Evolutionary Divergent Suppressor Mutations in Conformational Diseases


 , and
, and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Single Amino Acid Changes as Key Players in Molecular Evolution, Human Physiology and Genetic Diseases
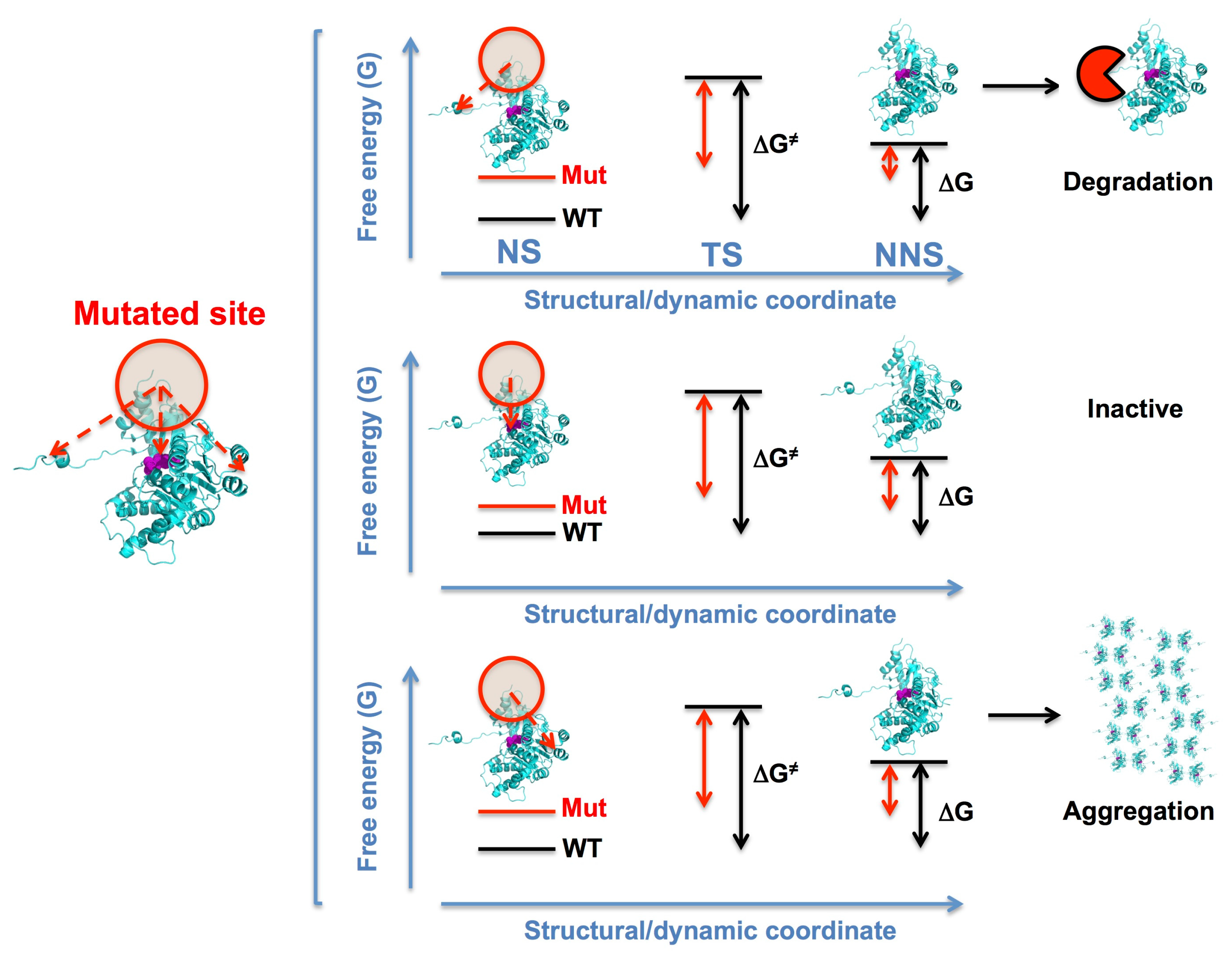
2. Control of Protein Function and Life Cycle through Structural Stability and Dynamics
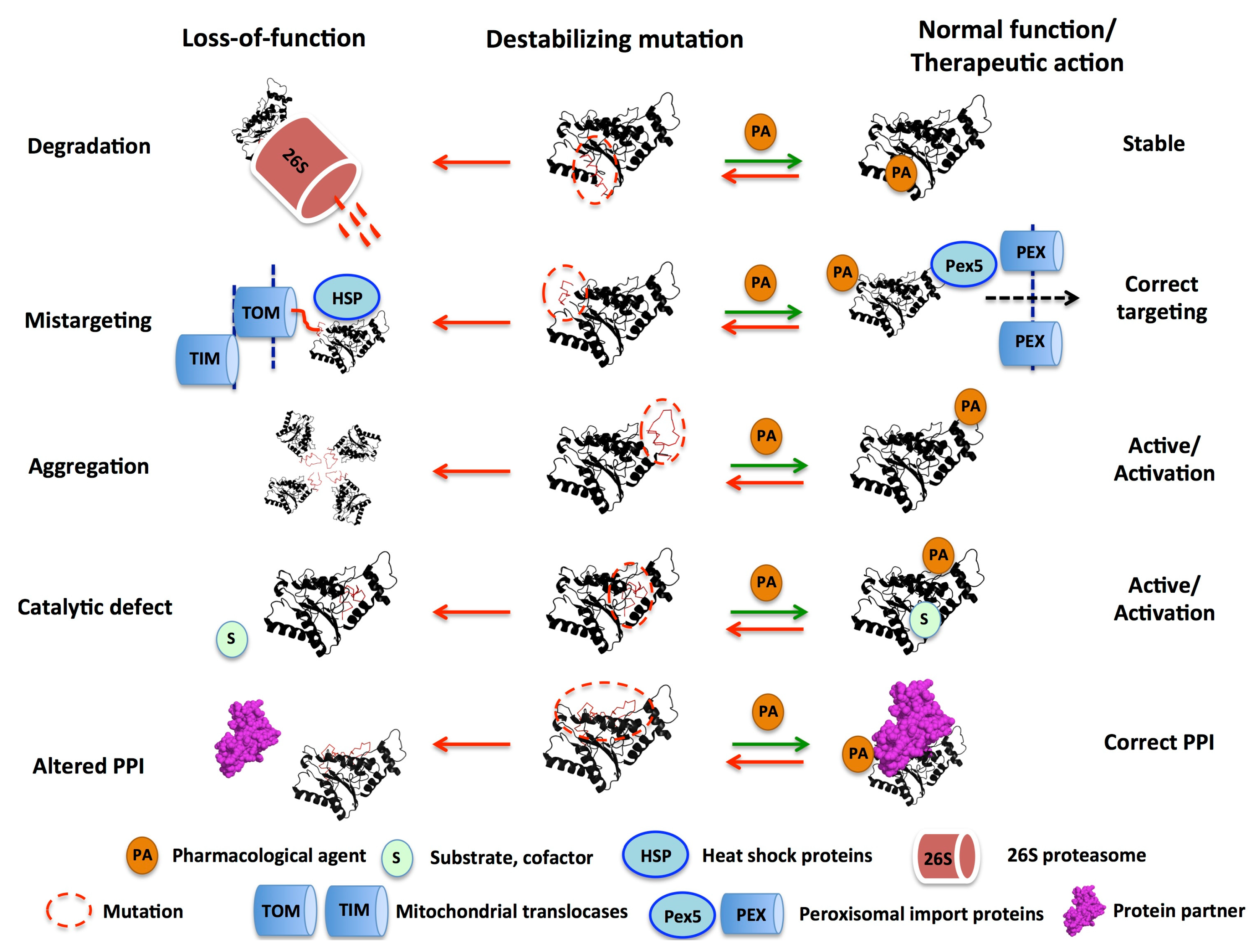
3. Mutational Effects in Loss-of-Function Disease Mechanisms
4. NADP(H):quinone Oxidoreductase 1 and Alanine:glyoxylate aminotransferase as Models of Genetic Diseases
4.1. NADP(H):quinone Oxidoreductase 1
4.2. Alanine:glyoxylate aminotransferase
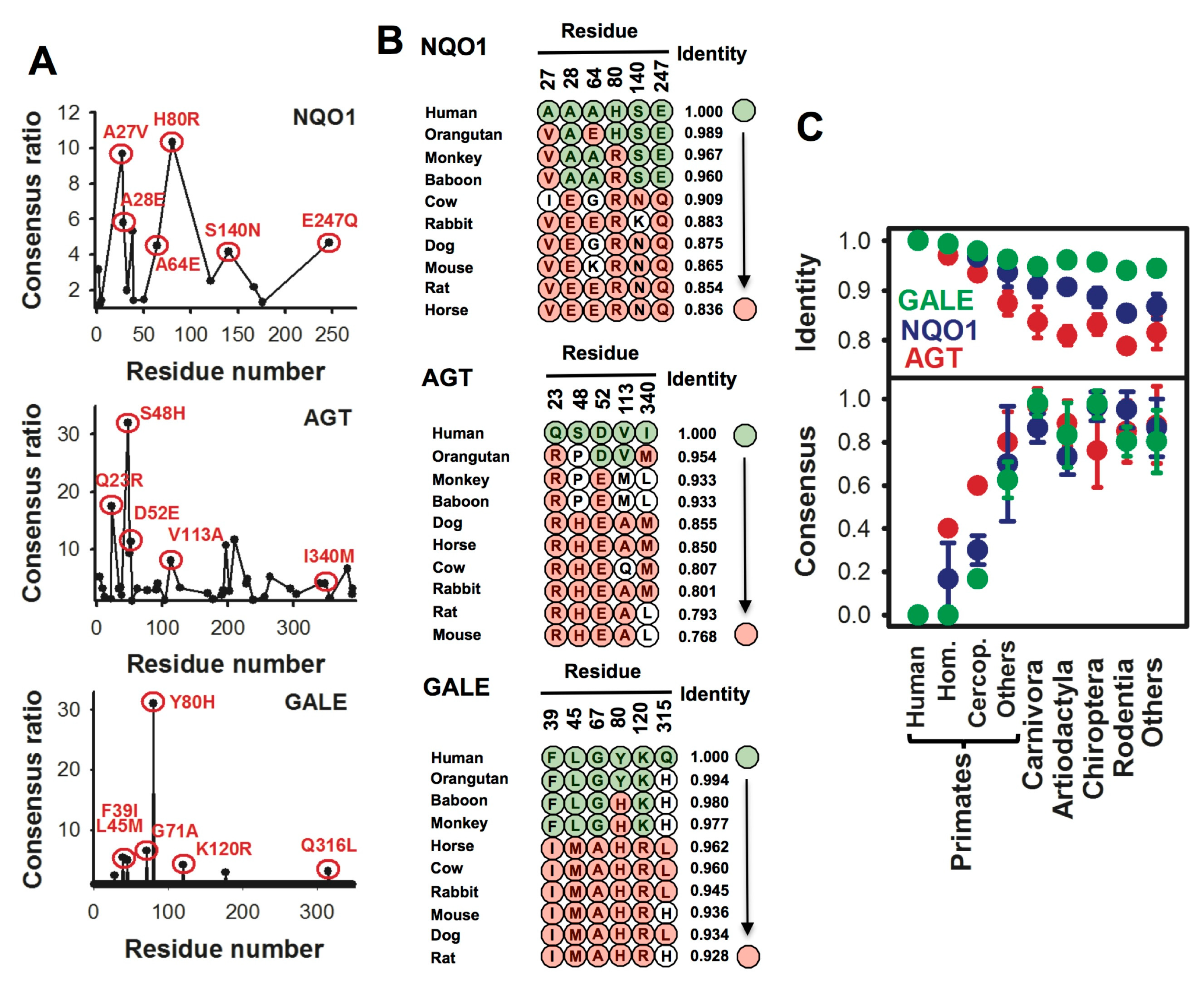
5. Evolutionary Divergence in Key Compensatory Consensus Amino Acids and its Potential Role in Species-dependent Disease Penetrance
5.1. Alanine:glyoxylate aminotransferase
5.2. NADP(H):quinone oxidoreductase 1
5.3. On the Different Sensitivity of Human and Non-Human Mammalian Enzymes against Disease-Associated Missense Mutations
6. Post-Translational Modifications: Potential Roles in Disease and Epistatic Interactions with Pathogenic and Compensatory Mutations
7. Concluding Remarks
Funding
Conflicts of Interest
Appendix A
Appendix B
References
- Anfinsen, C.B. Principles that govern the folding of protein chains. Science 1973, 181, 223–230. [Google Scholar] [CrossRef] [PubMed]
- Hartl, F.U. Protein misfolding diseases. Annu. Rev. Biochem. 2017, 86, 21–26. [Google Scholar] [CrossRef] [PubMed]
- Powers, E.T.; Balch, W.E. Diversity in the origins of proteostasis networks—A driver for protein function in evolution. Nat. Rev. Mol. Cell Biol. 2013, 14, 237–248. [Google Scholar] [CrossRef] [PubMed]
- Sontag, E.M.; Samant, R.S.; Frydman, J. Mechanisms and functions of spatial protein quality control. Annu. Rev. Biochem. 2017, 86, 97–122. [Google Scholar] [CrossRef] [PubMed]
- Hunter, T. The age of crosstalk: Phosphorylation, ubiquitination, and beyond. Mol. Cell 2007, 28, 730–738. [Google Scholar] [CrossRef] [PubMed]
- Shendure, J.; Akey, J.M. The origins, determinants, and consequences of human mutations. Science 2015, 349, 1478–1483. [Google Scholar] [CrossRef] [PubMed]
- Simons, Y.B.; Bullaughey, K.; Hudson, R.R.; Sella, G. A population genetic interpretation of GWAS findings for human quantitative traits. PLoS Biol. 2018, 16, e2002985. [Google Scholar] [CrossRef] [PubMed]
- Good, B.H.; McDonald, M.J.; Barrick, J.E.; Lenski, R.E.; Desai, M.M. The dynamics of molecular evolution over 60,000 generations. Nature 2017, 551, 45–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xue, K.S.; Stevens-Ayers, T.; Campbell, A.P.; Englund, J.A.; Pergam, S.A.; Boeckh, M.; Bloom, J.D. Parallel evolution of influenza across multiple spatiotemporal scales. Elife 2017, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jordan, D.M.; Frangakis, S.G.; Golzio, C.; Cassa, C.A.; Kurtzberg, J.; Davis, E.E.; Sunyaev, S.R.; Katsanis, N. Identification of cis-suppression of human disease mutations by comparative genomics. Nature 2015, 524, 225–229. [Google Scholar] [CrossRef] [PubMed]
