Construction of Red Fox Chromosomal Fragments from the Short-Read Genome Assembly

 ,
,  , ,
, , 
Abstract
:1. Introduction
2. Materials and Methods
3. Results
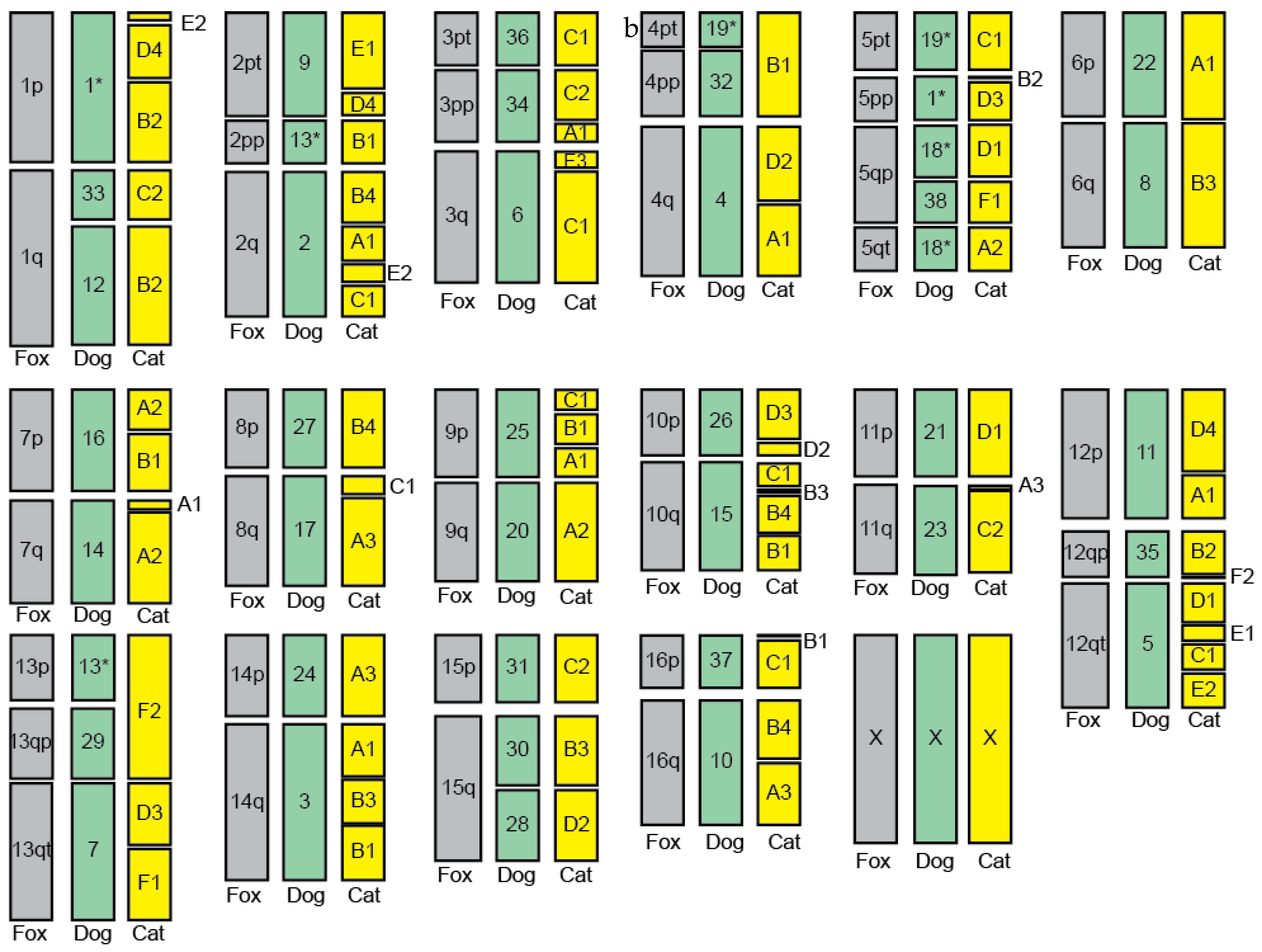
3.1. Interspecies Synteny
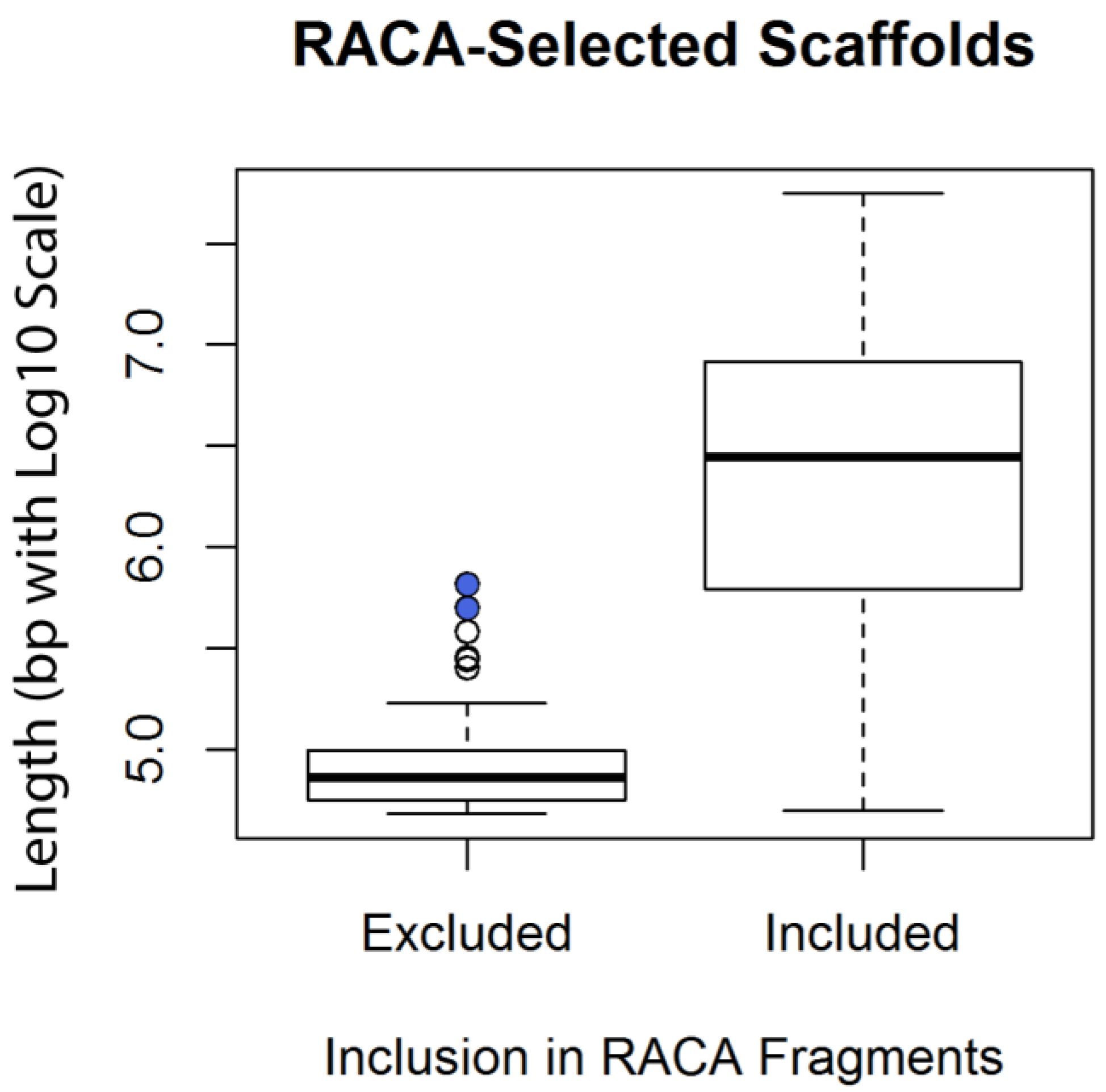
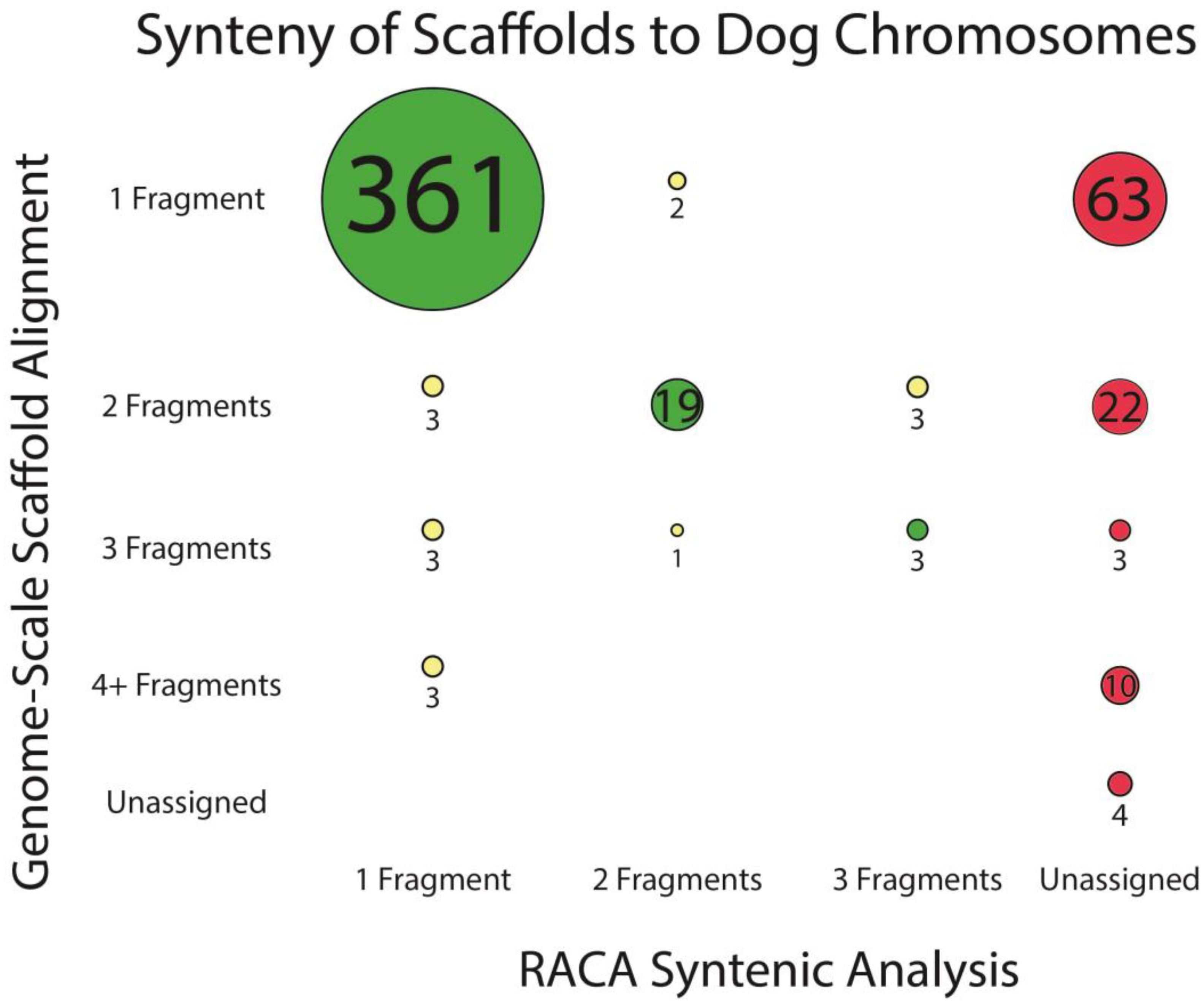
3.2. Reference-Assisted Chromosome Assembly
3.3. Assembly of Chromosome-Scale Fragments
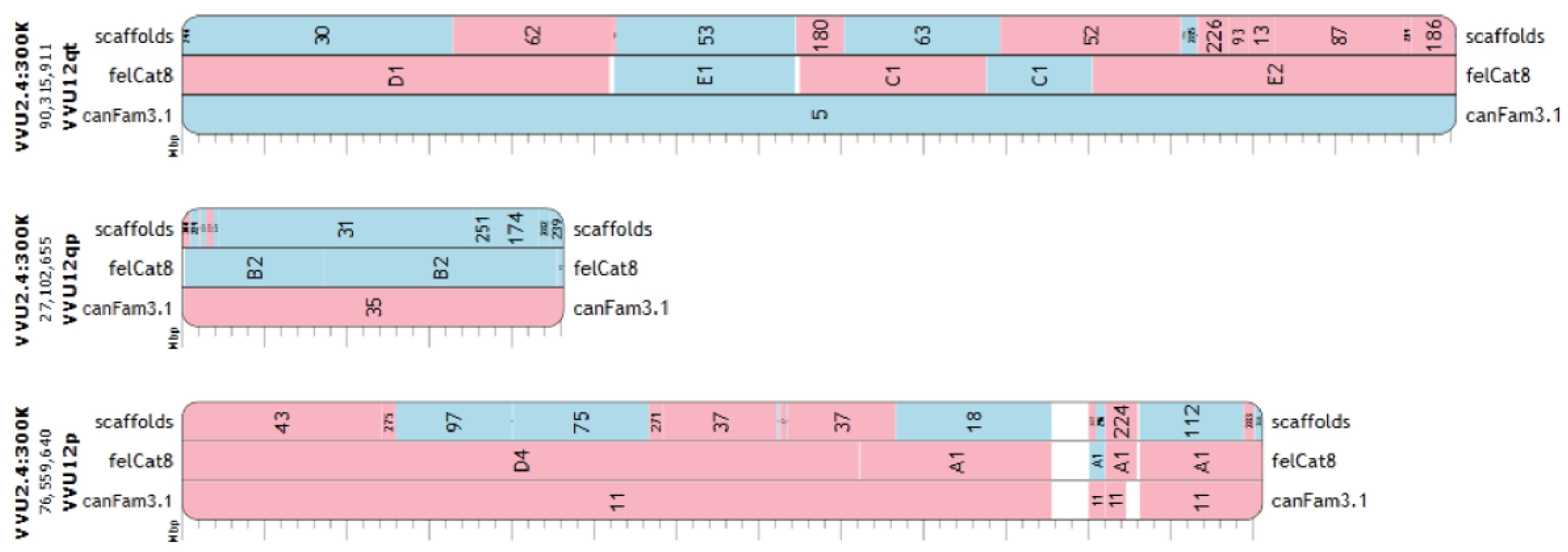
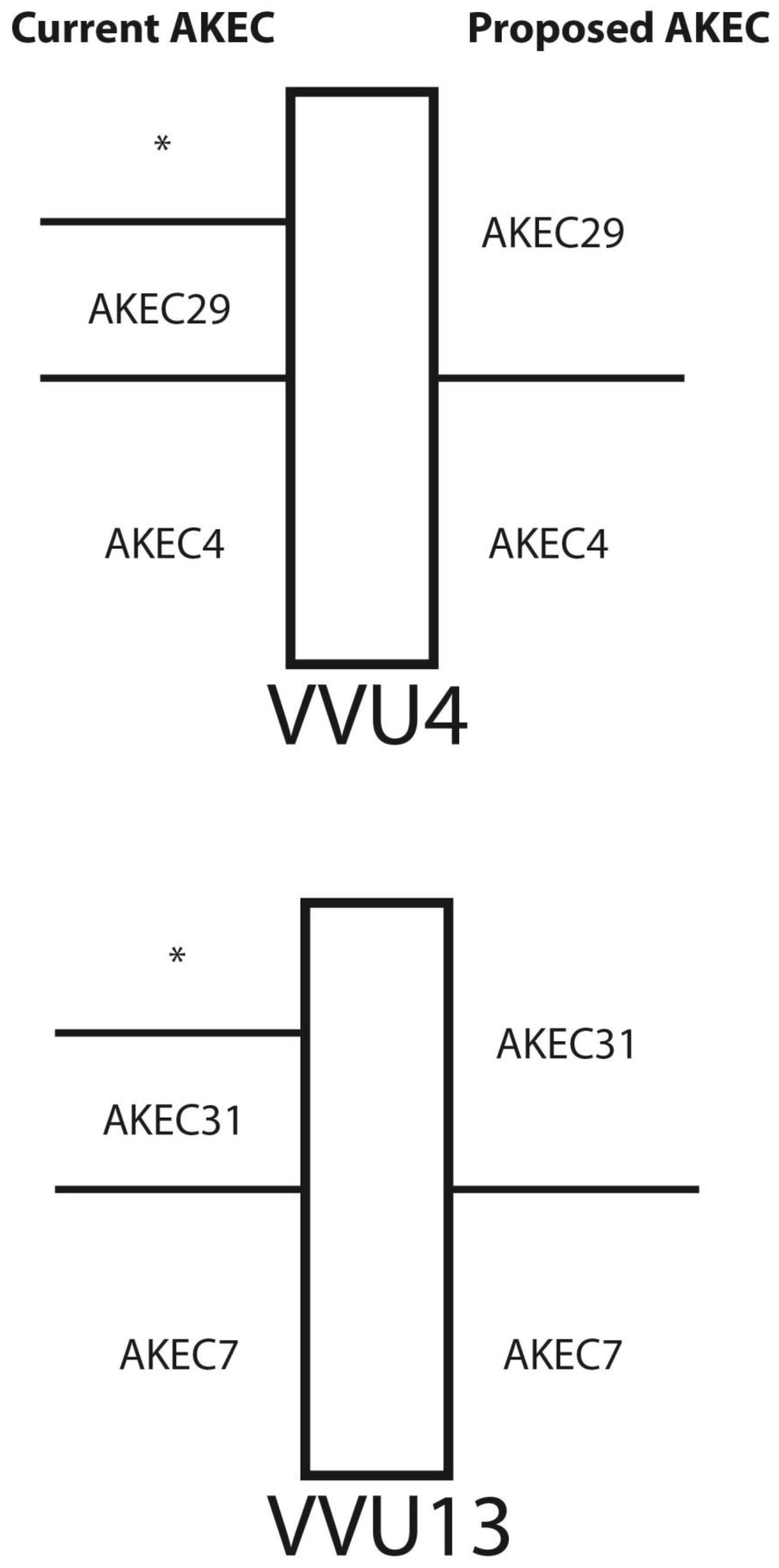
3.4. Refinement of Fox-Dog Synteny at Chromosomal Breakpoints
3.5. Assembled Chromosomes
4. Discussion
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
Appendix A

Appendix B
Appendix C

References
- O’Brien, S.J.; Menotti-Raymond, M.; Murphy, W.; Nash, W.G.; Wienberg, J.; Stanyon, R.; Copeland, N.G.; Jenkins, N.A.; Womack, J.E.; Marshall Graves, J.A. The promise of comparative genomics in mammals. Science 1999, 286, 458–462, 479–481. [Google Scholar] [CrossRef] [PubMed]
- Lander, E.S.; Linton, L.M.; Birren, B.W.; Nusbaum, C.; Zody, M.C.; Baldwin, J.; Devon, K.; Dewar, K.; Doyle, M.; FitzHugh, W.; et al. Initial sequencing and analysis of the human genome. Nature 2001, 409, 860–921. [Google Scholar] [CrossRef] [PubMed]
- Venter, J.C.; Adams, M.D.; Myers, E.W.; Li, P.W.; Mural, R.J.; Sutton, G.G.; Smith, H.O.; Yandell, M.; Evans, C.A.; Holt, R.A.; et al. The sequence of the human genome. Science 2001, 291, 1304–1351. [Google Scholar] [CrossRef] [PubMed]
- Waterston, R.H.; Lindblad-Toh, K.; Birney, E.; Rogers, J.; Abril, J.F.; Agarwal, P.; Agarwala, R.; Ainscough, R.; Alexandersson, M.; An, P.; et al. Initial sequencing and comparative analysis of the mouse genome. Nature 2002, 420, 520–562. [Google Scholar] [CrossRef] [PubMed]
- Lindblad-Toh, K.; Garber, M.; Zuk, O.; Lin, M.F.; Parker, B.J.; Washietl, S.; Kheradpour, P.; Ernst, J.; Jordan, G.; Mauceli, E.; et al. A high-resolution map of human evolutionary constraint using 29 mammals. Nature 2011, 478, 476–482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Genome 10K Consortium of Scientists. Genome 10K: A proposal to obtain whole-genome sequence for 10,000 vertebrate species. J. Hered. 2009, 100, 659–674. [Google Scholar] [CrossRef]
- Koepfli, K.-P.; Paten, B.; O’Brien, S.J. The Genome 10K Project: A way forward. Annu. Rev. Anim. Biosci. 2015, 3, 57–111. [Google Scholar] [CrossRef] [PubMed]
- Lariviere, S.; Pasitschniak-Arts, M. Vulpes vulpes. Mamm. Species 1996, 537, 1–11. [Google Scholar] [CrossRef]
