Epistatic Interactions in NS5A of Hepatitis C Virus Suggest Drug Resistance Mechanisms

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
3. Results
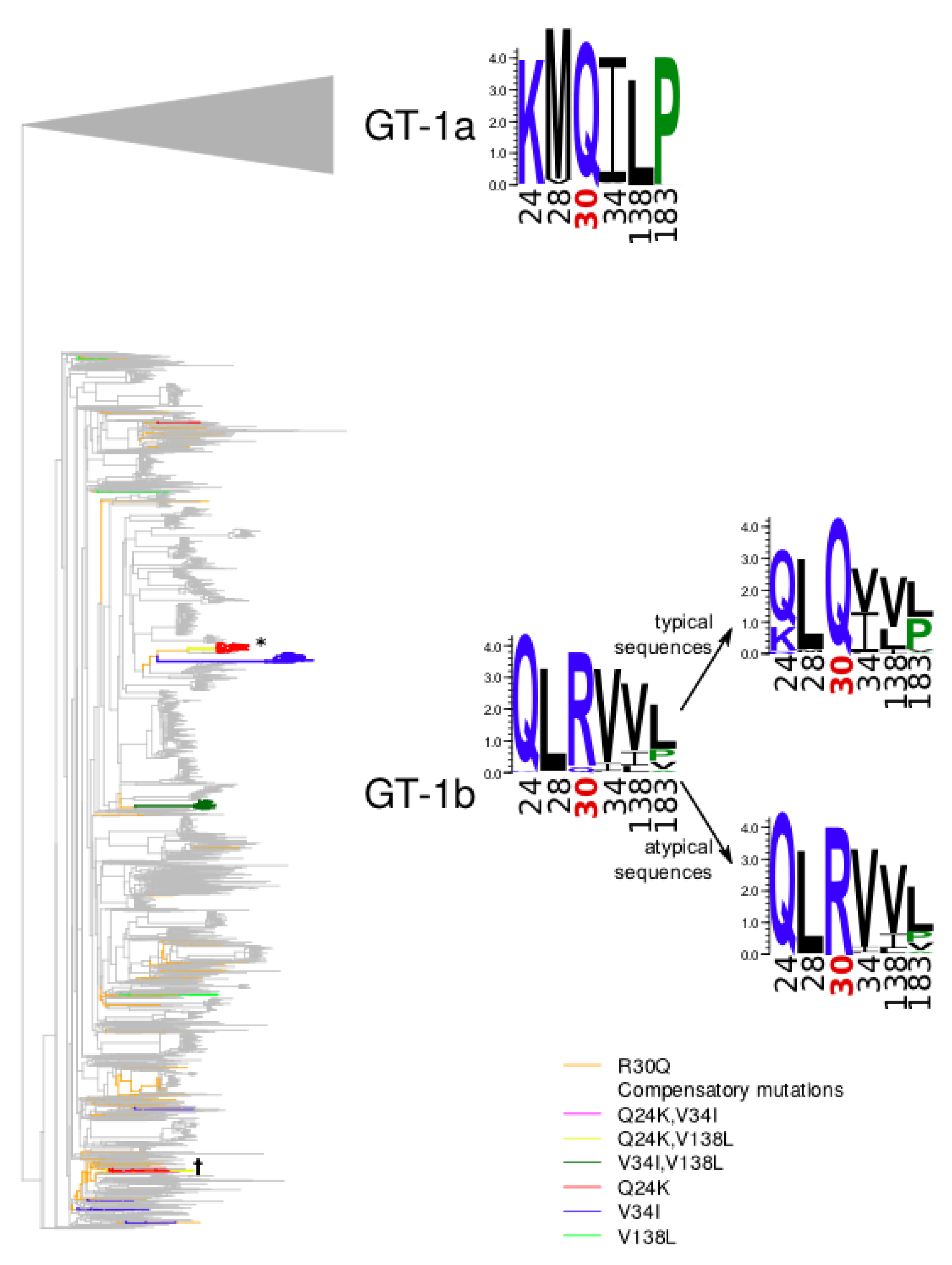
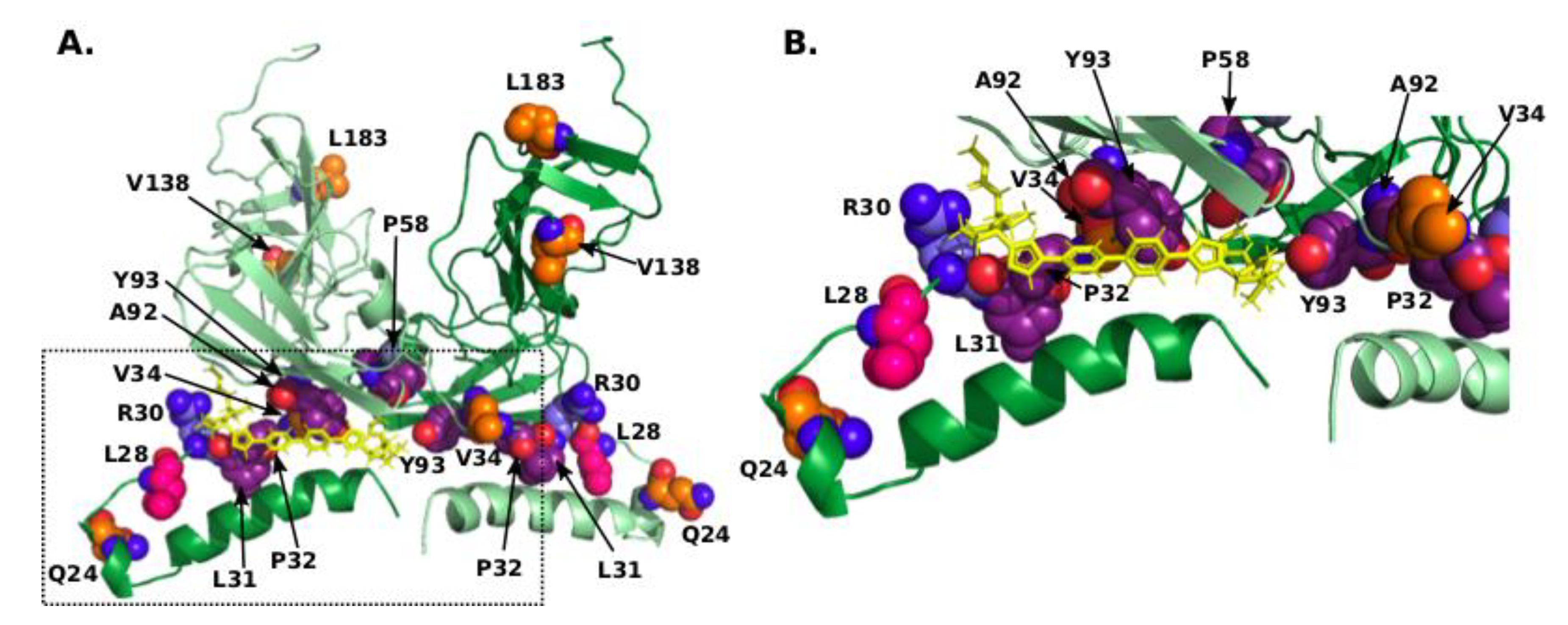
3.1. Identification of Mutations Associated with GT-1b R30Q
3.2. Analysis of Compensatory Mutations in GT1a-Failing Patients
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Manns, M.; Marcellin, P.; Poordad, F.; de Araujo, E.S.; Buti, M.; Horsmans, Y.; Janczewska, E.; Villamil, F.; Scott, J.; Peeters, M.; et al. Simeprevir with pegylated interferon ALFA 2a or 2b plus ribavirin in treatment-naive patients with chronic hepatitis C virus genotype 1 infection (QUEST-2): A randomised, double-blind, placebo-controlled phase 3 trial. Lancet 2014, 384, 414–426. [Google Scholar] [CrossRef]
- Spengler, U. Direct antiviral agents (DAAs)—A new age in the treatment of hepatitis C virus infection. Pharmacol. Ther. 2017, 183, 118–126. [Google Scholar] [CrossRef] [PubMed]
- WHO. Global Hepatitis Report 2017. Available online: http://apps.who.int/iris/bitstream/10665/255016/1/9789241565455-eng.pdf?ua=1 (accessed on 28 November 2017).