- Soskine, M.; Tawfik, D.S. Mutational effects and the evolution of new protein functions. Nat. Rev. Genet. 2010, 11, 572–582. [Google Scholar] [CrossRef] [PubMed]
- Tokuriki, N.; Tawfik, D.S. Stability effects of mutations and protein evolvability. Curr. Opin. Struct. Biol. 2009, 19, 596–604. [Google Scholar] [CrossRef] [PubMed]
- Tokuriki, N.; Tawfik, D.S. Protein dynamism and evolvability. Science 2009, 324, 203–207. [Google Scholar] [CrossRef] [PubMed]
- Mani, M.; Chen, C.; Amblee, V.; Liu, H.; Mathur, T.; Zwicke, G.; Zabad, S.; Patel, B.; Thakkar, J.; Jeffery, C.J. MoonProt: A database for proteins that are known to moonlight. Nucleic Acids Res. 2015, 43, D277–D282. [Google Scholar] [CrossRef] [PubMed]
- Diss, G.; Lehner, B. The genetic landscape of a physical interaction. Elife 2018, 7. [Google Scholar] [CrossRef] [PubMed]
- Lehner, B. Genotype to phenotype: Lessons from model organisms for human genetics. Nat. Rev. Genet. 2013, 14, 168–178. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, A.; Marguti, I.; Bechmann, I.; Jeney, V.; Chora, A.; Palha, N.R.; Rebelo, S.; Henri, A.; Beuzard, Y.; Soares, M.P. Sickle hemoglobin confers tolerance to Plasmodium infection. Cell 2011, 145, 398–409. [Google Scholar] [CrossRef] [PubMed]
- Alfonso-Sanchez, M.A.; Perez-Miranda, A.M.; Garcia-Obregon, S.; Pena, J.A. An evolutionary approach to the high frequency of the Delta F508 CFTR mutation in european populations. Med. Hypotheses 2010, 74, 989–992. [Google Scholar] [CrossRef] [PubMed]
- Storz, J.F. Compensatory mutations and epistasis for protein function. Curr. Opin. Struct. Biol. 2017, 50, 18–25. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Zhang, J. Why human disease-associated residues appear as the wild-type in other species: Genome-scale structural evidence for the compensation hypothesis. Mol. Biol. Evol. 2014, 31, 1787–1792. [Google Scholar] [CrossRef] [PubMed]
- Medina-Carmona, E.; Fuchs, J.E.; Gavira, J.A.; Mesa-Torres, N.; Neira, J.L.; Salido, E.; Palomino-Morales, R.; Burgos, M.; Timson, D.J.; Pey, A.L. Enhanced vulnerability of human proteins towards disease-associated inactivation through divergent evolution. Hum. Mol. Genet. 2017, 26, 3531–3544. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Ruiz, J.M. Protein kinetic stability. Biophys. Chem. 2010, 148, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Colon, W.; Church, J.; Sen, J.; Thibeault, J.; Trasatti, H.; Xia, K. Biological roles of protein kinetic stability. Biochemistry 2017, 56, 6179–6186. [Google Scholar] [CrossRef] [PubMed]
- Gaucher, E.A.; Govindarajan, S.; Ganesh, O.K. Palaeotemperature trend for Precambrian life inferred from resurrected proteins. Nature 2008, 451, 704–707. [Google Scholar] [CrossRef] [PubMed]
