A Similarity Regression Fusion Model for Integrating Multi-Omics Data to Identify Cancer Subtypes

Abstract
:1. Introduction
2. Materials and Methods
2.1. Datasets
2.2. Methods
2.2.1. Correlation Similarity
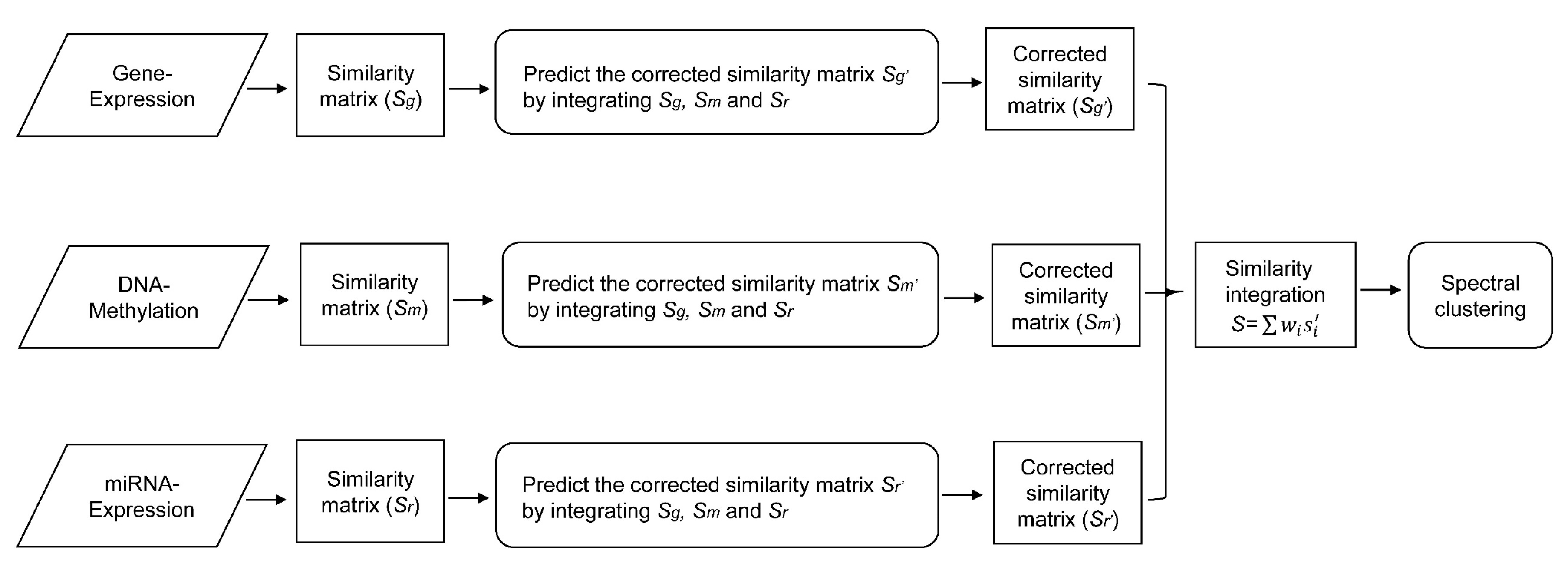
2.2.2. Similarity Regression Fusion
2.2.3. Parameter Learning
2.2.4. Similarity Integration and Cancer Subtype Prediction
3. Results
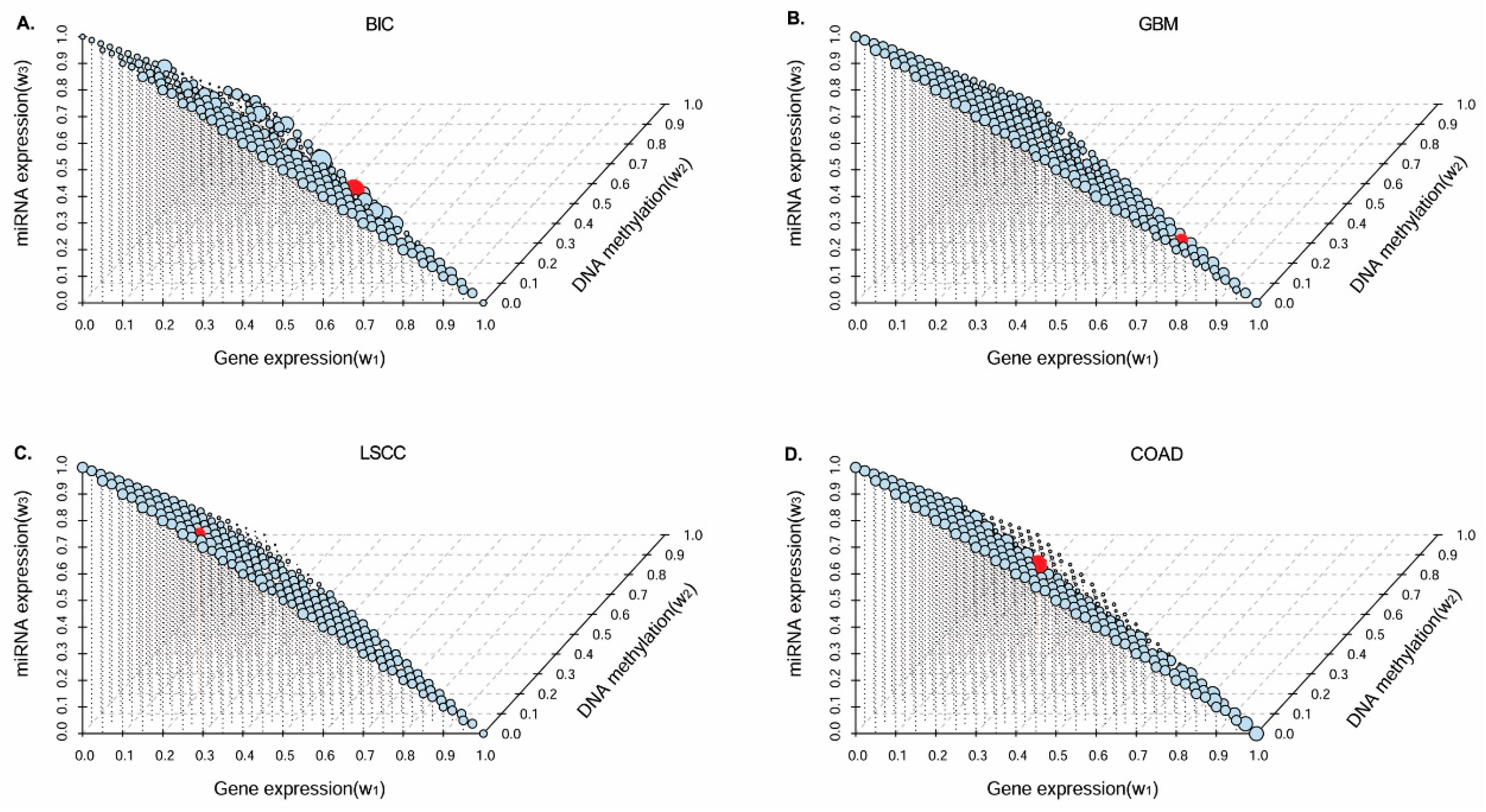
3.1. Parameter Selection
3.2. Performance Evaluation in Various Cancer Datasets
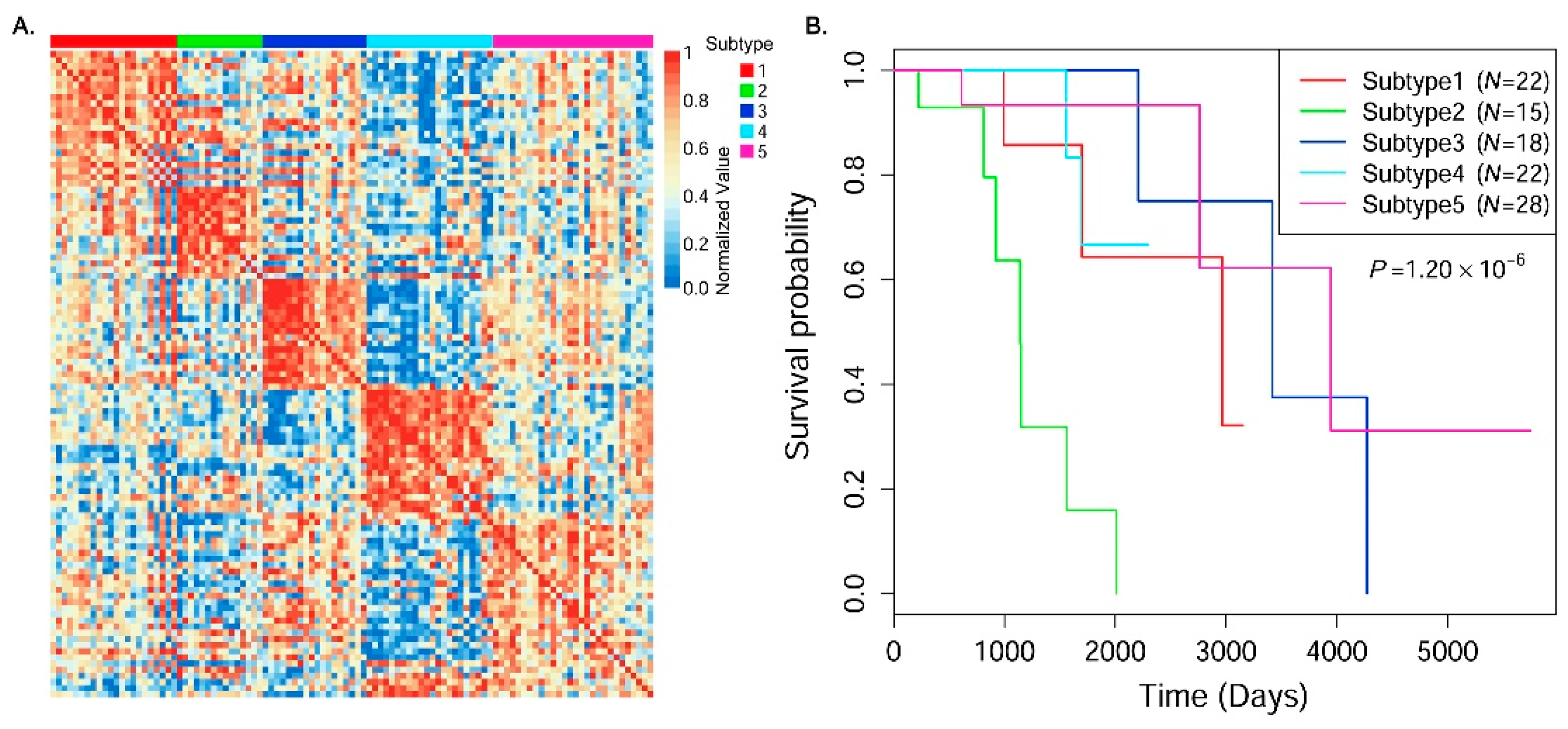
3.3. Cancer Subtype Clustering in Breast Cancer Data
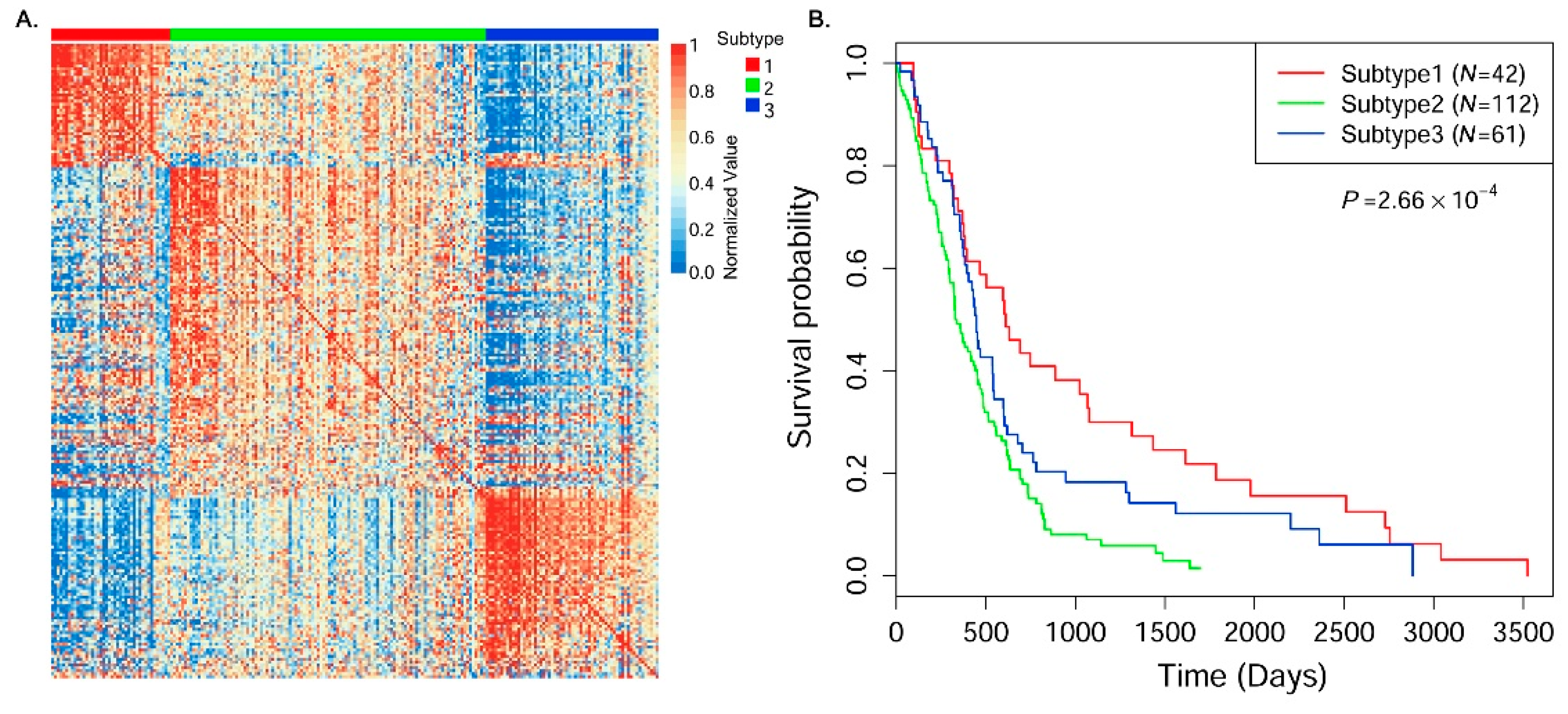
3.4. Cancer Subtype Clustering in Glioblastoma Multiforme Data
4. Discussion and Conclusions
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Stingl, J.; Caldas, C. Opinion-molecular heterogeneity of breast carcinomas and the cancer stem cell hypothesis. Nat. Rev. Cancer 2007, 7, 791–799. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.F.; Li, T.; Bai, Z.H.; Yang, Y.K.; Liu, X.X.; Zhan, J.L.; Shi, B.Z. Breast cancer intrinsic subtype classification, clinical use and future trends. Am. J. Cancer Res. 2015, 5, 2929–2943. [Google Scholar] [PubMed]
- Prat, A.; Pineda, E.; Adamo, B.; Galvan, P.; Fernandez, A.; Gaba, L.; Diez, M.; Viladot, M.; Arance, A.; Munoz, M. Clinical implications of the intrinsic molecular subtypes of breast cancer. Breast 2015, 24, S26–S35. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.Q.; Zhang, X.S.; Zhang, S.H. Breast tumor subgroups reveal diverse clinical prognostic power. Sci. Rep. 2014. [Google Scholar] [CrossRef] [PubMed]
- Rouzier, R.; Perou, C.M.; Symmans, W.F.; Ibrahim, N.; Cristofanilli, M.; Anderson, K.; Hess, K.R.; Stec, J.; Ayers, M.; Wagner, P.; et al. Breast cancer molecular subtypes respond differently to preoperative chemotherapy. Clin. Cancer Res. 2005, 11, 5678–5685. [Google Scholar] [CrossRef] [PubMed]
