RECTA: Regulon Identification Based on Comparative Genomics and Transcriptomics Analysis


Abstract
:1. Introduction
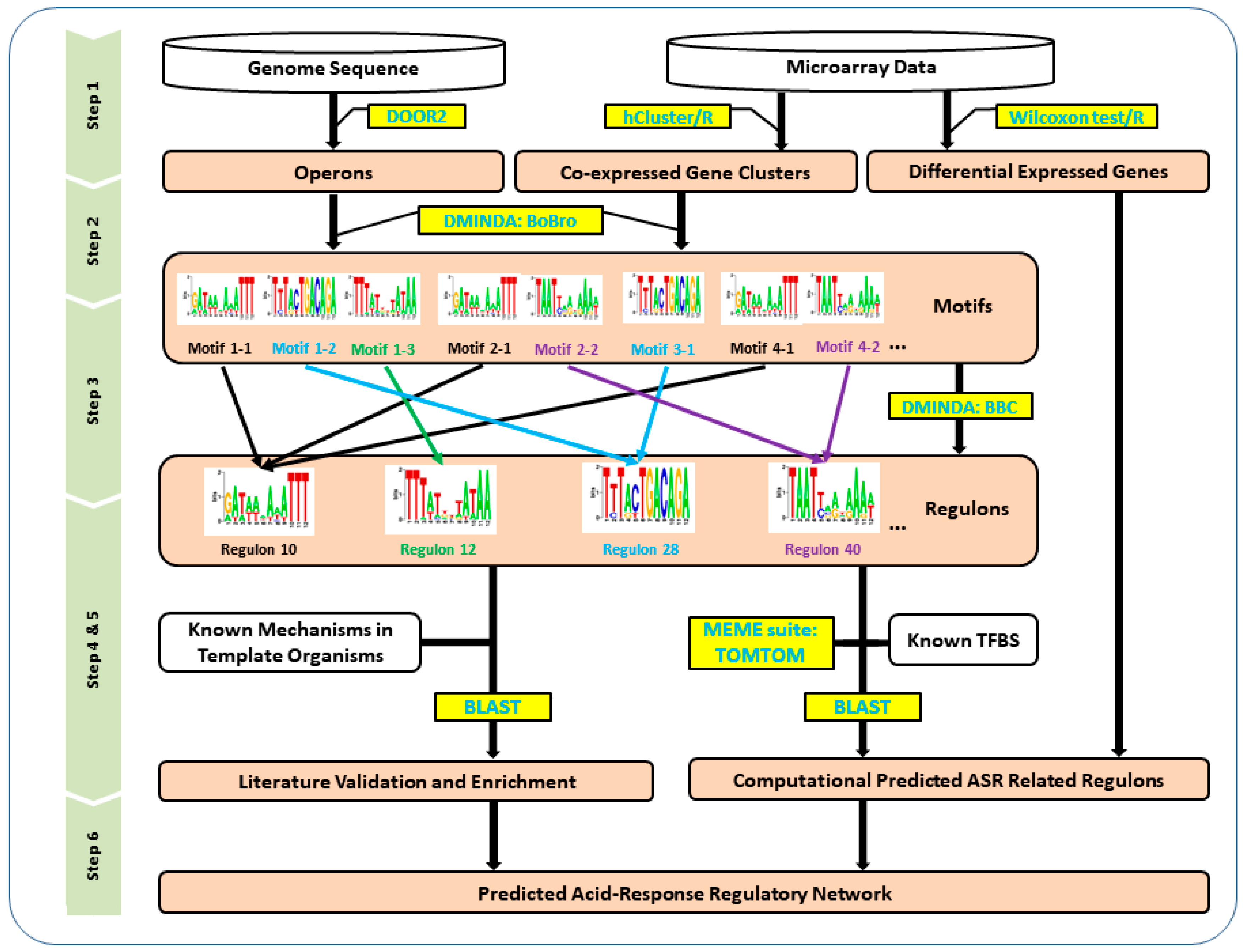
2. Materials and Methods
2.1. Data Acquisition
2.2. Operon Identification
2.3. Gene Differential Expression Analysis and Co-Expression Analysis
2.4. Motif finding and Regulon Prediction
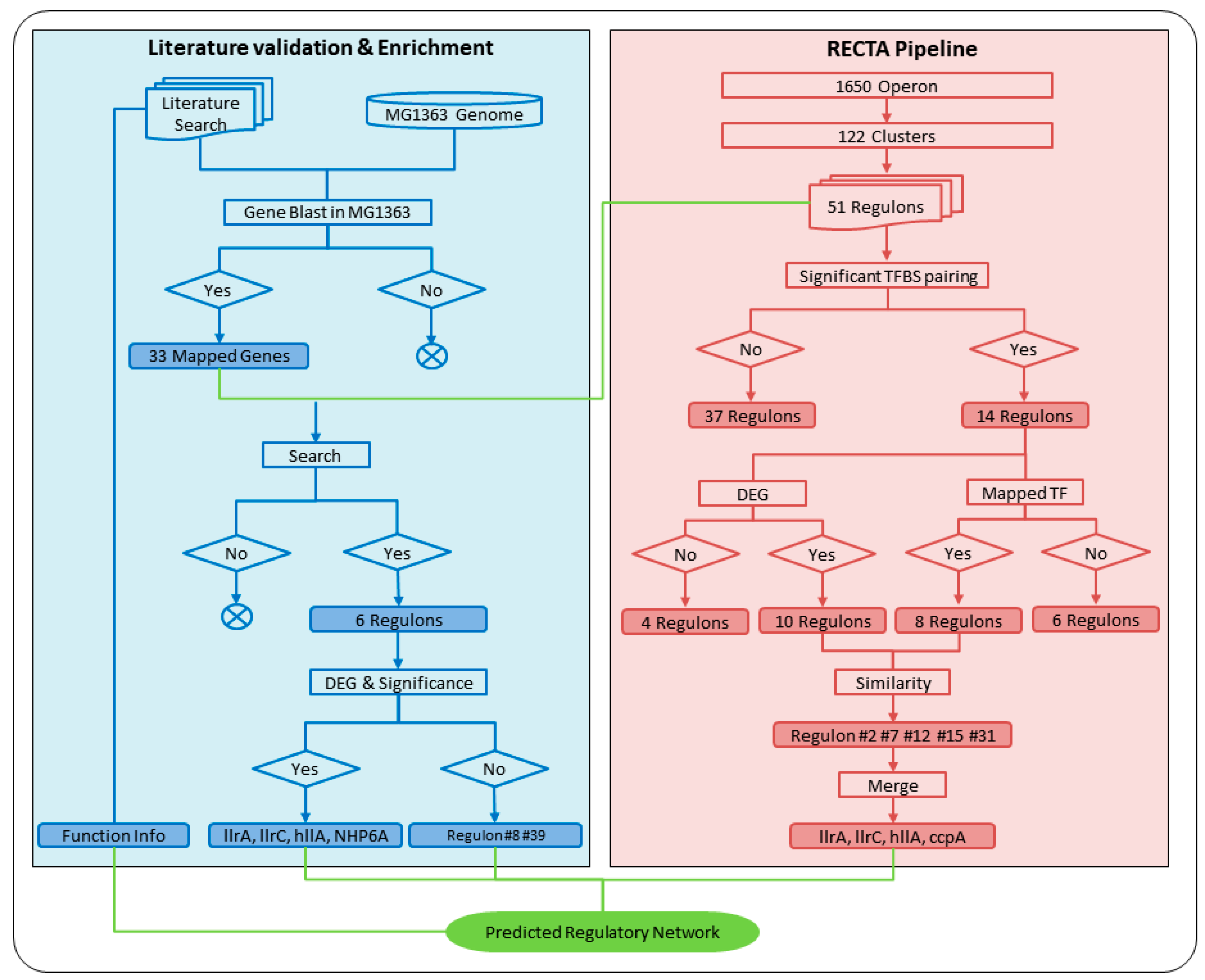
2.5. Regulon Validation Based on Transcription Factor BLAST and Differentially Expressed Gene Filtering
2.6. Regulon Validation Based on Known Acid Stress Response Proteins from the Literature
3. Results
3.1. Predicted Operons and Co-Expression Gene Module Generation
3.2. Predicted Regulons Based on Motif Finding and Clustering
3.3. Computationally-Verified Regulon Based on Transcription Factor BLAST and Differential Gene Expression Analysis
3.4. Verified Regulons Based on Literature Verification
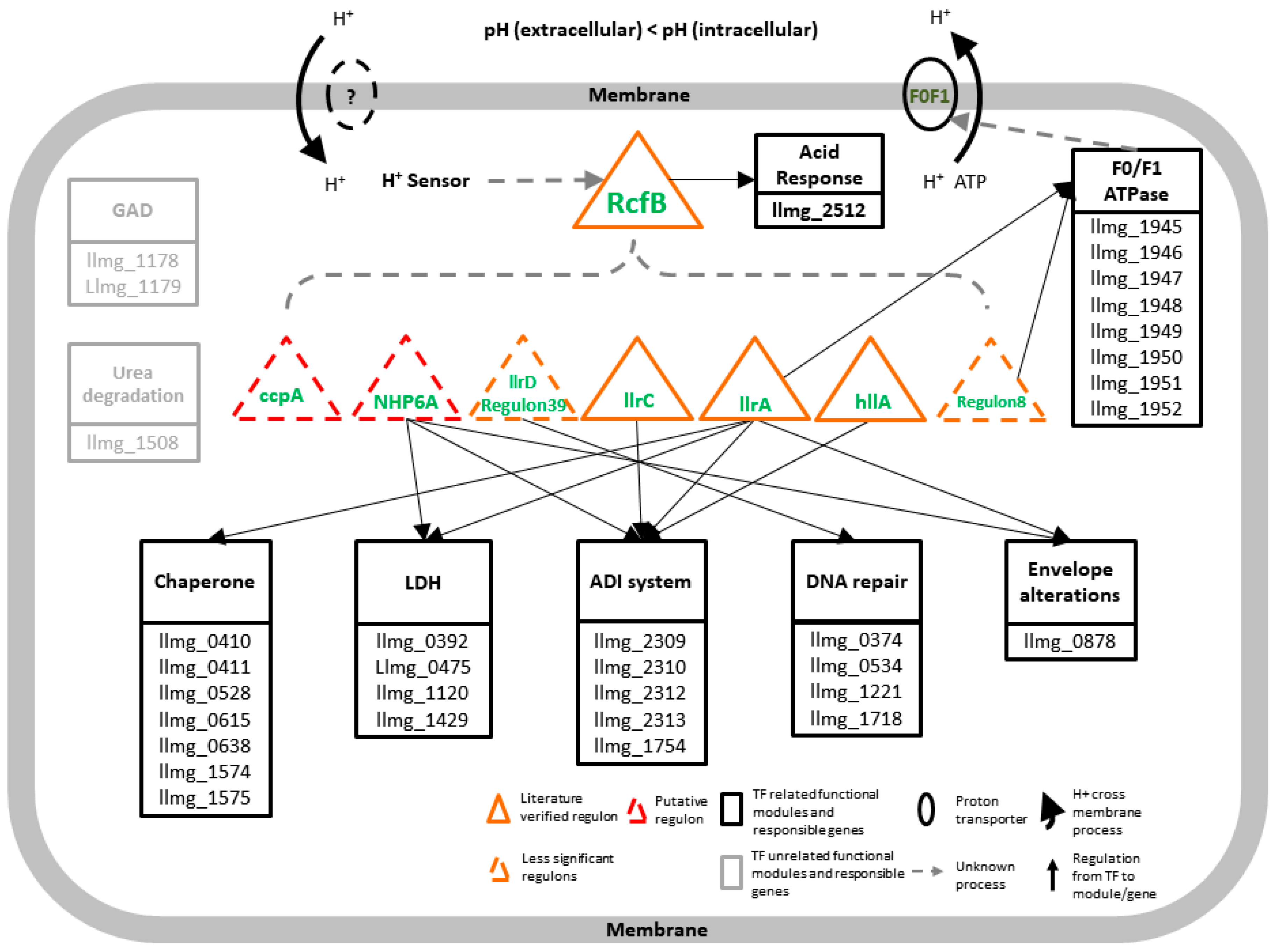
3.5. A Model of Regulatory Network in Response to pH Change
4. Discussion and Conclusions
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
Abbreviations
| ADI | Arginine deiminase |
| BBC | Bobro-based motif comparison |
| BoBro | Bottleneck broken |
| CEM | Co-expression (gene) module |
| DEG | Differentially expressed gene |
| DGE | Differential gene expression |
| E. coli | Escherichia coli |
| GAD | Glutamate decarboxylases |
| GRN | Gene regulatory network |
| LDH | Lactate dehydrogenase |
| L. lactis | Lactococcus lactis |
| MG1363 | Lactococcus lactis MG1363 |
| Motif | Cis-regulatory motif |
| RECTA | Regulon identification based on comparative genomics and transcriptomics analysis |
| TF | Transcription factors |
| TFBS | Transcriptional factor binding site |
References
- Carvalho, A.L.; Turner, D.L.; Fonseca, L.L.; Solopova, A.; Catarino, T.; Kuipers, O.P.; Voit, E.O.; Neves, A.R.; Santos, H. Metabolic and transcriptional analysis of acid stress in Lactococcus lactis, with a focus on the kinetics of lactic acid pools. PLoS ONE 2013, 8, e68470. [Google Scholar] [CrossRef] [PubMed]
- Locke, J.C.W.; Young, J.W.; Fontes, M.; Jiménez, M.J.H.; Elowitz, M.B. Stochastic pulse regulation in bacterial stress response. Science 2011, 334, 366–369. [Google Scholar] [CrossRef] [PubMed]
- Levine, J.H.; Lin, Y.; Elowitz, M.B. Functional roles of pulsing in genetic circuits. Science 2013, 342, 1193–1200. [Google Scholar] [CrossRef] [PubMed]
- Arnoldini, M.; Mostowy, R.; Bonhoeffer, S.; Ackermann, M. Evolution of stress response in the face of unreliable environmental signals. PLoS Comput. Biol. 2012, 8, e1002627. [Google Scholar] [CrossRef] [PubMed]
- Mao, X.; Ma, Q.; Liu, B.; Chen, X.; Zhang, H.; Xu, Y. Revisiting operons: An analysis of the landscape of transcriptional units in E. coli. BMC Bioinform. 2015, 16, 356. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Ma, Q.; Li, G. Elucidation of operon structures across closely related bacterial genomes. PLoS ONE 2014, 9, e100999. [Google Scholar] [CrossRef] [PubMed]