- Schipper, J.; Chanson, J.S.; Chiozza, F.; Cox, N.A.; Hoffmann, M.; Katariya, V.; Lamoreux, J.; Rodrigues, A.S.L.; Stuart, S.N.; Temple, H.J.; et al. The status of the world’s land and marine mammals: Diversity, threat and knowledge. Science 2008, 322, 225–230. [Google Scholar] [CrossRef] [PubMed]
- Dearborn, N. Silver Fox Farming in Eastern North America; US Department of Agriculture: Washington, DC, USA, 1915; pp. 1–35. [Google Scholar]
- Trut, L.N.; Oskina, I.; Kharlamova, A.V. Animal evolution during domestication: The domesticated fox as a model. Bioessays 2009, 31, 349–360. [Google Scholar] [CrossRef] [PubMed]
- Kukekova, A.V.; Johnson, J.L.; Xiang, X.; Feng, S.; Liu, S.; Rando, H.M.; Kharlamova, A.V.; Herbeck, Y.; Serdyukova, N.A.; Xiong, Z.; et al. The red fox genome assembly facilitates identification of the genomic regions associated with selection for tame and aggressive behavior. Nat. Ecol. Evol. 2018. [Google Scholar]
- Donaldson, M.E.; Rico, Y.; Hueffer, K.; Rando, H.M.; Kukekova, A.V.; Kyle, C.J. Development of a genotype-by-sequencing immunogenetic assay as exemplified by screening for variation in red fox with and without endemic rabies exposure. Ecol. Evol. 2018, 8, 572–583. [Google Scholar] [CrossRef] [PubMed]
- Rando, H.M.; Stutchman, J.T.; Bastounes, E.R.; Johnson, J.L.; Driscoll, C.A.; Barr, C.S.; Trut, L.N.; Sacks, B.N.; Kukekova, A.V. Y-chromosome markers for the red fox. J. Hered. 2017, 108, 678–685. [Google Scholar] [CrossRef] [PubMed]
- Metzker, M.L. Sequencing technologies—The next generation. Nat. Rev. Genet. 2010, 11, 31–46. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Chen, Y.; Mu, D.; Yuan, J.; Shi, Y.; Zhang, H.; Gan, J.; Li, N.; Hu, X.; Liu, B.; et al. Comparison of the two major classes of assembly algorithms: Overlap-layout-consensus and de-bruijn-graph. Brief. Funct. Genomics 2012, 11, 25–37. [Google Scholar] [CrossRef] [PubMed]
- Lindblad-Toh, K.; Wade, C.M.; Mikkelsen, T.S.; Karlsson, E.K.; Jaffe, D.B.; Kamal, M.; Clamp, M.; Chang, J.L.; Kulbokas, E.J.; Zody, M.C.; et al. Genome sequence, comparative analysis and haplotype structure of the domestic dog. Nature 2005, 438, 803–819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Batzoglou, S.; Jaffe, D.B.; Stanley, K.; Butler, J.; Gnerre, S.; Mauceli, E.; Berger, B.; Mesirov, J.P.; Lander, E.S. ARACHNE: A whole-genome shotgun assembler. Genome Res. 2002, 12, 177–189. [Google Scholar] [CrossRef] [PubMed]
- Jaffe, D.B.; Butler, J.; Gnerre, S.; Mauceli, E.; Lindblad-Toh, K.; Mesirov, J.P.; Zody, M.C.; Lander, E.S. Whole-genome sequence assembly for mammalian genomes: Arachne 2. Genome Res. 2003, 13, 91–96. [Google Scholar] [CrossRef] [PubMed]