- Xu, T.S.; Le, T.D.; Liu, L.; Wang, R.J.; Sun, B.Y.; Li, J.Y. Identifying cancer subtypes from miRNA-TF-mRNA regulatory networks and expression data. PLoS ONE 2016, 11. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Mezlini, A.M.; Demir, F.; Fiume, M.; Tu, Z.W.; Brudno, M.; Haibe-Kains, B.; Goldenberg, A. Similarity network fusion for aggregating data types on a genomic scale. Nat. Methods 2014, 11, 333–337. [Google Scholar] [CrossRef] [PubMed]
- Akbani, R.; Ng, K.S.; Werner, H.M.; Zhang, F.; Ju, Z.L.; Liu, W.B.; Yang, J.Y.; Lu, Y.L.; Weinstein, J.N.; Mills, G.B. A pan-cancer proteomic analysis of the cancer genome atlas (TCGA) project. Nat. Commun. 2014, 5, 3887. [Google Scholar] [CrossRef] [PubMed]
- Weinstein, J.N.; Collisson, E.A.; Mills, G.B.; Shaw, K.R.M.; Ozenberger, B.A.; Ellrott, K.; Shmulevich, I.; Sander, C.; Stuart, J.M.; Network, C.G.A.R. The cancer genome atlas pan-cancer analysis project. Nat. Genet. 2013, 45, 1113–1120. [Google Scholar] [CrossRef] [PubMed]
- Shen, S.H.; Wang, Y.Y.; Wang, C.Y.; Wu, Y.N.; Xing, Y. SURVIV for survival analysis of mRNA isoform variation. Nat. Commun. 2016, 7, 11548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bersanelli, M.; Mosca, E.; Remondini, D.; Giampieri, E.; Sala, C.; Castellani, G.; Milanesi, L. Methods for the integration of multi-omics data: Mathematical aspects. BMC Bioinform. 2016, 17, S15. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Liu, S.; Li, Z.; Shang, X. Bcdforest: A boosting cascade deep forest model towards the classification of cancer subtypes based on gene expression data. BMC Bioinform. 2018, 19, 118. [Google Scholar] [CrossRef] [PubMed]
