2.6) | 20 (13.3) | 0.204 | NA | NA |
| rs10043775 | 5 (6) | 24 (28.9) | 54 (65.1) | 132 (79.5) | 4 (5.4) | 26 (35.1) | 44 (59.4) | 114 (77) | 0.57 | 1.159 | 0.56 |
| rs4794067 | 8 (9.3) | 31 (36) | 47 (54.6) | 125 (72.6) | 3 (3.9) | 28 (36.8) | 45 (59.2) | 118 (77.6) | 0.367 | 3.216 | 0.2 |
| rs2891168 | 20 (29.8) | 31 (46.2) | 16 (23.9) | 63 (47) | 18 (28.6) | 33 (52.3) | 12 (19) | 57 (45.2) | 0.998 | 0.761 | 0.68 |
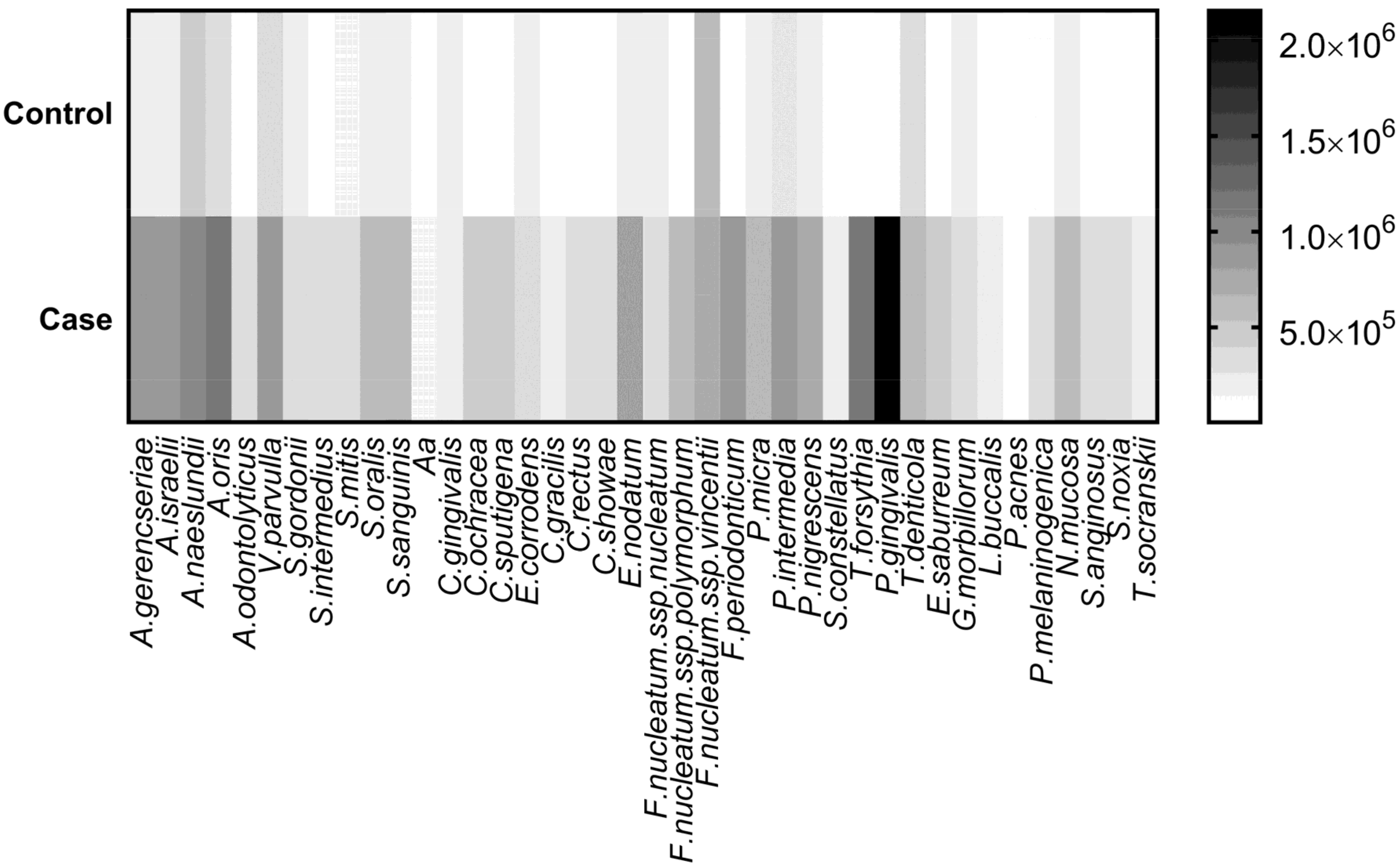
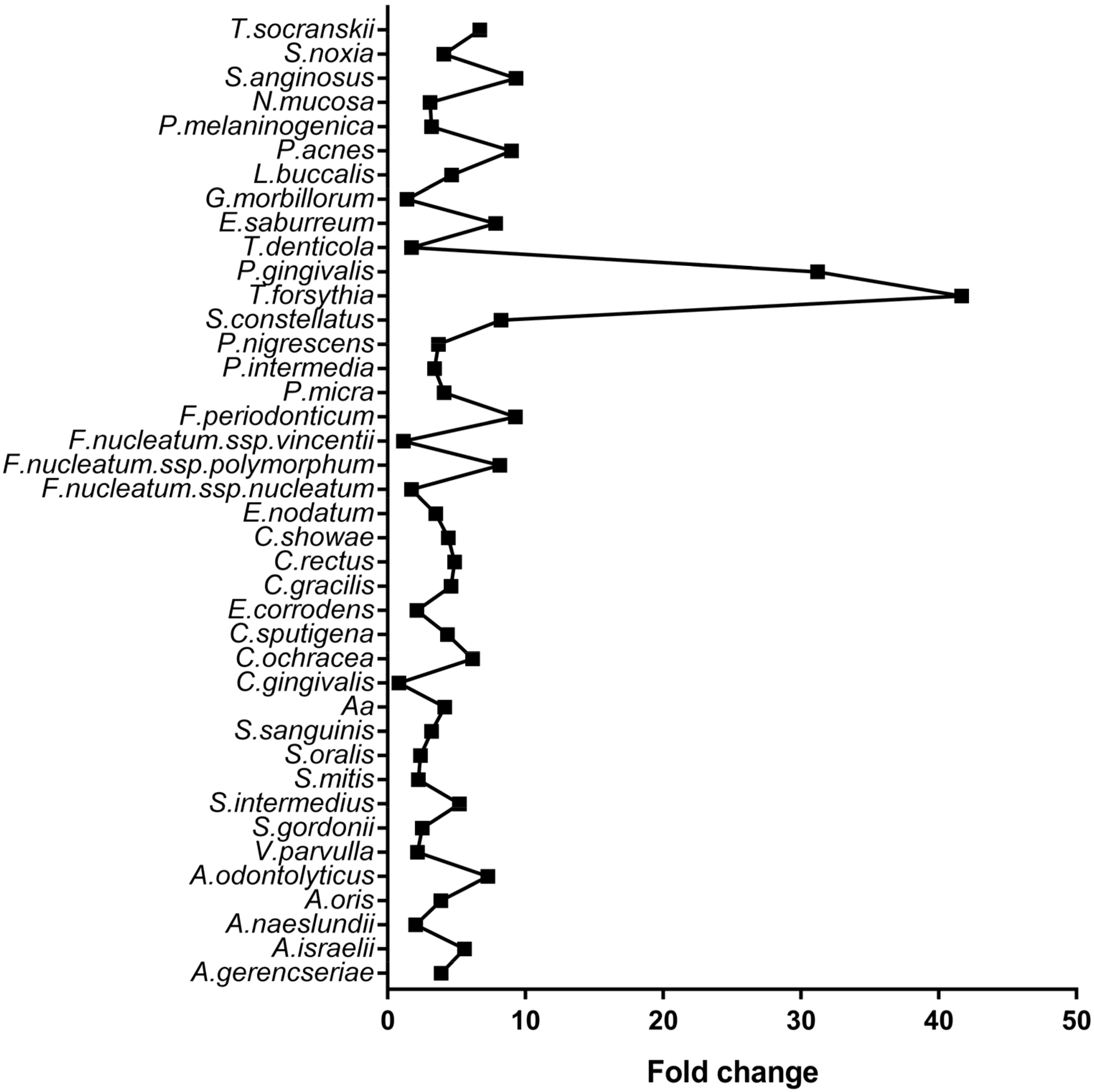
| Microorganism | Control | CP | Adjusted p-Value |
|---|---|---|---|
| A. israelii | 160,345 | 893,511 | <0.001 |
| P. gingivalis | 68,876 | 2,150,036 | <0.001 |
| A. oris | 302,836 | 1,171,655 | <0.001 |
| T. forsythia | 26,506 | 1,104,569 | <0.001 |
| F. periodonticum | 98,231 | 910,910 | <0.001 |
| A. gerencseriae | 235,808 | 913,498 | <0.001 |
| P. intermedia | 259,762 | 882,319 | <0.001 |
| E. nodatum | 228,546 | 795,966 | <0.001 |
| P. nigrescens | 188,205 | 692,758 | <0.001 |
| P. micra | 161,019 | 659,696 | <0.001 |
| A. naeslundii | 468,479 | 957,118 | <0.001 |
| F. nucleatum spp. polymorphum | 67,973 | 552,734 | <0.001 |
| V. parvulla | 385,381 | 837,456 | <0.001 |