- Cao, H.; Ma, Q.; Chen, X.; Xu, Y. DOOR: A prokaryotic operon database for genome analyses and functional inference. Brief. Bioinform. 2017. [Google Scholar] [CrossRef] [PubMed]
- Jacob, F.; Perrin, D.; Sanchez, C.; Monod, J. Operon: A group of genes with the expression coordinated by an operator. C. R. Hebd. Seances Acad. Sci. 1960, 250, 1727–1729. [Google Scholar] [PubMed]
- Liu, B.; Zhou, C.; Li, G.; Zhang, H.; Zeng, E.; Liu, Q.; Ma, Q. Bacterial regulon modeling and prediction based on systematic cis regulatory motif analyses. Sci. Rep. 2016, 6, 23030. [Google Scholar] [CrossRef] [PubMed]
- Kumka, J.E.; Bauer, C.E. Analysis of the FnrL regulon in Rhodobacter capsulatus reveals limited regulon overlap with orthologues from Rhodobacter sphaeroides and Escherichia coli. BMC Genom. 2015, 16, 895. [Google Scholar] [CrossRef] [PubMed]
- Tan, K.; Moreno-Hagelsieb, G.; Collado-Vides, J.; Stormo, G.D. A comparative genomics approach to prediction of new members of regulons. Genome Res. 2001, 11, 566–584. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Stamatoyannopoulos, J.A.; Bailey, T.L.; Noble, W.S. Quantifying similarity between motifs. Genome Biol. 2007, 8, R24. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q.; Liu, B.; Zhou, C.; Yin, Y.; Li, G.; Xu, Y. An integrated toolkit for accurate prediction and analysis of cis-regulatory motifs at a genome scale. Bioinformatics 2013, 29, 2261–2268. [Google Scholar] [CrossRef] [PubMed]
- Novichkov, P.S.; Rodionov, D.A.; Stavrovskaya, E.D.; Novichkova, E.S.; Kazakov, A.E.; Gelfand, M.S.; Arkin, A.P.; Mironov, A.A.; Dubchak, I. RegPredict: An integrated system for regulon inference in prokaryotes by comparative genomics approach. Nucleic Acids Res. 2010, 38, W299–W307. [Google Scholar] [CrossRef] [PubMed]
- Blanchette, M.; Schwikowski, B.; Tompa, M. Algorithms for phylogenetic footprinting. J. Comput. Biol. 2002, 9, 211–223. [Google Scholar] [CrossRef] [PubMed]
- Katara, P.; Grover, A.; Sharma, V. Phylogenetic footprinting: A boost for microbial regulatory genomics. Protoplasma 2012, 249, 901–907. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Zhang, H.; Zhou, C.; Li, G.; Fennell, A.; Wang, G.; Kang, Y.; Liu, Q.; Ma, Q. An integrative and applicable phylogenetic footprinting framework for cis-regulatory motifs identification in prokaryotic genomes. BMC Genom. 2016, 17, 578. [Google Scholar] [CrossRef] [PubMed]
- Mao, X.; Ma, Q.; Zhou, C.; Chen, X.; Zhang, H.; Yang, J.; Mao, F.; Lai, W.; Xu, Y. DOOR 2.0: Presenting operons and their functions through dynamic and integrated views. Nucleic Acids Res. 2014, 42, D654–D659. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Ma, Q.; Mao, X.; Yin, Y.; Zhu, X.; Xu, Y. Integration of sequence-similarity and functional association information can overcome intrinsic problems in orthology mapping across bacterial genomes. Nucleic Acids Res. 2011, 39, e150. [Google Scholar] [CrossRef] [PubMed]
- Aibar, S.; Gonzalez-Blas, C.B.; Moerman, T.; Huynh-Thu, V.A.; Imrichova, H.; Hulselmans, G.; Rambow, F.; Marine, J.C.; Geurts, P.; Aerts, J.; et al. SCENIC: Single-cell regulatory network inference and clustering. Nat. Methods 2017, 14, 1083–1086. [Google Scholar] [CrossRef] [PubMed]
