Morphological Stasis and Proteome Innovation in Cephalochordates


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
3. Results
3.1. Absence of Orthologs in Some Lancelet Species May Reflect Errors of Gene Prediction
Ryk Receptor Tyrosine Kinase
3.2. Domain Architecture Differences of Orthologs of Lancelet Species May Reflect Errors of Gene Prediction
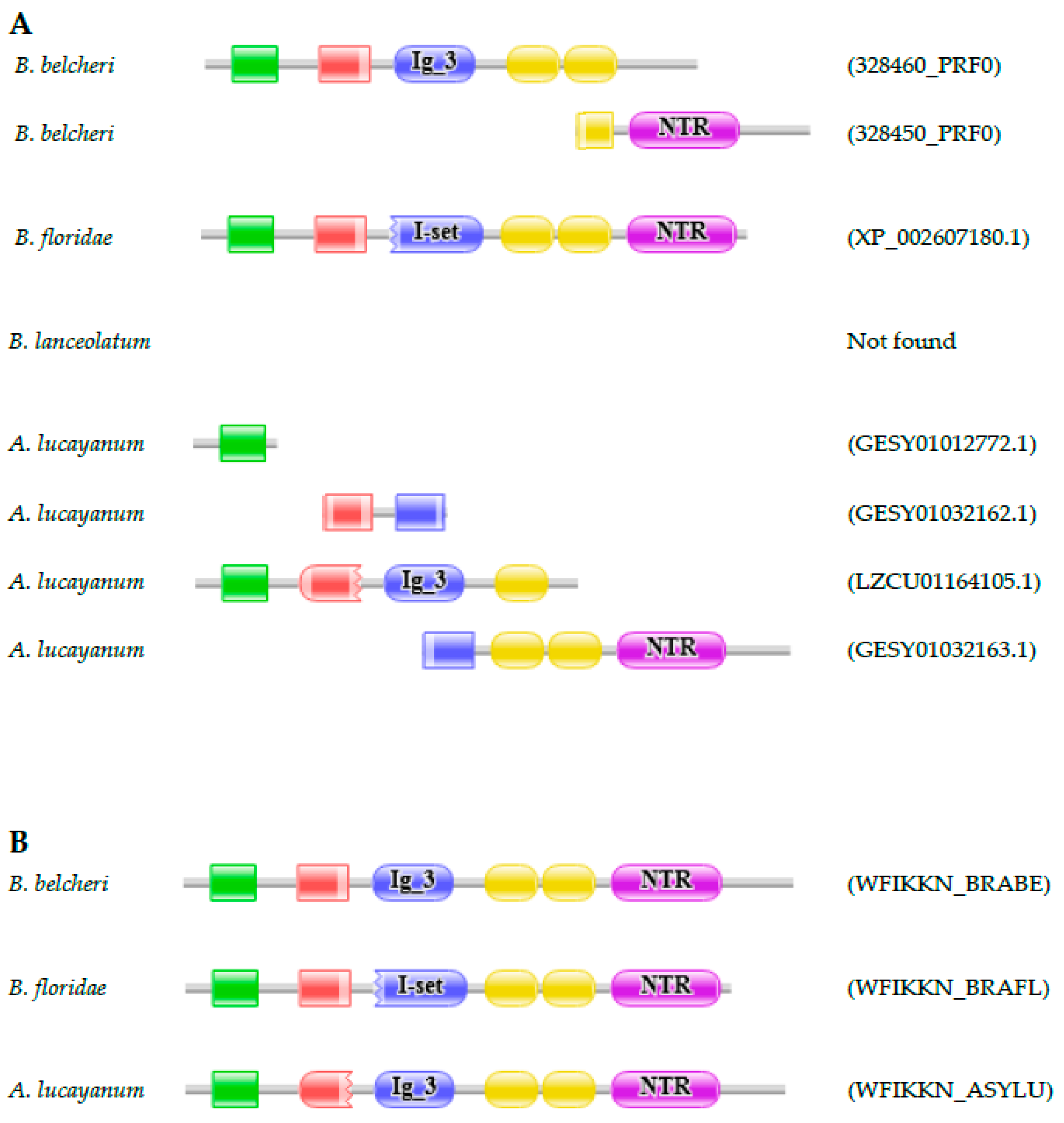
3.2.1. WAP, Follistatin, Immunoglobulin, Kunitz and NTR Domain-Containing (WFIKKN) Protein
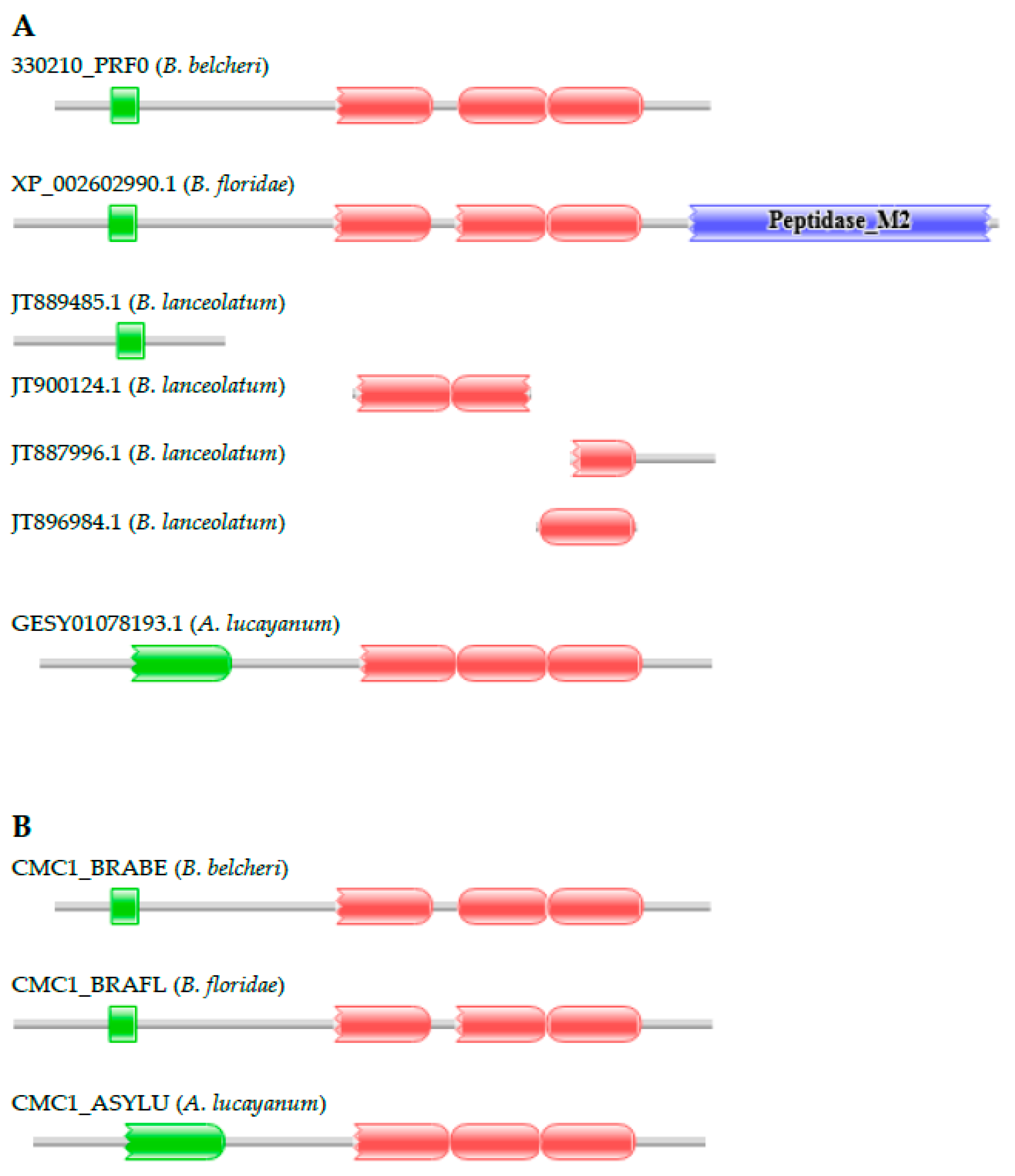
3.2.2. Calcium-Binding Mitochondrial Carrier Protein
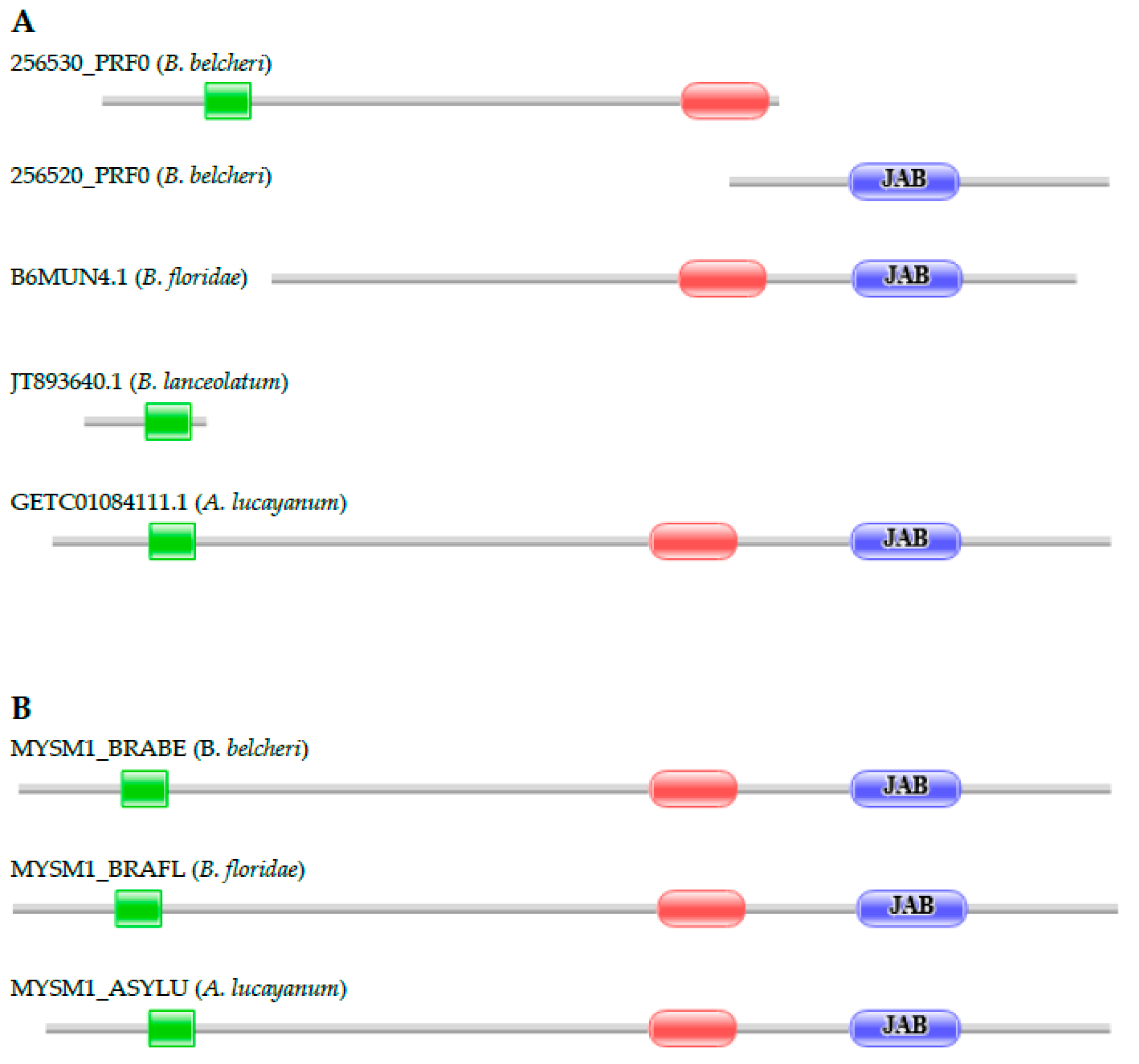
3.2.3. Histone H2A Deubiquitinase
3.2.4. Chordin
3.3. Lancelet Proteins with Domain Architectures Apparently Unique to Cephalochordates
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Blair, J.E.; Hedges, S.B. Molecular phylogeny and divergence times of deuterostome animals. Mol. Biol. Evol. 2005, 22, 2275–2284. [Google Scholar] [CrossRef] [PubMed]
- Delsuc, F.; Brinkmann, H.; Chourrout, D.; Philippe, H. Tunicates and not cephalochordates are the closest living relatives of vertebrates. Nature 2006, 439, 965–968. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.-Y.; Dzik, J.; Edgecombe, G.D.; Ramskold, L.; Zhou, G.-Q. A possible Early Cambrian chordate. Nature 1995, 377, 720–722. [Google Scholar] [CrossRef]