- Hart, K.M.; Harms, M.J.; Schmidt, B.H.; Elya, C.; Thornton, J.W.; Marqusee, S. Thermodynamic system drift in protein evolution. PLoS Biol. 2014, 12, e1001994. [Google Scholar] [CrossRef] [PubMed]
- Inobe, T.; Matouschek, A. Paradigms of protein degradation by the proteasome. Curr. Opin. Struct. Biol. 2014, 24, 156–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guharoy, M.; Bhowmick, P.; Sallam, M.; Tompa, P. Tripartite degrons confer diversity and specificity on regulated protein degradation in the ubiquitin-proteasome system. Nat. Commun. 2016, 7, 10239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van der Lee, R.; Lang, B.; Kruse, K.; Gsponer, J.; Sanchez de Groot, N.; Huynen, M.A.; Matouschek, A.; Fuxreiter, M.; Babu, M.M. Intrinsically disordered segments affect protein half-life in the cell and during evolution. Cell Rep. 2014, 8, 1832–1844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dikic, I. Proteasomal and autophagy degradation systems. Annu. Rev. Biochem. 2017, 86, 193–224. [Google Scholar] [CrossRef] [PubMed]
- Collins, G.A.; Goldberg, A.L. The Logic of the 26S Proteasome. Cell 2017, 169, 792–806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braten, O.; Livneh, I.; Ziv, T.; Admon, A.; Kehat, I.; Caspi, L.H.; Gonen, H.; Bercovich, B.; Godzik, A.; Jahandideh, S.; et al. Numerous proteins with unique characteristics are degraded by the 26S proteasome following monoubiquitination. Proc. Natl. Acad. Sci. USA 2016, 113, E4639–E4647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, H.; Matouschek, A. Recognition of client proteins by the proteasome. Annu. Rev. Biophys. 2017, 46, 149–173. [Google Scholar] [CrossRef] [PubMed]
- Bahar, I.; Lezon, T.R.; Yang, L.W.; Eyal, E. Global dynamics of proteins: Bridging between structure and function. Annu. Rev. Biophys. 2010, 39, 23–42. [Google Scholar] [CrossRef] [PubMed]
- Motlagh, H.N.; Wrabl, J.O.; Li, J.; Hilser, V.J. The ensemble nature of allostery. Nature 2014, 508, 331–339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tzeng, S.R.; Kalodimos, C.G. Protein activity regulation by conformational entropy. Nature 2012, 488, 236–240. [Google Scholar] [CrossRef] [PubMed]
- Campbell, E.; Kaltenbach, M.; Correy, G.J.; Carr, P.D.; Porebski, B.T.; Livingstone, E.K.; Afriat-Jurnou, L.; Buckle, A.M.; Weik, M.; Hollfelder, F.; et al. The role of protein dynamics in the evolution of new enzyme function. Nat. Chem. Biol. 2016, 12, 944–950. [Google Scholar] [CrossRef] [PubMed]
- Campbell, E.C.; Correy, G.J.; Mabbitt, P.D.; Buckle, A.M.; Tokuriki, N.; Jackson, C.J. Laboratory evolution of protein conformational dynamics. Curr.Opin. Struct. Biol. 2017, 50, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Otten, R.; Liu, L.; Kenner, L.R.; Clarkson, M.W.; Mavor, D.; Tawfik, D.S.; Kern, D.; Fraser, J.S. Rescue of conformational dynamics in enzyme catalysis by directed evolution. Nat. Commun. 2018, 9, 1314. [Google Scholar] [CrossRef] [PubMed]