- Pontius, J.U.; Mullikin, J.C.; Smith, D.R.; Agencourt, S.T.; Lindblad-Toh, K.; Gnerre, S.; Clamp, M.; Chang, J.L.; Stephens, R.; Neelam, B.; et al. Initial sequence and comparative analysis of the cat genome. Genome Res. 2007, 17, 1675–1689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mullikin, J.C.; Ning, Z. The phusion assembler. Genome Res. 2003, 13, 81–90. [Google Scholar] [CrossRef] [PubMed]
- Henson, J.; Tischler, G.; Ning, Z. Next-generation sequencing and large genome assemblies. Pharmacogenomics 2012, 13, 901–915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pevzner, P.A.; Tang, H.; Waterman, M.S. An Eulerian path approach to DNA fragment assembly. Proc. Natl. Acad. Sci. USA 2001, 98, 9748–9753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Compeau, P.E.C.; Pevzner, P.A.; Tesler, G. How to apply de Bruijn graphs to genome assembly. Nat. Biotechnol. 2011, 29, 987–991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pop, M. Genome assembly reborn: Recent computational challenges. Brief. Bioinform. 2009, 10, 354–366. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Zhu, H.; Ruan, J.; Qian, W.; Fang, X.; Shi, Z.; Li, Y.; Li, S.; Shan, G.; Kristiansen, K.; et al. De novo assembly of human genomes with massively parallel short read sequencing. Genome Res. 2010, 20, 265–272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, R.; Liu, B.; Xie, Y.; Li, Z.; Huang, W.; Yuan, J.; He, G.; Chen, Y.; Pan, Q.; Liu, Y.; et al. SOAPdenovo2: An empirically improved memory-efficient short-read de novo assembler. Gigascience 2012, 1, 18. [Google Scholar] [CrossRef] [PubMed]
- Yao, G.; Ye, L.; Gao, H.; Minx, P.; Warren, W.C.; Weinstock, G.M. Graph accordance of next-generation sequence assemblies. Bioinformatics 2012, 28, 13–16. [Google Scholar] [CrossRef] [PubMed]
- Eichler, E.E. Segmental duplications: What’s missing, misassigned, and misassembled—And should we care? Genome Res. 2001, 11, 653–656. [Google Scholar] [CrossRef] [PubMed]
- Kelley, D.R.; Salzberg, S.L. Detection and correction of false segmental duplications caused by genome mis-assembly. Genome Biol. 2010, 11, R28. [Google Scholar] [CrossRef] [PubMed]