- Golub, T.R.; Slonim, D.K.; Tamayo, P.; Huard, C.; Gaasenbeek, M.; Mesirov, J.P.; Coller, H.; Loh, M.L.; Downing, J.R.; Caligiuri, M.A.; et al. Molecular classification of cancer: Class discovery and class prediction by gene expression monitoring. Science 1999, 286, 531–537. [Google Scholar] [CrossRef] [PubMed]
- Sorlie, T.; Perou, C.M.; Tibshirani, R.; Aas, T.; Geisler, S.; Johnsen, H.; Hastie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc. Natl. Acad. Sci. USA 2001, 98, 10869–10874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lapointe, J.; Li, C.; Higgins, J.P.; van de Rijn, M.; Bair, E.; Montgomery, K.; Ferrari, M.; Egevad, L.; Rayford, W.; Bergerheim, U.; et al. Gene expression profiling identifies clinically relevant subtypes of prostate cancer. Proc. Natl. Acad. Sci. USA 2004, 101, 811–816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monti, S.; Tamayo, P.; Mesirov, J.; Golub, T. Consensus clustering: A resampling-based method for class discovery and visualization of gene expression microarray data. Mach. Learn. 2003, 52, 91–118. [Google Scholar] [CrossRef]
- Speicher, N.K.; Pfeifer, N. Integrating different data types by regularized unsupervised multiple kernel learning with application to cancer subtype discovery. Bioinformatics 2015, 31, 268–275. [Google Scholar] [CrossRef] [PubMed]
- Verhaak, R.G.W.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010, 17, 98–110. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research Network. Comprehensive genomic characterization of squamous cell lung cancers the cancer genome atlas research network. Nature 2012, 489, 519–525. [Google Scholar]
- Guo, Y.; Shang, X.; Li, Z. Identification of cancer subtypes by integrating multiple types of transcriptomics data with deep learning in breast cancer. Neurocomputing 2018. [Google Scholar] [CrossRef]