| E. saburreum | 61,402 | 480,620 | <0.001 |
| N. mucosa | 200,501 | 615,542 | <0.001 |
| S. sanguinis | 189,625 | 603,031 | <0.001 |
| C. ochracea | 68,674 | 423,281 | <0.001 |
| C. sputigena | 99,803 | 433,313 | <0.001 |
| S. anginosus | 39,551 | 368,112 | <0.001 |
| S. oralis | 232,754 | 552,347 | <0.001 |
| T. denticola | 374,527 | 648,599 | <0.001 |
| A. odontolyticus | 42,836 | 311,538 | <0.001 |
| C. showae | 73,988 | 325,362 | <0.001 |
| P. melaninogenica | 111,172 | 353,281 | 0.001 |
| S. noxia | 75,718 | 308,615 | 0.001 |
| S. intermedius | 54,897 | 285,286 | 0.001 |
| C. rectus | 59,601 | 289,257 | 0.001 |
| S. gordonii | 146,660 | 368,106 | 0.001 |
| E. corrodens | 183,801 | 388,124 | 0.002 |
| T.socranskii | 33,894 | 226,699 | 0.003 |
| L. buccalis | 52,565 | 244,344 | 0.003 |
| C. gracilis | 48,808 | 224,658 | 0.005 |
| S. mitis | 137,170 | 306,687 | 0.006 |
| S. constellatus | 23,077 | 190,087 | 0.007 |
| F.nucleatum spp. nucleatum | 162,832 | 280,366 | 0.025 |
| A. actinomycetemcomitans | 32,715 | 135,463 | 0.034 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cavalla, F.; Biguetti, C.C.; Lima Melchiades, J.; Tabanez, A.P.; De Campos Soriani Azevedo, M.; Favaro