- Jensen, S.T.; Shen, L.; Liu, J.S. Combining phylogenetic motif discovery and motif clustering to predict co-regulated genes. Bioinformatics 2005, 21, 3832–3839. [Google Scholar] [CrossRef] [PubMed]
- Wegmann, U.; O’Connell-Motherway, M.; Zomer, A.; Buist, G.; Shearman, C.; Canchaya, C.; Ventura, M.; Goesmann, A.; Gasson, M.J.; Kuipers, O.P.; et al. Complete genome sequence of the prototype lactic acid bacterium Lactococcus lactis subsp. cremoris MG1363. J. Bacteriol. 2007, 189, 3256–3270. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Liu, J.; Hou, J.; Dong, Y.; Lu, Y.; Jin, L.; Cao, R.; Li, T.; Wu, J. Oral administration of recombinant Lactococcus lactis expressing HSP65 and tandemly repeated P277 reduces the incidence of type I diabetes in non-obese diabetic mice. PLoS ONE 2014, 9, e105701. [Google Scholar] [CrossRef] [PubMed]
- Ramasamy, R.; Yasawardena, S.; Zomer, A.; Venema, G.; Kok, J.; Leenhouts, K. Immunogenicity of a malaria parasite antigen displayed by Lactococcus lactis in oral immunisations. Vaccine 2006, 24, 3900–3908. [Google Scholar] [CrossRef] [PubMed]
- Bermudez-Humaran, L.G.; Cortes-Perez, N.G.; Lefevre, F.; Guimaraes, V.; Rabot, S.; Alcocer-Gonzalez, J.M.; Gratadoux, J.J.; Rodriguez-Padilla, C.; Tamez-Guerra, R.S.; Corthier, G.; et al. A novel mucosal vaccine based on live Lactococci expressing E7 antigen and IL-12 induces systemic and mucosal immune responses and protects mice against human papillomavirus type 16-induced tumors. J. Immunol. 2005, 175, 7297–7302. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Li, A.; Zuo, F.; Yu, R.; Zeng, Z.; Ma, H.; Chen, S. Recombinant Lactococcus lactis NZ9000 secretes a bioactive kisspeptin that inhibits proliferation and migration of human colon carcinoma HT-29 cells. Microb. Cell Fact. 2016, 15, 102. [Google Scholar] [CrossRef] [PubMed]
- Hanniffy, S.B.; Carter, A.T.; Hitchin, E.; Wells, J.M. Mucosal delivery of a pneumococcal vaccine using Lactococcus lactis affords protection against respiratory infection. J. Infect. Dis. 2007, 195, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Hols, P.; Kleerebezem, M.; Schanck, A.N.; Ferain, T.; Hugenholtz, J.; Delcour, J.; de Vos, W.M. Conversion of Lactococcus lactis from homolactic to homoalanine fermentation through metabolic engineering. Nat. Biotechnol. 1999, 17, 588–592. [Google Scholar] [CrossRef] [PubMed]
- Million, M.; Maraninchi, M.; Henry, M.; Armougom, F.; Richet, H.; Carrieri, P.; Valero, R.; Raccah, D.; Vialettes, B.; Raoult, D. Obesity-associated gut microbiota is enriched in Lactobacillus reuteri and depleted in Bifidobacterium animalis and Methanobrevibacter smithii. Int. J. Obes. 2012, 36, 817–825. [Google Scholar] [CrossRef] [PubMed]
- Hutkins, R.W.; Nannen, N.L. pH homeostasis in lactic acid bacteria. J. Dairy Sci. 1993, 76, 2354–2365. [Google Scholar] [CrossRef]
- Van de Guchte, M.; Serror, P.; Chervaux, C.; Smokvina, T.; Ehrlich, S.D.; Maguin, E. Stress responses in lactic acid bacteria. Antonie Leeuwenhoek 2002, 82, 187–216. [Google Scholar] [CrossRef] [PubMed]