- Alkan, C.; Sajjadian, S.; Eichler, E.E. Limitations of next-generation genome sequence assembly. Nat. Methods 2011, 8, 61–65. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Larkin, D.M.; Cai, Q.; Asan; Zhang, Y.; Ge, R.-L.; Auvil, L.; Capitanu, B.; Zhang, G.; Lewin, H.A.; Ma, J. Reference-assisted chromosome assembly. Proc. Natl. Acad. Sci. USA 2013, 110, 1785–1790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ge, R.-L.; Cai, Q.; Shen, Y.-Y.; San, A.; Ma, L.; Zhang, Y.; Yi, X.; Chen, Y.; Yang, L.; Huang, Y.; He, R.; et al. Draft genome sequence of the Tibetan antelope. Nat. Commun. 2013, 4, 1858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, X.; Nevo, E.; Han, L.; Levanon, E.Y.; Zhao, J.; Avivi, A.; Larkin, D.; Jiang, X.; Feranchuk, S.; Zhu, Y.; et al. Genome-wide adaptive complexes to underground stresses in blind mole rats Spalax. Nat. Commun. 2014, 5, 3966. [Google Scholar] [CrossRef] [PubMed]
- Damas, J.; O’Connor, R.; Farré, M.; Lenis, V.P.E.; Martell, H.J.; Mandawala, A.; Fowler, K.; Joseph, S.; Swain, M.T.; Griffin, D.K.; Larkin, D.M. Upgrading short read animal genome assemblies to chromosome level using comparative genomics and a universal probe set. Genome Res. 2016, 27, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Wayne, R.K. Molecular evolution of the dog family. Trends Genet. 1993, 9, 218–224. [Google Scholar] [CrossRef]
- O’Brien, S.J.; Menninger, J.C.; Nash, W.G. Atlas of Mammalian Chromosomes; O’Brien, S.J., Menninger, J.C., Nash, W.G., Eds.; John Wiley and Sons: Hoboken, NJ, USA, 2006. [Google Scholar]
- Murphy, W.J.; Stanyon, R.; O’Brien, S.J. Evolution of mammalian genome organization inferred from comparative gene mapping. Genome Biol. 2001, 2, 0005.1–0005.8. [Google Scholar] [CrossRef]
- Nash, W.; Menninger, J.; Wienberg, J.; Padilla-Nash, H.; O’Brien, S. The pattern of phylogenomic evolution of the Canidae. Cytogenet. Cell Genet. 2001, 95, 210–224. [Google Scholar] [CrossRef] [PubMed]
- Graphodatsky, A.S.; Perelman, P.L.; Sokolovskaya, N.V.; Beklemisheva, V.R.; Serdukova, N.A.; Dobigny, G.; O’Brien, S.J.; Ferguson-Smith, M.A.; Yang, F. Phylogenomics of the dog and fox family (Canidae, Carnivora) revealed by chromosome painting. Chromosom. Res. 2008, 16, 129–143. [Google Scholar] [CrossRef] [PubMed]
- Graphodatsky, A.S.; Beklemisheva, V.R.; Dolf, G. High-resolution GTG-banding patterns of dog and silver fox chromosomes: Description and comparative analysis. Cytogenet. Cell Genet. 1995, 69, 226–231. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; O’Brien, P.C.; Milne, B.; Graphodatsky, A.S.; Solanky, N.; Trifonov, V.; Rens, W.; Sargan, D.R.; Ferguson-Smith, M.A. A complete comparative chromosome map for the dog, red fox, and human and its integration with canine genetic maps. Genomics 1999, 62, 189–202. [Google Scholar] [CrossRef] [PubMed]