- Kiel, C.; Benisty, H.; Llorens-Rico, V.; Serrano, L. The yin-yang of kinase activation and unfolding explains the peculiarity of Val600 in the activation segment of BRAF. Elife 2016, 5, e12814. [Google Scholar] [CrossRef] [PubMed]
- Tao, Y.X.; Conn, P.M. Pharmacoperones as novel therapeutics for diverse protein conformational diseases. Physiol. Rev. 2018, 98, 697–725. [Google Scholar] [CrossRef] [PubMed]
- Ben Bdira, F.; Gonzalez, E.; Pluta, P.; Lain, A.; Sanz-Parra, A.; Falcon-Perez, J.M.; Millet, O. Tuning intracellular homeostasis of human uroporphyrinogen III synthase by enzyme engineering at a single hotspot of congenital erythropoietic porphyria. Hum. Mol. Genet. 2014, 23, 5805–5813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajasekaran, N.; Sekhar, A.; Naganathan, A.N. A Universal pattern in the percolation and dissipation of protein structural perturbations. J. Phys. Chem. Lett. 2017, 8, 4779–4784. [Google Scholar] [CrossRef] [PubMed]
- Tang, Q.; Fenton, A.W. Whole-protein alanine-scanning mutagenesis of allostery: A large percentage of a protein can contribute to mechanism. Hum. Mutat. 2017, 38, 1132–1143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valentini, G.; Maggi, M.; Pey, A.L. Protein stability, folding and misfolding in human PGK1 deficiency. Biomolecules 2013, 3, 1030–1052. [Google Scholar] [CrossRef] [PubMed]
- Pey, A.L.; Majtan, T.; Sanchez-Ruiz, J.M.; Kraus, J.P. Human cystathionine beta-synthase (CBS) contains two classes of binding sites for S-adenosylmethionine (SAM): Complex regulation of CBS activity and stability by SAM. Biochem.J. 2013, 449, 109–121. [Google Scholar] [CrossRef] [PubMed]
- Vega, S.; Abian, O.; Velazquez-Campoy, A. On the link between conformational changes, ligand binding and heat capacity. Biochim. Biophys. Acta 2016, 1860, 868–878. [Google Scholar] [CrossRef] [PubMed]
- Munoz, I.G.; Morel, B.; Medina-Carmona, E.; Pey, A.L. A mechanism for cancer-associated inactivation of NQO1 due to P187S and its reactivation by the consensus mutation H80R. FEBS Lett. 2017, 591, 2826–2835. [Google Scholar] [CrossRef] [PubMed]
- Pey, A.L.; Megarity, C.F.; Medina-Carmona, E.; Timson, D.J. Natural small molecules as stabilizers and activators of cancer-associated NQO1 polymorphisms. Curr. Drug Targets 2016, 17, 1506–1514. [Google Scholar] [CrossRef] [PubMed]
- Faig, M.; Bianchet, M.A.; Talalay, P.; Chen, S.; Winski, S.; Ross, D.; Amzel, L.M. Structures of recombinant human and mouse NAD(P)H:quinone oxidoreductases: Species comparison and structural changes with substrate binding and release. Proc. Natl. Acad. Sci. USA 2000, 97, 3177–3182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pey, A.L.; Megarity, C.F.; Timson, D.J. FAD binding overcomes defects in activity and stability displayed by cancer-associated variants of human NQO1. Biochim. Biophys. Acta 2014, 1842, 2163–2173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moscovitz, O.; Tsvetkov, P.; Hazan, N.; Michaelevski, I.; Keisar, H.; Ben-Nissan, G.; Shaul, Y.; Sharon, M. A mutually inhibitory feedback loop between the 20S proteasome and its regulator, NQO1. Mol. Cell 2012, 47, 76–86. [Google Scholar] [CrossRef] [PubMed]