- Xu, T.S.; Le, T.D.; Liu, L.; Su, N.; Wang, R.J.; Sun, B.Y.; Colaprico, A.; Bontempi, G.; Li, J.Y. Cancersubtypes: An R/Bioconductor package for molecular cancer subtype identification, validation and visualization. Bioinformatics 2017, 33, 3131–3133. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.Y.; Zheng, H.R.; Wang, J.X.; Wang, C.Y.; Wu, F.X. Integrating omics data with a multiplex network-based approach for the identification of cancer subtypes. IEEE Trans. Nanobiosci. 2016, 15, 335–342. [Google Scholar] [CrossRef] [PubMed]
- Shen, R.L.; Olshen, A.B.; Ladanyi, M. Integrative clustering of multiple genomic data types using a joint latent variable model with application to breast and lung cancer subtype analysis. Bioinformatics 2009, 25, 2906–2912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, R.; Mo, Q.X.; Schultz, N.; Seshan, V.E.; Olshen, A.B.; Huse, J.; Ladanyi, M.; Sander, C. Integrative subtype discovery in glioblastoma using iCluster. PLoS ONE 2012. [Google Scholar] [CrossRef] [PubMed]
- Brunet, J.P.; Tamayo, P.; Golub, T.R.; Mesirov, J.P. Metagenes and molecular pattern discovery using matrix factorization. Proc. Natl. Acad. Sci. USA 2004, 101, 4164–4169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaujoux, R.; Seoighe, C. A flexible r package for nonnegative matrix factorization. BMC Bioinform. 2010, 11, 367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, T.; Zhang, A. Integrate multi-omic data using affinity network fusion (ANF) for cancer patient clustering. arXiv, 2017; arXiv:1708.07136. [Google Scholar] [Green Version]