- Graphodatsky, A.S.; Yang, F.; O’Brien, P.C.; Serdukova, N.A.; Milne, B.; Trifonov, V.; Ferguson-Smith, M.A. A comparative chromosome map of the Arctic fox, red fox and dog defined by chromosome painting and high resolution G-banding. Chromosom. Res. 2000, 8, 253–263. [Google Scholar] [CrossRef]
- Becker, S.E.D.; Thomas, R.; Trifonov, V.A.; Wayne, R.K.; Graphodatsky, A.S.; Breen, M. Anchoring the dog to its relatives reveals new evolutionary breakpoints across 11 species of the Canidae and provides new clues for the role of B chromosomes. Chromosom. Res. 2011, 19, 685–708. [Google Scholar] [CrossRef] [PubMed]
- Kukekova, A.V.; Trut, L.N.; Oskina, I.N.; Kharlamova, A.V.; Shikhevich, S.G.; Kirkness, E.F.; Aguirre, G.D.; Acland, G.M. A marker set for construction of a genetic map of the silver fox (Vulpes vulpes). J. Hered. 2004, 95, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Kukekova, A.V.; Trut, L.N.; Oskina, I.N.; Johnson, J.L.; Temnykh, S.V.; Kharlamova, A.V.; Shepeleva, D.V.; Gulievich, R.G.; Shikhevich, S.G.; Graphodatsky, A.S.; et al. A meiotic linkage map of the silver fox, aligned and compared to the canine genome. Genome Res. 2007, 17, 387–399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kukekova, A.V.; Trut, L.N.; Chase, K.; Kharlamova, A.V.; Johnson, J.L.; Temnykh, S.V.; Oskina, I.N.; Gulevich, R.G.; Vladimirova, A.V.; Klebanov, S.; et al. Mapping loci for fox domestication: Deconstrution/reconstruction of a behavioral phenotype. Behav. Genet. 2011, 41, 593–606. [Google Scholar] [CrossRef] [PubMed]
- Kukekova, A.V.; Temnykh, S.V.; Johnson, J.L.; Trut, L.N.; Acland, G.M. Genetics of behavior in the silver fox. Mamm. Genome 2012, 23, 164–177. [Google Scholar] [CrossRef] [PubMed]
- Nelson, R.M.; Temnykh, S.V.; Johnson, J.L.; Kharlamova, A.V.; Vladimirova, A.V.; Gulevich, R.G.; Shepeleva, D.V.; Oskina, I.N.; Acland, G.M.; Rönnegård, L.; Trut, L.N.; Carlborg, Ö.; et al. Genetics of interactive behavior in silver foxes (Vulpes vulpes). Behav. Genet. 2017, 47, 88–101. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Graphodatsky, A.S.; O’Brien, P.C.; Colabella, A.; Solanky, N.; Squire, M.; Sargan, D.R.; Ferguson-Smith, M.A. Reciprocal chromosome painting illuminates the history of genome evolution of the domestic cat, dog and human. Chromosom. Res. 2000, 8, 393–404. [Google Scholar] [CrossRef]
- Davis, B.W.; Raudsepp, T.; Pearks Wilkerson, A.J.; Agarwala, R.; Schäffer, A.A.; Houck, M.; Chowdhary, B.P.; Murphy, W.J. A high-resolution cat radiation hybrid and integrated FISH mapping resource for phylogenomic studies across Felidae. Genomics 2009, 93, 299–304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harris, R.S. Improved Pairwise Alignment of Genomic DNA; The Pennsylvania State University: State College, PA, USA, 2007. [Google Scholar]