- Lienhart, W.D.; Gudipati, V.; Uhl, M.K.; Binter, A.; Pulido, S.A.; Saf, R.; Zangger, K.; Gruber, K.; Macheroux, P. Collapse of the native structure caused by a single amino acid exchange in human NAD(P)H:quinone oxidoreductase 1. FEBS J. 2014, 281, 4691–4704. [Google Scholar] [CrossRef] [PubMed]
- Lienhart, W.D.; Strandback, E.; Gudipati, V.; Koch, K.; Binter, A.; Uhl, M.K.; Rantasa, D.M.; Bourgeois, B.; Madl, T.; Zangger, K.; et al. Catalytic competence, structure and stability of the cancer-associated R139W variant of the human NAD(P)H:quinone oxidoreductase 1 (NQO1). FEBS J. 2017, 284, 1233–1245. [Google Scholar] [CrossRef] [PubMed]
- Medina-Carmona, E.; Palomino-Morales, R.J.; Fuchs, J.E.; Padín-Gonzalez, E.; Mesa-Torres, N.; Salido, E.; Timson, D.J.; Pey, A.L. Conformational dynamics is key to understanding loss-of-function of NQO1 cancer-associated polymorphisms and its correction by pharmacological ligands. Sci. Rep. 2016, 6, 20331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Medina-Carmona, E.; Neira, J.L.; Salido, E.; Fuchs, J.E.; Palomino-Morales, R.; Timson, D.J.; Pey, A.L. Site-to-site interdomain communication may mediate different loss-of-function mechanisms in a cancer-associated NQO1 polymorphism. Sci. Rep. 2017, 7, 44352. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Limon, A.; Alriquet, M.; Lang, W.H.; Calloni, G.; Wittig, I.; Vabulas, R.M. Recognition of enzymes lacking bound cofactor by protein quality control. Proc. Natl. Acad. Sci. USA 2016, 113, 12156–12161. [Google Scholar] [CrossRef] [PubMed]
- Tsvetkov, P.; Adamovich, Y.; Elliott, E.; Shaul, Y. E3 ligase STUB1/CHIP regulates NAD(P)H:quinone oxidoreductase 1 (NQO1) accumulation in aged brain, a process impaired in certain Alzheimer disease patients. J. Biol.Chem. 2011, 286, 8839–8845. [Google Scholar] [CrossRef] [PubMed]
- Salido, E.; Pey, A.L.; Rodriguez, R.; Lorenzo, V. Primary hyperoxalurias: Disorders of glyoxylate detoxification. Biochim. Biophys. Acta 2012, 1822, 1453–1464. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Roe, S.M.; Hou, Y.; Bartlam, M.; Rao, Z.; Pearl, L.H.; Danpure, C.J. Crystal structure of alanine:glyoxylate aminotransferase and the relationship between genotype and enzymatic phenotype in primary hyperoxaluria type 1. J. Mol. Biol. 2003, 331, 643–652. [Google Scholar] [CrossRef]
- Oppici, E.; Dindo, M.; Conter, C.; Borri Voltattorni, C.; Cellini, B. Folding defects leading to primary hyperoxaluria. Handb. Exp. Pharmacol. 2018, 245, 313–343. [Google Scholar] [PubMed]