- Smith, D.B.; Bukh, J.; Kuiken, C.; Muerhoff, A.S.; Rice, C.M.; Stapleton, J.T.; Simmonds, P. Expanded classification of hepatitis C virus into 7 genotypes and 67 subtypes: Updated criteria and genotype assignment web resource. Hepatology 2014, 59, 318–327. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Valera, L.; Jia, L.; Kirk, M.J.; Gao, M.; Fridell, R.A. In vitro activity of daclatasvir on hepatitis C virus genotype 3 NS5A. Antimicrob. Agents Chemother. 2013, 57, 611–613. [Google Scholar] [CrossRef] [PubMed]
- Cordek, D.G.; Bechtel, J.T.; Maynard, A.T.; Kazmierski, W.M.; Cameron, C.E. Targeting the NS5A protein of HCV: An emerging option. Drugs Future 2011, 36, 691–711. [Google Scholar] [CrossRef] [PubMed]
- Kohler, J.J.; Nettles, J.H.; Amblard, F.; Hurwitz, S.J.; Bassit, L.; Stanton, R.A.; Ehteshami, M.; Schinazi, R.F. Approaches to hepatitis C treatment and cure using NS5A inhibitors. Infect. Drug Resist. 2014, 7, 41–56. [Google Scholar] [PubMed]
- European Association for the Study of the Liver. EASL recommendations on treatment of hepatitis C 2016. J. Hepatol. 2017, 66, 153–194. [Google Scholar]
- Gitto, S.; Gamal, N.; Andreone, P. NS5A inhibitors for the treatment of hepatitis C infection. J. Viral Hepat. 2017, 24, 180–186. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Curry, S.; McMonagle, P.; Yeh, W.W.; Ludmerer, S.W.; Jumes, P.A.; Marshall, W.L.; Kong, S.; Ingravallo, P.; Black, S.; et al. Susceptibilities of genotype 1a, 1b, and 3 hepatitis C virus variants to the NS5A inhibitor elbasvir. Antimicrob. Agents Chemother. 2015, 59, 6922–6929. [Google Scholar] [CrossRef] [PubMed]
- Paolucci, S.; Premoli, M.; Novati, S.; Gulminetti, R.; Maserati, R.; Barbarini, G.; Sacchi, P.; Piralla, A.; Sassera, D.; De Marco, L.; et al. Baseline and breakthrough resistance mutations in HCV patients failing DAAs. Sci. Rep. 2017, 7, 16017. [Google Scholar] [CrossRef] [PubMed]
- Dietz, J.; Susser, S.; Berkowski, C.; Perner, D.; Zeuzem, S.; Sarrazin, C. Consideration of viral resistance for optimization of direct antiviral therapy of hepatitis C virus genotype 1-infected patients. PLoS ONE 2015, 10, e0134395. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, D.; Zhou, N.; Ueland, J.; Monikowski, A.; McPhee, F. Natural prevalence of NS5A polymorphisms in subjects infected with hepatitis C virus genotype 3 and their effects on the antiviral activity of NS5A inhibitors. J. Clin. Virol. 2013, 57, 13–18. [Google Scholar] [CrossRef] [PubMed]
- Walker, A.; Siemann, H.; Groten, S.; Ross, R.S.; Scherbaum, N.; Timm, J. Natural prevalence of resistance-associated variants in hepatitis C virus NS5A in genotype 3a-infected people who inject drugs in Germany. J. Clin. Virol. 2015, 70, 43–45. [Google Scholar] [CrossRef] [PubMed]
- Wyles, D.; Poordad, F.; Wang, S.; Alric, L.; Felizarta, F.; Kwo, P.Y.; Maliakkal, B.; Agarwal, K.; Hassanein, T.; Weilert, F.; et al. Surveyor-II, Part 3: Efficacy and Safety of Glecaprevir/Pibrentasvir (abt-493/abt-530) in Patients with Hepatitis C Virus Genotype 3 Infection with Prior Treatment Experience And/Or Cirrhosis; The Liver Meeting from the American Association for the Study of Liver Diseases (AASLD); AASLD: Boston, MA, USA, 2016; p. #113. [Google Scholar]
- Suzuki, F.; Sezaki, H.; Akuta, N.; Suzuki, Y.; Seko, Y.; Kawamura, Y.; Hosaka, T.; Kobayashi, M.; Saito, S.; Arase, Y.; et al. Prevalence of hepatitis C virus variants resistant to NS3 protease inhibitors or the NS5A inhibitor (BMS-790052) in hepatitis patients with genotype 1b. J. Clin. Virol. 2012, 54, 352–354. [Google Scholar] [CrossRef] [PubMed]
- Lok, A.S.; Gardiner, D.F.; Hezode, C.; Lawitz, E.J.; Bourliere, M.; Everson, G.T.; Marcellin, P.; Rodriguez-Torres, M.; Pol, S.; Serfaty, L.; et al. Randomized trial of daclatasvir and asunaprevir with or without PegIFN/RBV for hepatitis C virus genotype 1 null responders. J. Hepatol. 2014, 60, 490–499. [Google Scholar] [CrossRef] [PubMed]