- Hauryliuk, V.; Atkinson, G.C.; Murakami, K.S.; Tenson, T.; Gerdes, K. Recent functional insights into the role of (p)ppGpp in bacterial physiology. Nat. Rev. Micro 2015, 13, 298–309. [Google Scholar] [CrossRef] [PubMed]
- Rallu, F.; Gruss, A.; Ehrlich, S.D.; Maguin, E. Acid- and multistress-resistant mutants of Lactococcus lactis: Identification of intracellular stress signals. Mol. Microbiol. 2000, 35, 517–528. [Google Scholar] [CrossRef] [PubMed]
- Koebmann, B.J.; Nilsson, D.; Kuipers, O.P.; Jensen, P.R. The membrane-bound H+-ATPase complex is essential for growth of Lactococcus lactis. J. Bacteriol. 2000, 182, 4738–4743. [Google Scholar] [CrossRef] [PubMed]
- Lund, P.; Tramonti, A.; De Biase, D. Coping with low pH: Molecular strategies in neutralophilic bacteria. FEMS Microbiol. Rev. 2014, 38, 1091–1125. [Google Scholar] [CrossRef] [PubMed]
- Shabayek, S.; Spellerberg, B. Acid Stress Response Mechanisms of Group B Streptococci. Front. Cell Infect. Microbiol. 2017, 7, 395. [Google Scholar] [CrossRef] [PubMed]
- Frees, D.; Vogensen, F.K.; Ingmer, H. Identification of proteins induced at low pH in Lactococcus lactis. Int. J. Food Microbiol. 2003, 87, 293–300. [Google Scholar] [CrossRef]
- Jayaraman, G.C.; Penders, J.E.; Burne, R.A. Transcriptional analysis of the Streptococcus mutans hrcA, grpE and dnaK genes and regulation of expression in response to heat shock and environmental acidification. Mol. Microbiol. 1997, 25, 329–341. [Google Scholar] [CrossRef] [PubMed]
- Kern, R.; Malki, A.; Abdallah, J.; Tagourti, J.; Richarme, G. Escherichia coli HdeB is an acid stress chaperone. J. Bacteriol. 2007, 189, 603–610. [Google Scholar] [CrossRef] [PubMed]
- Mujacic, M.; Baneyx, F. Chaperone Hsp31 contributes to acid resistance in stationary-phase Escherichia coli. Appl. Environ. Microbiol. 2007, 73, 1014–1018. [Google Scholar] [CrossRef] [PubMed]
- Budin-Verneuil, A.; Maguin, E.; Auffray, Y.; Ehrlich, D.S.; Pichereau, V. Genetic structure and transcriptional analysis of the arginine deiminase (ADI) cluster in Lactococcus lactis MG1363. Can. J. Microbiol. 2006, 52, 617–622. [Google Scholar] [CrossRef] [PubMed]
- Ryan, S.; Begley, M.; Gahan, C.G.; Hill, C. Molecular characterization of the arginine deiminase system in Listeria monocytogenes: Regulation and role in acid tolerance. Environ. Microbiol. 2009, 11, 432–445. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Fukamachi, T.; Saito, H.; Kobayashi, H. Adenosine deamination increases the survival under acidic conditions in Escherichia coli. J. Appl. Microbiol. 2012, 112, 775–781. [Google Scholar] [CrossRef] [PubMed]
- Zuniga, M.; Perez, G.; Gonzalez-Candelas, F. Evolution of arginine deiminase (ADI) pathway genes. Mol. Phylogenet. Evol. 2002, 25, 429–444. [Google Scholar] [CrossRef]
- Nomura, M.; Nakajima, I.; Fujita, Y.; Kobayashi, M.; Kimoto, H.; Suzuki, I.; Aso, H. Lactococcus lactis contains only one glutamate decarboxylase gene. Microbiology 1999, 145, 1375–1380. [Google Scholar] [CrossRef] [PubMed]