- Vaske, C.J.; Benz, S.C.; Sanborn, J.Z.; Earl, D.; Szeto, C.; Zhu, J.C.; Haussler, D.; Stuart, J.M. Inference of patient-specific pathway activities from multi-dimensional cancer genomics data using PARADIGM. Bioinformatics 2010, 26, i237–i245. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.; Sun, Z.; da Silveira, W.A.; Yu, Z.; Lawson, A.; Hardiman, G.; Kelemen, L.E.; Chung, D. Semi-supervised identification of cancer subgroups using survival outcomes and overlapping grouping information. Stat. Methods Med. Res. 2018. [Google Scholar] [CrossRef] [PubMed]
- Yan, S.C.; Xu, D.; Zhang, B.Y.; Zhang, H.J.; Yang, Q.; Lin, S. Graph embedding and extensions: A general framework for dimensionality reduction. IEEE Trans. Pattern Anal. Mach. Intell. 2007, 29, 40–51. [Google Scholar] [CrossRef] [PubMed]
- Hu, R.; Qiu, X.; Glazko, G.; Klebanov, L.; Yakovlev, A. Detecting intergene correlation changes in microarray analysis: A new approach to gene selection. BMC Bioinform. 2009, 10, 20. [Google Scholar] [CrossRef] [PubMed]
- Hu, P.; Shen, Z.; Tu, H.; Zhang, L.; Shi, T. Integrating multiple resources to identify specific transcriptional cooperativity with a Bayesian approach. Bioinformatics 2014, 30, 823–830. [Google Scholar] [CrossRef] [PubMed]
- Mantel, N. Evaluation of survival data and two new rank order statistics arising in its consideration. Cancer Chemother. Rep. 1966, 50, 163–170. [Google Scholar] [PubMed]
- Goel, M.K.; Khanna, P.; Kishore, J. Understanding survival analysis: Kaplan-Meier estimate. Int. J. Ayurveda Res. 2010, 1, 274–278. [Google Scholar] [PubMed]
- Parker, J.S.; Mullins, M.; Cheang, M.C.; Leung, S.; Voduc, D.; Vickery, T.; Davies, S.; Fauron, C.; He, X.; Hu, Z.; et al. Supervised risk predictor of breast cancer based on intrinsic subtypes. J. Clin. Oncol. 2009, 27, 1160–1167. [Google Scholar] [CrossRef] [PubMed]
- Mohammed, H.; Russell, I.A.; Stark, R.; Rueda, O.M.; Hickey, T.E.; Tarulli, G.A.; Serandour, A.A.A.; Birrell, S.N.; Bruna, A.; Saadi, A.; et al. Progesterone receptor modulates ER alpha action in breast cancer. Nature 2015, 523, 313–317. [Google Scholar] [CrossRef] [PubMed]