- Birdsey, G.M.; Lewin, J.; Holbrook, J.D.; Simpson, V.R.; Cunningham, A.A.; Danpure, C.J. A comparative analysis of the evolutionary relationship between diet and enzyme targeting in bats, marsupials and other mammals. Proc. Biol. Sci. 2005, 272, 833–840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holbrook, J.D.; Birdsey, G.M.; Yang, Z.; Bruford, M.W.; Danpure, C.J. Molecular adaptation of alanine:glyoxylate aminotransferase targeting in primates. Mol. Biol. Evol. 2000, 17, 387–400. [Google Scholar] [CrossRef] [PubMed]
- Martin, W. Evolutionary origins of metabolic compartmentalization in eukaryotes. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2010, 365, 847–855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mesa-Torres, N.; Calvo, A.C.; Oppici, E.; Titelbaum, N.; Montioli, R.; Miranda-Vizuete, A.; Cellini, B.; Salido, E.; Pey, A.L. Caenorhabditis elegans AGXT-1 is a mitochondrial and temperature-adapted ortholog of peroxisomal human AGT1: New insights into between-species divergence in glyoxylate metabolism. Biochim. Biophys. Acta 2016, 1864, 1195–1205. [Google Scholar] [CrossRef] [PubMed]
- Williams, E.L.; Acquaviva, C.; Amoroso, A.; Chevalier, F.; Coulter-Mackie, M.; Monico, C.G.; Giachino, D.; Owen, T.; Robbiano, A.; Salido, E.; et al. Primary hyperoxaluria type 1: Update and additional mutation analysis of the AGXT gene. Hum. Mutat. 2009, 30, 910–917. [Google Scholar] [CrossRef] [PubMed]
- Pey, A.L.; Albert, A.; Salido, E. Protein homeostasis defects of alanine-glyoxylate aminotransferase: New therapeutic strategies in primary hyperoxaluria type I. Biomed. Res. Int. 2013, 2013, 687658. [Google Scholar] [CrossRef] [PubMed]
- Mesa-Torres, N.; Salido, E.; Pey, A.L. The lower limits for protein stability and foldability in primary hyperoxaluria type I. Biochim. Biophys. Acta 2014, 1844, 2355–2365. [Google Scholar] [CrossRef] [PubMed]
- Pey, A.L.; Salido, E.; Sanchez-Ruiz, J.M. Role of low native state kinetic stability and interaction of partially unfolded states with molecular chaperones in the mitochondrial protein mistargeting associated with primary hyperoxaluria. Amino Acids 2011, 41, 1233–1245. [Google Scholar] [CrossRef] [PubMed]
- Mesa-Torres, N.; Fabelo-Rosa, I.; Riverol, D.; Yunta, C.; Albert, A.; Salido, E.; Pey, A.L. The role of protein denaturation energetics and molecular chaperones in the aggregation and mistargeting of mutants causing primary hyperoxaluria type I. PLoS ONE 2013, 8, e71963. [Google Scholar] [CrossRef] [PubMed]
- Santana, A.; Salido, E.; Torres, A.; Shapiro, L.J. Primary hyperoxaluria type 1 in the Canary Islands: A conformational disease due to I244T mutation in the P11L-containing alanine:glyoxylate aminotransferase. Proc. Natl. Acad. Sci. USA 2003, 100, 7277–7282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albert, A.; Yunta, C.; Arranz, R.; Pena, A.; Salido, E.; Valpuesta, J.M.; Martin-Benito, J. Structure of GroEL in complex with an early folding intermediate of alanine glyoxylate aminotransferase. J. Biol.Chem. 2010, 285, 6371–6376. [Google Scholar] [CrossRef] [PubMed]