- Harris, R.S. Miller Lab, LASTZ. Available online: http://www.bx.psu.edu/~rsharris/lastz/ (accessed on 17 March 2015).
- Kent, J. kentUtils. Available online: https://github.com/ENCODE-DCC/kentUtils (accessed on 19 February 2015).
- Kent, W.J.; Baertsch, R.; Hinrichs, A.; Miller, W.; Haussler, D. Evolution’s cauldron: Duplication, deletion, and rearrangement in the mouse and human genomes. Proc. Natl. Acad. Sci. USA 2003, 100, 11484–11489. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J. RACA: Reference-Assisted Chromosome Assembly. Available online: http://bioen-compbio.bioen.illinois.edu/RACA/ (accessed on 17 April 2014).
- R Core Team. R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. 2014. Available online: http://www.R-project.org/ (accessed on 5 June 2018).
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolmogorov, M.; Raney, B.; Paten, B.; Pham, S. Ragout—A reference-assisted assembly tool for bacterial genomes. Bioinformatics 2014, 30, 302–309. [Google Scholar] [CrossRef] [PubMed]
- Evolution Highway (fox). Available online: http://eh-demo.ncsa.illinois.edu/fox (accessed on 5 February 2018).
- Murphy, W.J.; Larkin, D.M.; Everts-van der Wind, A.; Bourque, G.; Tesler, G.; Auvil, L.; Beever, J.E.; Chowdhary, B.P.; Galibert, F.; Gatzke, L.; et al. Dynamics of mammalian chromosome evolution inferred from multispecies comparative maps. Science 2005, 309, 613–617. [Google Scholar] [CrossRef] [PubMed]
- Larkin, D.M.; Pape, G.; Donthu, R.; Auvil, L.; Welge, M.; Lewin, H.A. Breakpoint regions and homologous synteny blocks in chromosomes have different evolutionary histories. Genome Res. 2009, 19, 770–777. [Google Scholar] [CrossRef] [PubMed]
- Sankoff, D. The where and wherefore of evolutionary breakpoints. J. Biol. 2009, 8, 66. [Google Scholar] [CrossRef] [PubMed]
- Wayne, R.K.; Nash, W.; O’Brien, S.J. Chromosomal evolution of the Canidae. I. Species with high diploid numbers. Cytogenet. Cell Genet. 1987, 44, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Wayne, R.K.; Nash, W.; O’Brien, S.J. Chromosome evolution of the Canidae. II. Divergence from the primitive carnivore karyotype. Cytogenet. Cell Genet. 1987, 44, 134–141. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.L.; Wittgenstein, H.; Mitchell, S.E.; Hyma, K.E.; Temnykh, S.V.; Kharlamova, A.V.; Gulevich, R.G.; Vladimirova, A.V.; Fong, H.W.F.; Acland, G.M.; et al. Genotyping-by-sequencing (GBS) detects genetic structure and confirms behavioral QTL in tame and aggressive foxes (Vulpes vulpes). PLoS ONE 2015, 10, e0127013. [Google Scholar] [CrossRef] [PubMed]