- Patel, M.A.; Kim, J.E.; Ruzevick, J.; Li, G.; Lim, M. The future of glioblastoma therapy: Synergism of standard of care and immunotherapy. Cancers 2014, 6, 1953–1985. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.J.; Zhang, X.S.; Hui, W.W.; Lu, J.Y.; Li, Q.Q.; Liu, S.H.; Shang, X.Q. Improving the measurement of semantic similarity by combining gene ontology and co-functional network: A random walk based approach. BMC Syst. Biol. 2018, 12. [Google Scholar]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using david bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef] [PubMed]
- Noushmehr, H.; Weisenberger, D.J.; Diefes, K.; Phillips, H.S.; Pujara, K.; Berman, B.P.; Pan, F.; Pelloski, C.E.; Sulman, E.P.; Bhat, K.P.; et al. Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer Cell 2010, 17, 510–522. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.L.; Shang, X.Q. Detection of network motif based on a novel graph canonization algorithm from transcriptional regulation networks. Molecules 2017, 22. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Landskron, G.; De la Fuente, M.; Thuwajit, P.; Thuwajit, C.; Hermoso, M.A. Chronic inflammation and cytokines in the tumor microenvironment. J. Immunol. Res. 2014, 2014, 149185. [Google Scholar] [CrossRef] [PubMed]
- Colotta, F.; Allavena, P.; Sica, A.; Garlanda, C.; Mantovani, A. Cancer-related inflammation, the seventh hallmark of cancer: Links to genetic instability. Carcinogenesis 2009, 30, 1073–1081. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Datasets | Gene Expression (Features) | DNA Methylation (Features) | miRNA (Features) | Patients |
|---|---|---|---|---|
| BIC | 17,814 | 23,094 | 354 | 105 |