- Chen, J.-Y.; Huang, D.-Y.; Li, C.-W. An early Cambrian craniate-like chordate. Nature 1999, 402, 518–522. [Google Scholar] [CrossRef]
- Lacalli, T. The Middle Cambrian fossil Pikaia and the evolution of chordate swimming. EvoDevo 2012, 3, 12. [Google Scholar] [CrossRef] [PubMed]
- Morris, S.C.; Whittington, H.B. The animals of the Burgess Shale. Sci. Am. 1979, 241, 122–135. [Google Scholar] [CrossRef]
- Igawa, T.; Nozawa, M.; Suzuki, D.G.; Reimer, J.D.; Morov, A.R.; Wang, Y.; Henmi, Y.; Yasui, K. Evolutionary history of the extant amphioxus lineage with shallow-branching diversification. Sci. Rep. 2017, 7, 1157. [Google Scholar] [CrossRef] [PubMed]
- Somorjai, I.; Bertrand, S.; Camasses, A.; Haguenauer, A.; Escriva, H. Evidence for stasis and not genetic piracy in developmental expression patterns of Branchiostoma lanceolatum and Branchiostoma floridae, two amphioxus species that have evolved independently over the course of 200 Myr. Dev. Genes Evol. 2008, 218, 703–713. [Google Scholar] [CrossRef] [PubMed]