- Ye, C.; Hill, C.M.; Wu, S.; Ruan, J.; Ma, Z. DBG2OLC: Efficient assembly of large genomes using long erroneous reads of the third generation sequencing technologies. Sci. Rep. 2016, 6, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Jain, M.; Koren, S.; Miga, K.H.; Quick, J.; Rand, A.C.; Sasani, T.A.; Tyson, J.R.; Beggs, A.D.; Dilthey, A.T.; Fiddes, I.T.; et al. Nanopore sequencing and assembly of a human genome with ultra-long reads. Nat. Biotechnol. 2018, 36, 338–345. [Google Scholar] [CrossRef] [PubMed]
- Jiao, W.B.; Schneeberger, K. The impact of third generation genomic technologies on plant genome assembly. Curr. Opin. Plant Biol. 2017, 36, 64–70. [Google Scholar] [CrossRef] [PubMed]
- Lieberman-Aiden, E.; van Berkum, N.; Williams, L.; Imakaev, M.; Ragoczy, T.; Telling, A.; Amit, I.; Lajoie, B.R.; Sabo, P.J.; Dorschner, M.O.; et al. Comprehensive mapping of long range interactions reveals folding principles of the human genome. Science 2009, 326, 289–293. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, D.; Li, X.; Hernandez, L.; Ramnarain, S.; Huff, E.; Wang, Y. Ordered restriction maps of Saccharomyces cerevisiae chromosomes constructed by optical mapping. Science 1993, 262, 110–114. [Google Scholar] [CrossRef] [PubMed]
- Koren, S.; Rhie, A.; Walenz, B.P.; Dilthey, A.T.; Bickhart, D.M.; Kingan, S.B.; Hiendleder, S.; Williams, J.L.; Smith, T.P.L.; Phillippy, A. Complete assembly of parental haplotypes with trio binning. bioRxiv 2018, 271486. [Google Scholar] [CrossRef] [Green Version]
- Hubisz, M.J.; Pollard, K.S.; Siepel, A. PHAST and RPHAST: Phylogenetic analysis with space/time models. Brief. Bioinform. 2011, 12, 41–51. [Google Scholar] [CrossRef] [PubMed]
- Tavare, S. Some probabilistic and statistical problems in the analysis of DNA sequences. Lect. Math. Life Sci. 1986, 17, 57–86. [Google Scholar]
- Yang, Z. Estimating the pattern of nucleotide substitution. J. Mol. Evol. 1994, 39, 105–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilbert, D.G. Polydendron version 0.8d. 1999. Available online: http://iubio.bio.indiana.edu/treeapp/treeprint-form.html (accessed on 6 July 2016).
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wurster-Hill, D.H.; Gray, C.W. The interrelationships of chromosome banding patterns in procyonids, viverrids, and felids. Cytogenet. Cell Genet. 1975, 15, 306–331. [Google Scholar] [CrossRef] [PubMed]