| GBM | 12,042 | 1,305 | 534 | 215 |
| LSCC | 12,042 | 23,074 | 352 | 106 |
| COAD | 17,814 | 23,088 | 312 | 92 |
| Datasets (# Subtypes) | Gene Expression | DNA Methylation | miRNA Expression | Integrative Data (SRF) |
|---|---|---|---|---|
| BIC (5) | 1.59 × 10−2 (0.19) | 3.67 × 10−1 (0.17) | 3.56 × 10−2 (0.19) | 1.20 × 10−6 (0.15) |
| GBM (3) | 2.49 × 10−3 (0.20) | 5.71 × 10−3 (0.22) | 1.50 × 10−3 (0.23) | 2.66 × 10−4 (0.21) |
| LSCC (4) | 2.13 × 10−2 (0.26) | 5.62 × 10−1 (0.19) | 4.64 × 10−3 (0.25) | 1.98 × 10−3 (0.26) |
| COAD (3) | 6.64 × 10−3 (0.27) | 3.84 × 10−1 (0.26) | 2.61 × 10−2 (0.30) | 1.96 × 10−3 (0.26) |
| Datasets (#Subtypes) | CNMF | iCluster | SNF | CC | SNF-CC | ANF | SRF |
|---|---|---|---|---|---|---|---|
| BIC (5) | 9.74 × 10−2 | 2.04 × 10−2 | 1.35 × 10−3 | 2.81 × 10−5 | 2.94 × 10−4 | 7.60 × 10−4 | 1.20 × 10−6 |
| GBM (3) | 3.65 × 10−1 | 2.68 × 10−3 | 3.87 × 10−4 | 7.49 × 10−1 | 7.93 × 10−4 | 1.75 × 10−2 | 2.66 × 10−4 |
| LSCC (4) | 2.33 × 10−2 | 7.62 × 10−3 | 1.78 × 10−2 | 1.03 × 10−2 | 1.64 × 10−2 | 1.83 × 10−2 | 1.98 × 10−3 |
| COAD (3) | 2.22 × 10−2 | 2.46 × 10−3 | 3.60 × 10−2 | 3.72 × 10−2 | 3.80 × 10−2 | 3.92 × 10−2 | 1.96 × 10−3 |
| Subtype ID | C1 (N = 22) | C2 (N = 15) | C3 (N = 18) | C4 (N = 22) | C5 (N = 28) |
|---|---|---|---|---|---|
| Average age (years) | 55.4 | 62.7 | 56.1 | 50.8 | 57.8 |
| Average survival time (days) | 1885.7 | 1116.7 | 3299.3 | 1623.5 | 2439.3 |
| Subtype ID | C1 (N = 22) | C2 (N = 15) | C3 (N = 18) | C4 (N = 22) | C5 (N = 28) |
|---|---|---|---|---|---|
| Basal (23) | 0 (0.0%) | 1 (4.3%) | 0 (0.0%) | 18 (78.3%) | 4 (17.4%) |
| Her2 (11) | 0 (0.0%) | 2 (18.2%) | 0 (0.0%) | 3 (27.3%) | 6 (54.5%) |
| LumA (55) | 17 (30.9%) | 6 (10.9%) | 16 (29.1%) | 0 (0.0%) | 16 (29.1%) |
| LumB (12) | 4 (33.3%) | 6 (50.0%) | 0 (0.0%) | 0 (0.0%) | 2 (16.6%) |