- Holland, N.D.; Holland, L.Z.; Heimberg, A. Hybrids between the Florida amphioxus (Branchiostoma floridae) and the Bahamas lancelet (Asymmetron lucayanum): Developmental morphology and chromosome counts. Biol. Bull. 2015, 228, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Wilson, A.C.; Maxson, L.R.; Sarich, V.M. Two types of molecular evolution. Evidence from studies of interspecific hybridization. Proc. Natl. Acad. Sci. USA 1974, 71, 2843–2847. [Google Scholar] [CrossRef] [PubMed]
- Prager, E.M.; Wilson, A.C. Slow evolutionary loss of the potential for interspecific hybridization in birds: A manifestation of slow regulatory evolution. Proc. Natl. Acad. Sci. USA 1975, 72, 200–204. [Google Scholar] [CrossRef] [PubMed]
- Fitzpatrick, B.M. Rates of evolution of hybrid inviability in birds and mammals. Evolution 2004, 58, 1865–1870. [Google Scholar] [CrossRef] [PubMed]
- Estes, S.; Arnold, S.J. Resolving the paradox of stasis: Models with stabilizing selection explain evolutionary divergence on all timescales. Am. Nat. 2007, 169, 227–244. [Google Scholar] [CrossRef] [PubMed]
- Hendry, A. Evolutionary biology: The Elvis paradox. Nature 2007, 446, 147–149. [Google Scholar] [CrossRef] [PubMed]
- Haller, B.C.; Hendry, A.P. Solving the paradox of stasis: Squashed stabilizing selection and the limits of detection. Evolution 2014, 68, 483–500. [Google Scholar] [CrossRef] [PubMed]