- Sanders, J.W.; Leenhouts, K.; Burghoorn, J.; Brands, J.R.; Venema, G.; Kok, J. A chloride-inducible acid resistance mechanism in Lactococcus lactis and its regulation. Mol. Microbiol. 1998, 27, 299–310. [Google Scholar] [CrossRef] [PubMed]
- Hoskins, J.; Alborn, W.E., Jr.; Arnold, J.; Blaszczak, L.C.; Burgett, S.; DeHoff, B.S.; Estrem, S.T.; Fritz, L.; Fu, D.J.; Fuller, W.; et al. Genome of the bacterium Streptococcus pneumoniae strain R6. J. Bacteriol. 2001, 183, 5709–5717. [Google Scholar] [CrossRef] [PubMed]
- Cotter, P.D.; Hill, C. Surviving the acid test: Responses of gram-positive bacteria to low pH. Microbiol. Mol. Biol. Rev. 2003, 67, 429–453. [Google Scholar] [CrossRef] [PubMed]
- Linares, D.M.; Kok, J.; Poolman, B. Genome sequences of Lactococcus lactis MG1363 (revised) and NZ9000 and comparative physiological studies. J. Bacteriol. 2010, 192, 5806–5812. [Google Scholar] [CrossRef] [PubMed]
- Sanders, J.W.; Venema, G.; Kok, J. Environmental stress responses in Lactococcus lactis. FEMS Microbiol. Rev. 1999, 23, 483–501. [Google Scholar] [CrossRef] [Green Version]
- Hartke, A.; Bouché, S.; Giard, J.C.; Benachour, A.; Boutibonnes, P.; Auffray, Y. The lactic acid stress response of Lactococcus lactis subsp. lactis. Curr. Microbiol. 1996, 33, 194–199. [Google Scholar] [CrossRef] [PubMed]
- Antoine Lucas, S.J. Using AMAP and CTC Packages for Huge Clustering. R News 2006, 6, 58–60. [Google Scholar]
- Bauer, D.F. Constructing Confidence Sets Using Rank Statistics. J. Am. Stat. Assoc. 1972, 67, 687–690. [Google Scholar] [CrossRef]
- Bailey, T.L.; Boden, M.; Buske, F.A.; Frith, M.; Grant, C.E.; Clementi, L.; Ren, J.; Li, W.W.; Noble, W.S. MEME Suite: Tools for motif discovery and searching. Nucleic Acids Res. 2009, 37, W202–W208. [Google Scholar] [CrossRef] [PubMed]
- DPInteract. Available online: http://arep.med.harvard.edu/dpinteract (accessed on 30 May 2018).
- Khan, A.; Fornes, O.; Stigliani, A.; Gheorghe, M.; Castro-Mondragon, J.A.; van der Lee, R.; Bessy, A.; Cheneby, J.; Kulkarni, S.R.; Tan, G.; et al. JASPAR 2018: Update of the open-access database of transcription factor binding profiles and its web framework. Nucleic Acids Res. 2018, 46, D260–D266. [Google Scholar] [CrossRef] [PubMed]
- Kazakov, A.E.; Cipriano, M.J.; Novichkov, P.S.; Minovitsky, S.; Vinogradov, D.V.; Arkin, A.; Mironov, A.A.; Gelfand, M.S.; Dubchak, I. RegTransBase—A database of regulatory sequences and interactions in a wide range of prokaryotic genomes. Nucleic Acids Res. 2007, 35, D407–D412. [Google Scholar] [CrossRef] [PubMed]
- Munch, R.; Hiller, K.; Barg, H.; Heldt, D.; Linz, S.; Wingender, E.; Jahn, D. PRODORIC: Prokaryotic database of gene regulation. Nucleic Acids Res. 2003, 31, 266–269. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, M.C.; Monteiro, P.T.; Guerreiro, J.F.; Goncalves, J.P.; Mira, N.P.; dos Santos, S.C.; Cabrito, T.R.; Palma, M.; Costa, C.; Francisco, A.P.; et al. The YEASTRACT database: An upgraded information system for the analysis of gene and genomic transcription regulation in Saccharomyces cerevisiae. Nucleic Acids Res. 2014, 42, D161–D166. [Google Scholar] [CrossRef] [PubMed]