| Normal (2) | 0 (0.0%) | 0 (0.0%) | 1 (50.0%) | 1 (50.0%) | 0 (0.0%) |
| ER+ (80) | 22 (27.5%) | 13 (16.25%) | 18 (22.5%) | 5 (6.25%) | 22 (27.5%) |
| ER− (24) | 0 (0.0%) | 1 (4.2%) | 0 (0.0%) | 17 (70.8%) | 6 (25.0%) |
| PR+ (71) | 19 (26.8%) | 11 (15.5%) | 18 (25.3%) | 3 (4.2%) | 20 (28.2%) |
| PR− (34) | 3 (8.8%) | 4 (11.8%) | 0 (0.0%) | 19 (55.9%) | 8 (23.5%) |
| Subtype ID | C1 (N = 42) | C2 (N = 112) | C3 (N = 61) |
|---|---|---|---|
| Patients (Male:Female) | (24:18) | (69:43) | (41:20) |
| Average age (years) | 46.4 | 58.8 | 54.8 |
| Average survival time (days) | 931.9 | 402.5 | 564.9 |
| Subtype ID | C1 (N = 42) | C2 (N = 112) | C3 (N = 61) |
|---|---|---|---|
| Classical (58) | 2 (3.4%) | 44 (75.9%) | 12 (20.7%) |
| Mesenchymal (66) | 0 (0.0%) | 28 (42.4%) | 38 (57.6%) |
| Neural (34) | 3 (8.8%) | 25 (73.5%) | 6 (17.7%) |
| Proneural (57) | 37 (64.9%) | 15 (26.3%) | 5 (8.8%) |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guo, Y.; Zheng, J.; Shang, X.; Li, Z. A Similarity Regression Fusion Model for Integrating Multi-Omics Data to Identify Cancer Subtypes. Genes 2018, 9, 314. https://doi.org/10.3390/genes9070314
Guo Y, Zheng J, Shang X, Li Z. A Similarity Regression Fusion Model for Integrating Multi-Omics Data to Identify Cancer Subtypes. Genes. 2018; 9(7):314. https://doi.org/10.3390/genes9070314
Chicago/Turabian StyleGuo, Yang, Jianning Zheng, Xuequn Shang, and Zhanhuai Li. 2018. "A Similarity Regression Fusion Model for Integrating Multi-Omics Data to Identify Cancer Subtypes" Genes 9, no. 7: 314. https://doi.org/10.3390/genes9070314
APA StyleGuo, Y., Zheng, J., Shang, X., & Li, Z. (2018). A Similarity Regression Fusion Model for Integrating Multi-Omics Data to Identify Cancer Subtypes. Genes, 9(7), 314. https://doi.org/10.3390/genes9070314