- Putnam, N.H.; Butts, T.; Ferrier, D.E.; Furlong, R.F.; Hellsten, U.; Kawashima, T.; Robinson-Rechavi, M.; Shoguchi, E.; Terry, A.; Yu, J.K.; et al. The amphioxus genome and the evolution of the chordate karyotype. Nature 2008, 453, 1064–1071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yue, J.X.; Yu, J.K.; Putnam, N.H.; Holland, L.Z. The transcriptome of an amphioxus, Asymmetron lucayanum, from the Bahamas: A window into chordate evolution. Genome Biol. Evol. 2014, 6, 2681–2696. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Chen, Z.; Yan, X.; Yu, T.; Huang, G.; Yan, Q.; Pontarotti, P.A.; Zhao, H.; Li, J.; Yang, P.; et al. Decelerated genome evolution in modern vertebrates revealed by analysis of multiple lancelet genomes. Nat. Commun. 2014, 5, 5896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patthy, L. Modular assembly of genes and the evolution of new functions. Genetica 2003, 118, 217–231. [Google Scholar] [CrossRef] [PubMed]
- Tordai, H.; Nagy, A.; Farkas, K.; Bányai, L.; Patthy, L. Modules, multidomain proteins and organismic complexity. FEBS J. 2005, 272, 5064–5078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bányai, L.; Patthy, L. Putative extremely high rate of proteome innovation in lancelets might be explained