- Dam, P.; Olman, V.; Harris, K.; Su, Z.; Xu, Y. Operon prediction using both genome-specific and general genomic information. Nucleic Acids Res. 2007, 35, 288–298. [Google Scholar] [CrossRef] [PubMed]
- Pachter, L. Models for transcript quantification from RNA-Seq. arXiv, 2011; arXiv:1104.3889. [Google Scholar]
- Chou, W.C.; Ma, Q.; Yang, S.; Cao, S.; Klingeman, D.M.; Brown, S.D.; Xu, Y. Analysis of strand-specific RNA-seq data using machine learning reveals the structures of transcription units in Clostridium thermocellum. Nucleic Acids Res. 2015, 43, e67. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Chen, X.; McDermaid, A.; Ma, Q. DMINDA 2.0: Integrated and systematic views of regulatory DNA motif identification and analyses. Bioinformatics 2017. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q.; Zhang, H.; Mao, X.; Zhou, C.; Liu, B.; Chen, X.; Xu, Y. DMINDA: An integrated web server for DNA motif identification and analyses. Nucleic Acids Res. 2014. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Yang, J.; Li, Y.; McDermaid, A.; Ma, Q. An algorithmic perspective of de novo cis-regulatory motif finding based on ChIP-seq data. Brief. Bioinform. 2017. [Google Scholar] [CrossRef] [PubMed]
- Zomer, A.L.; Buist, G.; Larsen, R.; Kok, J.; Kuipers, O.P. Time-resolved determination of the CcpA regulon of Lactococcus lactis subsp. cremoris MG1363. J. Bacteriol. 2007, 189, 1366–1381. [Google Scholar] [CrossRef] [PubMed]
- Abranches, J.; Nascimento, M.M.; Zeng, L.; Browngardt, C.M.; Wen, Z.T.; Rivera, M.F.; Burne, R.A. CcpA regulates central metabolism and virulence gene expression in Streptococcus mutans. J. Bacteriol. 2008, 190, 2340–2349. [Google Scholar] [CrossRef] [PubMed]
- O’Connell-Motherway, M.; van Sinderen, D.; Morel-Deville, F.; Fitzgerald, G.F.; Ehrlich, S.D.; Morel, P. Six putative two-component regulatory systems isolated from Lactococcus lactis subsp. cremoris MG1363. Microbiology 2000, 146, 935–947. [Google Scholar] [CrossRef] [PubMed]
- Bolotin, A.; Mauger, S.; Malarme, K.; Ehrlich, S.D.; Sorokin, A. Low-redundancy sequencing of the entire Lactococcus lactis IL1403 genome. Antonie Leeuwenhoek 1999, 76, 27–76. [Google Scholar] [CrossRef] [PubMed]
- Kolodrubetz, D.; Burgum, A. Duplicated NHP6 genes of Saccharomyces cerevisiae encode proteins homologous to bovine high mobility group protein 1. J. Biol. Chem. 1990, 265, 3234–3239. [Google Scholar] [PubMed]
- Stillman, D.J. Nhp6: A small but powerful effector of chromatin structure in Saccharomyces cerevisiae. Biochim. Biophys. Acta 2010, 1799, 175–180. [Google Scholar] [CrossRef] [PubMed]
- Madsen, S.M.; Arnau, J.; Vrang, A.; Givskov, M.; Israelsen, H. Molecular characterization of the pH-inducible and growth phase-dependent promoter P170 of Lactococcus lactis. Mol. Microbiol. 1999, 32, 75–87. [Google Scholar] [CrossRef] [PubMed]