- Dutrillaux, B.; Couturier, J. The ancestral karyotype of Carnivora: Comparison with that of platyrrhine monkeys. Cytogenet. Cell Genet. 1983, 35, 200–208. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Fox Chromosomes | Dog Chromosome | Previously Unassigned Positions on Dog Chromosome (bp) | Previous Gap Size (Kbp) | Dog Chromosome Positions Still Unassigned (bp) | Updated Gap Size (Kbp) |
|---|---|---|---|---|---|
| 1 & 5 | 1 | 24,600,000–25,700,000 | 1100 | 24,988,836–25,579,247 | 590.4 |
| 2 & 13 | 13 | 37,800,000–38,600,000 | 800 | 38,258,211–38,277,954 | 19.7 |
| 5 (2 fragments) | 18 | 24,400,000–26,000,000 | 1600 | 25,259,332–25,332,083 | 72.8 |
| 4 & 5 | 19 | 18,800,000–22,100,000 | 3300 | 19,878,341–20,333,685 | 455.3 |
| Fox Fragment | Dog Chromosome | Dog Positions (Mbp) | Direction | Cat Chromosome | Cat Position (Mbp) |
|---|---|---|---|---|---|
| 4pt | 19 * | 0–19.9 | Forward | B1 | 84.0–144.6 |
| 4pp | 32 | 0–38.7 | Reverse | ||
| 4q | 4 | 0–88.3 | Forward | D2 | 8.6–48.2 |
| A1 | 172.6–227.4 | ||||
| 13p | 13 * | 0–38.3 | Reverse | F2 | 0.6–82.8 |
| 13qp | 29 | 0–41.7 | Reverse | ||
| 13qt | 7 | 0–80.9 | Reverse | D3 | 33.3–71.5 |
| F1 | 0.4–41.3; 65.7–68.7 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rando, H.M.; Farré, M.; Robson, M.P.; Won, N.B.; Johnson, J.L.; Buch, R.; Bastounes, E.R.; Xiang, X.; Feng, S.; Liu, S.; et al. Construction of Red Fox Chromosomal Fragments from the Short-Read Genome Assembly. Genes 2018, 9, 308. https://doi.org/10.3390/genes9060308
Rando HM, Farré M, Robson MP, Won NB, Johnson JL, Buch R, Bastounes ER, Xiang X, Feng S, Liu S, et al. Construction of Red Fox Chromosomal Fragments from the Short-Read Genome Assembly. Genes. 2018; 9(6):308. https://doi.org/10.3390/genes9060308
Chicago/Turabian StyleRando, Halie M., Marta Farré, Michael P. Robson, Naomi B. Won, Jennifer L. Johnson, Ronak Buch, Estelle R. Bastounes, Xueyan Xiang, Shaohong Feng, Shiping Liu, and et al. 2018. "Construction of Red Fox Chromosomal Fragments from the Short-Read Genome Assembly" Genes 9, no. 6: 308. https://doi.org/10.3390/genes9060308
APA StyleRando, H. M., Farré, M., Robson, M. P., Won, N. B., Johnson, J. L., Buch, R., Bastounes, E. R., Xiang, X., Feng, S., Liu, S., Xiong, Z., Kim, J., Zhang, G., Trut, L. N., Larkin, D. M., & Kukekova, A. V. (2018). Construction of Red Fox Chromosomal Fragments from the Short-Read Genome Assembly. Genes, 9(6), 308. https://doi.org/10.3390/genes9060308