Trombone, A.P.; Faveri, M.; Feres, M.; Pompermaier Garlet, G. Genetic Association with Subgingival Bacterial Colonization in Chronic Periodontitis. Genes 2018, 9, 271. https://doi.org/10.3390/genes9060271
Cavalla F, Biguetti CC, Lima Melchiades J, Tabanez AP, De Campos Soriani Azevedo M, Favaro Trombone AP, Faveri M, Feres M, Pompermaier Garlet G. Genetic Association with Subgingival Bacterial Colonization in Chronic Periodontitis. Genes. 2018; 9(6):271. https://doi.org/10.3390/genes9060271
Chicago/Turabian StyleCavalla, Franco, Claudia Cristina Biguetti, Jessica Lima Melchiades, Andre Pantenuci Tabanez, Michelle De Campos Soriani Azevedo, Ana Paula Favaro Trombone, Marcelo Faveri, Magda Feres, and Gustavo Pompermaier Garlet. 2018. "Genetic Association with Subgingival Bacterial Colonization in Chronic Periodontitis" Genes 9, no. 6: 271. https://doi.org/10.3390/genes9060271
APA StyleCavalla, F., Biguetti, C. C., Lima Melchiades, J., Tabanez, A. P., De Campos Soriani Azevedo, M., Favaro Trombone, A. P., Faveri, M., Feres, M., & Pompermaier Garlet, G. (2018). Genetic Association with Subgingival Bacterial Colonization in Chronic Periodontitis. Genes, 9(6), 271. https://doi.org/10.3390/genes9060271