- Rince, A.; Dufour, A.; Le Pogam, S.; Thuault, D.; Bourgeois, C.M.; Le Pennec, J.P. Cloning, expression, and nucleotide sequence of genes involved in production of lactococcin DR, a bacteriocin from Lactococcus lactis subsp. lactis. Appl. Environ. Microbiol. 1994, 60, 1652–1657. [Google Scholar] [PubMed]
- Hindre, T.; Le Pennec, J.P.; Haras, D.; Dufour, A. Regulation of lantibiotic lacticin 481 production at the transcriptional level by acid pH. FEMS Microbiol. Lett. 2004, 231, 291–298. [Google Scholar] [CrossRef]
- Madsen, S.M.; Hindre, T.; Le Pennec, J.P.; Israelsen, H.; Dufour, A. Two acid-inducible promoters from Lactococcus lactis require the cis-acting ACiD-box and the transcription regulator RcfB. Mol. Microbiol. 2005, 56, 735–746. [Google Scholar] [CrossRef] [PubMed]
- Akyol, I.; Comlekcioglu, U.; Karakas, A.; Serdaroglu, K.; Ekinci, M.S.; Ozkose, E. Regulation of the acid inducible rcfB promoter in Lactococcus lactis subsp. lactis. Ann. Microbiol. 2008, 58, 269. [Google Scholar] [CrossRef]
- Dennis, D.; Kaplan, N.O. d- and l-lactic acid dehydrogenases in Lactobacillus plantarum. J. Biol. Chem. 1960, 235, 810–818. [Google Scholar] [PubMed]
- Amachi, S.; Ishikawa, K.; Toyoda, S.; Kagawa, Y.; Yokota, A.; Tomita, F. Characterization of a mutant of Lactococcus lactis with reduced membrane-bound ATPase activity under acidic conditions. Biosci. Biotechnol. Biochem. 1998, 62, 1574–1580. [Google Scholar] [CrossRef] [PubMed]
- O’Sullivan, E.; Condon, S. Relationship between acid tolerance, cytoplasmic pH, and ATP and H+-ATPase levels in chemostat cultures of Lactococcus lactis. Appl. Environ. Microbiol. 1999, 65, 2287–2293. [Google Scholar] [PubMed]
- Cao, H.; Wei, D.; Yang, Y.; Shang, Y.; Li, G.; Zhou, Y.; Ma, Q.; Xu, Y. Systems-level understanding of ethanol-induced stresses and adaptation in E. coli. Sci. Rep. 2017, 7, 44150. [Google Scholar] [CrossRef] [PubMed]
- Alsaker, K.V.; Paredes, C.; Papoutsakis, E.T. Metabolite stress and tolerance in the production of biofuels and chemicals: Gene-expression-based systems analysis of butanol, butyrate, and acetate stresses in the anaerobe Clostridium acetobutylicum. Biotechnol. Bioeng. 2010, 105, 1131–1147. [Google Scholar] [CrossRef] [PubMed]
- Cumley, N.J.; Smith, L.M.; Anthony, M.; May, R.C. The CovS/CovR acid response regulator is required for intracellular survival of group B Streptococcus in macrophages. Infect. Immun. 2012, 80, 1650–1661. [Google Scholar] [CrossRef] [PubMed]
- Budin-Verneuil, A.; Maguin, E.; Auffray, Y.; Ehrlich, S.D.; Pichereau, V. Transcriptional analysis of the cyclopropane fatty acid synthase gene of Lactococcus lactis MG1363 at low pH. FEMS Microbiol. Lett. 2005, 250, 189–194. [Google Scholar] [CrossRef] [PubMed]
- Santi, I.; Grifantini, R.; Jiang, S.M.; Brettoni, C.; Grandi, G.; Wessels, M.R.; Soriani, M. CsrRS regulates group B Streptococcus virulence gene expression in response to environmental pH: A new perspective on vaccine development. J. Bacteriol. 2009, 191, 5387–5397. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Regulon ID | No. of Operons | DEG | TF Template | TF (Gene) BLAST in MG1363 |
|---|---|---|---|---|
| Regulon #2 | 82 | Y | spo0A | llrC (llmg_0414) |