- Montioli, R.; Fargue, S.; Lewin, J.; Zamparelli, C.; Danpure, C.J.; Borri Voltattorni, C.; Cellini, B. The N-terminal extension is essential for the formation of the active dimeric structure of liver peroxisomal alanine:glyoxylate aminotransferase. Int. J. Biochem. Cell Biol. 2012, 44, 536–546. [Google Scholar] [CrossRef] [PubMed]
- Fargue, S.; Lewin, J.; Rumsby, G.; Danpure, C.J. Four of the most common mutations in primary hyperoxaluria type 1 unmask the cryptic mitochondrial targeting sequence of alanine:glyoxylate aminotransferase encoded by the polymorphic minor allele. J. Biol. Chem. 2013, 288, 2475–2484. [Google Scholar] [CrossRef] [PubMed]
- Mesa-Torres, N.; Yunta, C.; Fabelo-Rosa, I.; Gonzalez-Rubio, J.M.; Sanchez-Ruiz, J.M.; Salido, E.; Albert, A.; Pey, A.L. The consensus-based approach for gene/enzyme replacement therapies and crystallization strategies: The case of human alanine:glyoxylate aminotransferase. Biochem. J. 2014, 462, 453–463. [Google Scholar] [CrossRef] [PubMed]
- Kukreja, A.; Lasaro, M.; Cobaugh, C.; Forbes, C.; Tang, J.P.; Gao, X.; Martin-Higueras, C.; Pey, A.L.; Salido, E.; Sobolov, S.B.; et al. Development of an mRNA therapeutic for the treatment of primary hyperoxaluria type 1. Nucleic Acid Ther. 2018. submitted for publication. [Google Scholar]
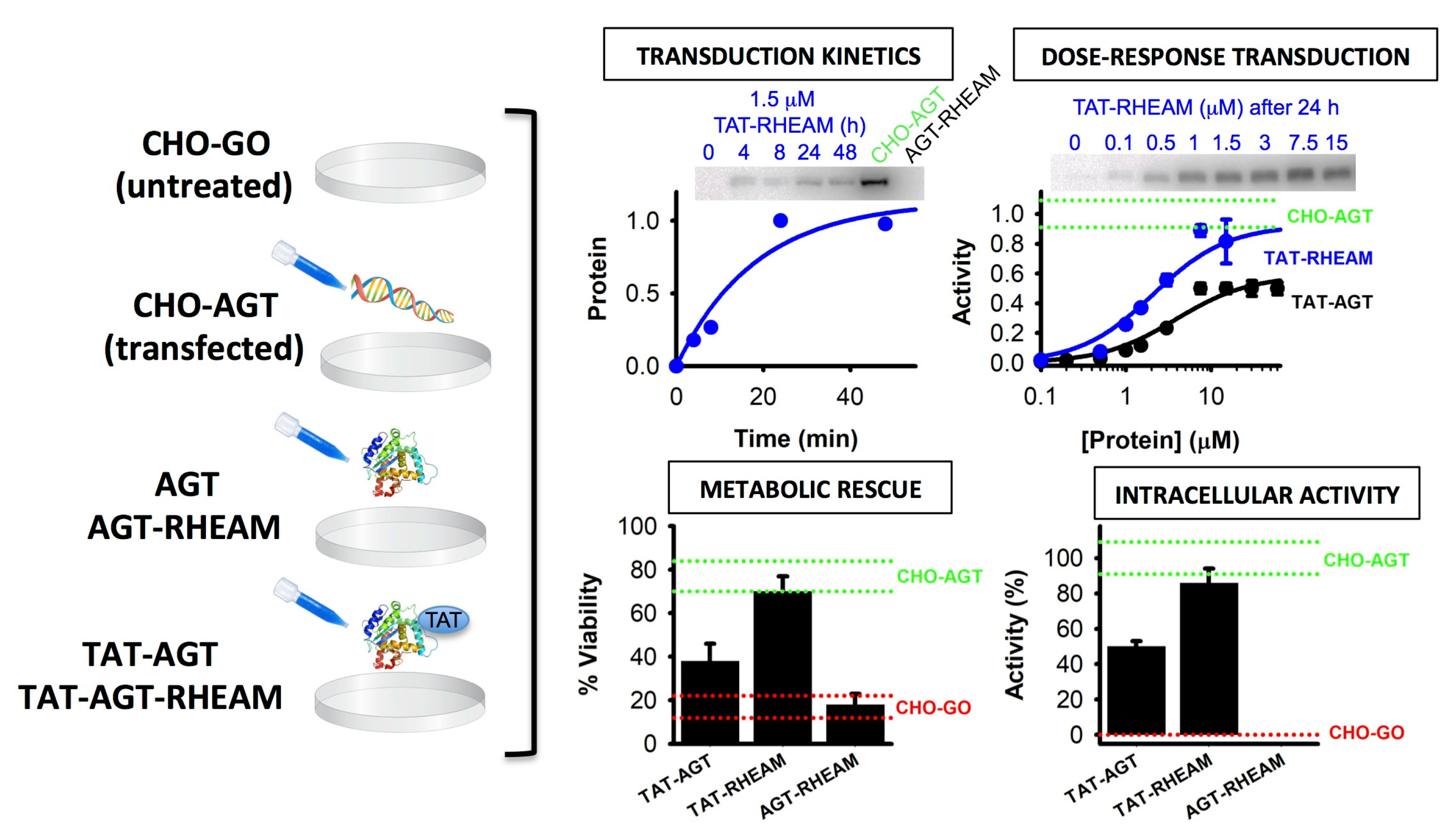
- Roncador, A.; Oppici, E.; Montioli, R.; Maset, F.; Cellini, B. TAT-mediated delivery of human alanine:glyoxylate aminotransferase in a cellular model of primary hyperoxaluria type I. Int. J. Pept. Res. Ther. 2013, 19, 175–184. [Google Scholar] [CrossRef]
- Schymkowitz, J.; Borg, J.; Stricher, F.; Nys, R.; Rousseau, F.; Serrano, L. The FoldX web server: An online force field. Nucleic Acids Res. 2005, 33, W382–W388. [Google Scholar] [CrossRef] [PubMed]
- Pey, A.L.; Padin-Gonzalez, E.; Mesa-Torres, N.; Timson, D.J. The metastability of human UDP-galactose 4’-epimerase (GALE) is increased by variants associated with type III galactosemia but decreased by substrate and cofactor binding. Arch. Biochem. Biophys. 2014, 562, 103–114. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, J.E.; Muñoz, I.G.; Timson, D.J.; Pey, A.L. Experimental and computational evidence on conformational fluctuations as a source of catalytic defects in genetic diseases. RSC Adv. 2016, 6, 58604. [Google Scholar] [CrossRef] [Green Version]
- Timson, D.J. The molecular basis of galactosemia—Past, present and future. Gene 2016, 589, 133–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erana, H.; Fernandez-Borges, N.; Elezgarai, S.R.; Harrathi, C.; Charco, J.M.; Chianini, F.; Dagleish, M.P.; Ortega, G.; Millet, O.; Castilla, J. In vitro approach to identify key amino acids in low susceptibility of rabbit prion protein to misfolding. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [PubMed]