by high rate of gene prediction errors. Sci. Rep. 2016, 6, 30700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harrow, J.; Nagy, A.; Reymond, A.; Alioto, T.; Patthy, L.; Antonarakis, S.E.; Guigó, R. Identifying protein-coding genes in genomic sequences. Genome Biol. 2009, 10, 201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagy, A.; Szláma, G.; Szarka, E.; Trexler, M.; Bányai, L.; Patthy, L. Reassessing domain architecture evolution of metazoan proteins: Major impact of gene prediction errors. Genes 2011, 2, 449–501. [Google Scholar] [CrossRef] [PubMed]
- Yue, J.X.; Kozmikova, I.; Ono, H.; Nossa, C.W.; Kozmik, Z.; Putnam, N.H.; Yu, J.K.; Holland, L.Z. Conserved noncoding elements in the most distant genera of Cephalochordates: The goldilocks principle. Genome Biol. Evol. 2016, 8, 2387–2405. [Google Scholar] [CrossRef] [PubMed]
- Oulion, S.; Bertrand, S.; Belgacem, M.R.; Le Petillon, Y.; Escriva, H. Sequencing and analysis of the Mediterranean amphioxus (Branchiostoma lanceolatum) transcriptome. PLoS ONE 2012, 7, e36554. [Google Scholar] [CrossRef] [PubMed]
- The Lancelet (Branchiostoma belcheri) Genome Sequencing and Annotation Project Database. Available online: http://genome.bucm.edu.cn/lancelet/ (accessed on 1 March 2018).