| Regulon #3 | 32 | Y | FoxQ1 | N/A |
| Regulon #4 | 20 | Y | SPT2 | N/A |
| Regulon #7 | 49 | Y | lhfB | hllA (llmg_0496) |
| Regulon #10 | 5 | N | GAL80 | llmg_0271 |
| Regulon #12 | 259 | Y | CovR | llrA (llmg_0908) |
| Regulon #15 | 19 | Y | c4494 | ccpA (llmg_0775) |
| Regulon #20 | 79 | Y | NHP6A | N/A |
| Regulon #28 | 5 | Y | 1Z916 | N/A |
| Regulon #31 | 65 | Y | ihfA | hllA (llmg_0496) |
| Regulon #37 | 10 | N | CovR | llrA (llmg_0908) |
| Regulon #40 | 7 | Y | Awh | N/A |
| Regulon #44 | 12 | N | YBR182C | N/A |
| Regulon #47 | 5 | N | RHE_PF00288 | ccpA (llmg_0775) |
| Template Organisms | MG1363 | |||
|---|---|---|---|---|
| Organisms | Transporters | Functions/Pathways | Mapped Genes (Locus Tag) | Regulons |
| Lactococcus lactis | ldh | LDH | ldh (llmg_1120) | NHP6A, llrA |
| ldhB | ldhB (llmg_0392, llmg_0475) | |||
| ldhX | ldhX (llmg_1429) | |||
| Lactococcus lactis | gadB | GAD | gadB (llmg_1179) | N/A |
| gadC | gadC (llmg_1178) | |||
| L actococcus lactis | arcA | ADI pathway | arcA (llmg_2313) | NHP6A, llrA, llrC, hllA |
| arcB | arcB (llmg_2312) | |||
| arcC1 | arcC1 (llmg_2310) | |||
| arcC2 | arcC2 (llmg_2309) | |||
| argF | argF (llmg_1754) | |||
| Bacteria | ureA/B/C$ | Urea degradation | pyrC (llmg_1508) | N/A |
| L actococcus lactis | atpEBFHAGDC$$ | F0/F1ATPase | llmg_1952, llmg_1951, llmg_1950, llmg_1949, llmg_1948, llmg_1947, llmg_1946, llmg_1945 | llrA, (Regulon8, llmg_1803) $$$ |
| Lactococcus lactis | rcfB | Acid response | rcfB (llmg_2512) | (Regulon39, llrD) $$$ |
| Lactococcus lactis, Escherichia coli K12 | dnak | Chaperone, Protein repair and protease | dnaK (llmg_1574) | llrA |
| groEL | groEL2 (llmg_0411) | |||
| groES | groES (llmg_0410) | |||
| grpE | grpE (llmg_1575) | |||
| clpE | clpE (llmg_0528) | |||
| clpC | clpC (llmg_0615) | |||
| clpP | clpP (llmg_0638) | |||
| Lactococcus lactis, Bacillus subtilis | dltC, agK, SGP, ffh | Envelope alterations | llmg_0878 | NHP6A, llrA |
| Lactococcus lactis | recA, uvr, smn | DNA repair | llmg_0374, llmg_0534, llmg_1718 llmg_1221 | (Regulon39, llrD) $$$ |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, X.; Ma, A.; McDermaid, A.; Zhang, H.; Liu, C.; Cao, H.; Ma, Q. RECTA: Regulon Identification Based on Comparative Genomics and Transcriptomics Analysis. Genes 2018, 9, 278. https://doi.org/10.3390/genes9060278
Chen X, Ma A, McDermaid A, Zhang H, Liu C, Cao H, Ma Q. RECTA: Regulon Identification Based on Comparative Genomics and Transcriptomics Analysis. Genes. 2018; 9(6):278. https://doi.org/10.3390/genes9060278
Chicago/Turabian StyleChen, Xin, Anjun Ma, Adam McDermaid, Hanyuan Zhang, Chao Liu, Huansheng Cao, and Qin Ma. 2018. "RECTA: Regulon Identification Based on Comparative Genomics and Transcriptomics Analysis" Genes 9, no. 6: 278. https://doi.org/10.3390/genes9060278
APA StyleChen, X., Ma, A., McDermaid, A., Zhang, H., Liu, C., Cao, H., & Ma, Q. (2018). RECTA: Regulon Identification Based on Comparative Genomics and Transcriptomics Analysis. Genes, 9(6), 278. https://doi.org/10.3390/genes9060278