- Henriques, S.F.; Patissier, C.; Bourg, N.; Fecchio, C.; Sandona, D.; Marsolier, J.; Richard, I. Different outcome of sarcoglycan missense mutation between human and mouse. PLoS ONE 2018, 13, e0191274. [Google Scholar] [CrossRef] [PubMed]
- Hornbeck, P.V.; Zhang, B.; Murray, B.; Kornhauser, J.M.; Latham, V.; Skrzypek, E. PhosphoSitePlus, 2014: Mutations, PTMs and recalibrations. Nucleic Acids Res. 2015, 43, D512–D520. [Google Scholar] [CrossRef] [PubMed]
- Lumb, M.J.; Drake, A.F.; Danpure, C.J. Effect of N-terminal alpha-helix formation on the dimerization and intracellular targeting of alanine:glyoxylate aminotransferase. J. Biol.Chem. 1999, 274, 20587–20596. [Google Scholar] [CrossRef] [PubMed]
- Starr, T.N.; Thornton, J.W. Epistasis in protein evolution. Prot. Sci. 2016, 25, 1204–1218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oppici, E.; Roncador, A.; Montioli, R.; Bianconi, S.; Cellini, B. Gly161 mutations associated with primary hyperoxaluria type I induce the cytosolic aggregation and the intracellular degradation of the apo-form of alanine:glyoxylate aminotransferase. Biochim. Biophys. Acta 2013, 1832, 2277–2288. [Google Scholar] [CrossRef] [PubMed]
- Cellini, B.; Bertoldi, M.; Montioli, R.; Paiardini, A.; Borri Voltattorni, C. Human wild-type alanine:glyoxylate aminotransferase and its naturally occurring G82E variant: Functional properties and physiological implications. Biochem. J. 2007, 408, 39–50. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Rahman, S.; Choudhry, H.; Zamzami, M.A.; Sarwar Jamal, M.; Islam, A.; Ahmad, F.; Hassan, M.I. Computing disease-linked SOD1 mutations: Deciphering protein stability and patient-phenotype relations. Sci. Rep. 2017, 7, 4678. [Google Scholar] [CrossRef] [PubMed]







© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mesa-Torres, N.; Betancor-Fernández, I.; Oppici, E.; Cellini, B.; Salido, E.; Pey, A.L. Evolutionary Divergent Suppressor Mutations in Conformational Diseases. Genes 2018, 9, 352. https://doi.org/10.3390/genes9070352
Mesa-Torres N, Betancor-Fernández I, Oppici E, Cellini B, Salido E, Pey AL. Evolutionary Divergent Suppressor Mutations in Conformational Diseases. Genes. 2018; 9(7):352. https://doi.org/10.3390/genes9070352
Chicago/Turabian StyleMesa-Torres, Noel, Isabel Betancor-Fernández, Elisa Oppici, Barbara Cellini, Eduardo Salido, and Angel L. Pey. 2018. "Evolutionary Divergent Suppressor Mutations in Conformational Diseases" Genes 9, no. 7: 352. https://doi.org/10.3390/genes9070352
APA StyleMesa-Torres, N., Betancor-Fernández, I., Oppici, E., Cellini, B., Salido, E., & Pey, A. L. (2018). Evolutionary Divergent Suppressor Mutations in Conformational Diseases. Genes, 9(7), 352. https://doi.org/10.3390/genes9070352