- Schilling, T.F.; Knight, R.D. Origins of anteroposterior patterning and HOX gene regulation during chordate evolution. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2001, 356, 1599–1613. [Google Scholar] [CrossRef] [PubMed]
- Kozmikova, I.; Yu, J.K. Dorsal-ventral patterning in amphioxus: Current understanding, unresolved issues, and future directions. Int. J. Dev. Biol. 2017, 61, 601–610. [Google Scholar] [CrossRef] [PubMed]
- Soukup, V.; Kozmik, Z. The Bmp signaling pathway regulates development of left-right asymmetry in amphioxus. Dev. Biol. 2018, 434, 164–174. [Google Scholar] [CrossRef] [PubMed]
- Soukup, V.; Yong, L.W.; Lu, T.M.; Huang, S.W.; Kozmik, Z.; Yu, J.K. The Nodal signaling pathway controls left-right asymmetric development in amphioxus. EvoDevo 2015, 6, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finn, R.D.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Mistry, J.; Mitchell, A.L.; Potter, S.C.; Punta, M.; Qureshi, M.; Sangrador-Vegas, A.; et al. The Pfam protein families database: Towards a more sustainable future. Nucleic Acids Res. 2016, 44, D279–D285. [Google Scholar] [CrossRef] [PubMed]
- Sievers, F.; Wilm, A.; Dineen, D.G.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Söding, J.; et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef] [PubMed]
- Nagy, A.; Hegyi, H.; Farkas, K.; Tordai, H.; Kozma, E.; Bányai, L.; Patthy, L. Identification and correction of abnormal, incomplete and mispredicted proteins in public databases. BMC Bioinform. 2008, 9, 353. [Google Scholar] [CrossRef] [PubMed]
- Nagy, A.; Patthy, L. MisPred: A resource for identification of erroneous protein sequences in public databases. Database (Oxf.) 2013, 2013, bat053. [Google Scholar] [CrossRef] [PubMed]
- Nagy, A.; Patthy, L. FixPred: A resource for correction of erroneous protein sequences. Database 2014, 2014, bau032. [Google Scholar] [CrossRef] [PubMed]
- Patthy, L. The WIF module. Trends Biochem. Sci. 2000, 25, 12–13. [Google Scholar] [CrossRef]
- Putnam, N.H.; Srivastava, M.; Hellsten, U.; Dirks, B.; Chapman, J.; Salamov, A.; Terry, A.; Shapiro, H.; Lindquist, E.; Kapitonov, V.V.; et al. Sea anemone genome reveals ancestral eumetazoan gene repertoire and genomic organization. Science 2007, 317, 86–94. [Google Scholar] [CrossRef] [PubMed]
- Nagy, A.; Bányai, L.; Patthy, L. Reassessing domain architecture evolution of metazoan proteins: Major impact of errors caused by confusing paralogs and epaktologs. Genes 2011, 2, 516–561. [Google Scholar] [CrossRef] [PubMed]
- Guigó, R.; Flicek, P.; Abril, J.F.; Reymond, A.; Lagarde, J.; Denoeud, F.; Antonarakis, S.; Ashburner, M.; Bajic, V.B.; Birney, E.; et al. EGASP: The human ENCODE Genome Annotation Assessment Project. Genome Biol. 2006, 7, S2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, J.K.; Wang, M.C.; Shin, T.; Kohara, Y.; Holland, L.Z.; Satoh, N.; Satou, Y. A cDNA resource for the cephalochordate amphioxus Branchiostoma floridae. Dev. Genes Evol. 2008, 218, 723–727. [Google Scholar] [CrossRef] [PubMed]
- Denton, J.F.; Lugo-Martinez, J.; Tucker, A.E.; Schrider, D.R.; Warren, W.C.; Hahn, M.W. Extensive error in the number of genes inferred from draft genome assemblies. PLoS Comput. Biol. 2014, 10, e1003998. [Google Scholar] [CrossRef] [PubMed]
- Francis, W.R.; Wörheide, G. similar ratios of introns to intergenic sequence across animal genomes. Genome Biol. Evol. 2017, 9, 1582–1598. [Google Scholar] [CrossRef] [PubMed]





© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bányai, L.; Kerekes, K.; Trexler, M.; Patthy, L. Morphological Stasis and Proteome Innovation in Cephalochordates. Genes 2018, 9, 353. https://doi.org/10.3390/genes9070353
Bányai L, Kerekes K, Trexler M, Patthy L. Morphological Stasis and Proteome Innovation in Cephalochordates. Genes. 2018; 9(7):353. https://doi.org/10.3390/genes9070353
Chicago/Turabian StyleBányai, László, Krisztina Kerekes, Mária Trexler, and László Patthy. 2018. "Morphological Stasis and Proteome Innovation in Cephalochordates" Genes 9, no. 7: 353. https://doi.org/10.3390/genes9070353
APA StyleBányai, L., Kerekes, K., Trexler, M., & Patthy, L. (2018). Morphological Stasis and Proteome Innovation in Cephalochordates. Genes, 9(7), 353. https://doi.org/10.3390/genes9070353