- McPhee, F.; Hernandez, D.; Zhou, N.; Yu, F.; Ueland, J.; Monikowski, A.; Chayama, K.; Toyota, J.; Izumi, N.; Yokosuka, O.; et al. Virological escape in HCV genotype-1-infected patients receiving daclatasvir plus ribavirin and peginterferon alfa-2a or alfa-2b. Antivir. Ther. 2014, 19, 479–490. [Google Scholar] [CrossRef] [PubMed]
- Murakami, E.; Imamura, M.; Hayes, C.N.; Abe, H.; Hiraga, N.; Honda, Y.; Ono, A.; Kosaka, K.; Kawaoka, T.; Tsuge, M.; et al. Ultradeep sequencing study of chronic hepatitis C virus genotype 1 infection in patients treated with daclatasvir, peginterferon, and ribavirin. Antimicrob. Agents Chemother. 2014, 58, 2105–2112. [Google Scholar] [CrossRef] [PubMed]
- Coburn, C.A.; Meinke, P.T.; Chang, W.; Fandozzi, C.M.; Graham, D.J.; Hu, B.; Huang, Q.; Kargman, S.; Kozlowski, J.; Liu, R.; et al. Discovery of MK-8742: An HCV NS5A inhibitor with broad genotype activity. ChemMedChem 2013, 8, 1930–1940. [Google Scholar] [CrossRef] [PubMed]
- Forns, X.; Gordon, S.C.; Zuckerman, E.; Lawitz, E.; Calleja, J.L.; Hofer, H.; Gilbert, C.; Palcza, J.; Howe, A.Y.; DiNubile, M.J.; et al. Grazoprevir and elbasvir plus ribavirin for chronic HCV genotype-1 infection after failure of combination therapy containing a direct-acting antiviral agent. J. Hepatol. 2015, 63, 564–572. [Google Scholar] [CrossRef] [PubMed]
- Sulkowski, M.S.; Eron, J.J.; Wyles, D.; Trinh, R.; Lalezari, J.; Wang, C.; Slim, J.; Bhatti, L.; Gathe, J.; Ruane, P.J.; et al. Ombitasvir, paritaprevir co-dosed with ritonavir, dasabuvir, and ribavirin for hepatitis C in patients co-infected with HIV-1: A randomized trial. JAMA 2015, 313, 1223–1231. [Google Scholar] [CrossRef] [PubMed]
- Zeuzem, S.; Ghalib, R.; Reddy, K.R.; Pockros, P.J.; Ben Ari, Z.; Zhao, Y.; Brown, D.D.; Wan, S.; DiNubile, M.J.; Nguyen, B.Y.; et al. Grazoprevir-Elbasvir combination therapy for treatment-naive cirrhotic and noncirrhotic patients with chronic hepatitis C virus genotype 1, 4, or 6 infection: A randomized trial. Ann. Intern. Med. 2015, 163, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, P.; Tripathi, R.; Schnell, G.; Reisch, T.; Beyer, J.; Irvin, M.; Xie, W.; Larsen, L.; Cohen, D.; Podsadecki, T.; et al. Resistance analysis of baseline and treatment-emergent variants in hepatitis C virus genotype 1 in the AVIATOR study with paritaprevir-ritonavir, ombitasvir, and dasabuvir. Antimicrob. Agents Chemother. 2015, 59, 5445–5454. [Google Scholar] [CrossRef] [PubMed]
- Sulkowski, M.; Ghalib, R.; Rodriguez-Torres, M.; Younoss, I.Z.; Corregidor, A.; Fevery, B.; Callewaert, K.; Symonds, B.; De La Rosa, G.; Picchio, G.; et al. Once-daily simeprevir (TMC435) plus sofosbuvir (GS-7977) with or without ribavirin in HCV genotype 1 prior null responders with metavir F0-2: Cosmos study subgroup analysis. J. Hepatol. 2014, 60, S4. [Google Scholar] [CrossRef]
- Wong, K.A.; Worth, A.; Martin, R.; Svarovskaia, E.; Brainard, D.M.; Lawitz, E.; Miller, M.D.; Mo, H. Characterization of Hepatitis C virus resistance from a multiple-dose clinical trial of the novel NS5A inhibitor GS-5885. Antimicrob. Agents Chemother. 2013, 57, 6333–6340. [Google Scholar] [CrossRef] [PubMed]
- Tellinghuisen, T.L.; Marcotrigiano, J.; Rice, C.M. Structure of the zinc-binding domain of an essential component of the hepatitis C virus replicase. Nature 2005, 435, 374–379. [Google Scholar] [CrossRef] [PubMed]
- Love, R.A.; Brodsky, O.; Hickey, M.J.; Wells, P.A.; Cronin, C.N. Crystal structure of a novel dimeric form of NS5A domain I protein from hepatitis C virus. J. Virol. 2009, 83, 4395–4403. [Google Scholar] [CrossRef] [PubMed]
- Penin, F.; Brass, V.; Appel, N.; Ramboarina, S.; Montserret, R.; Ficheux, D.; Blum, H.E.; Bartenschlager, R.; Moradpour, D. Structure and function of the membrane anchor domain of hepatitis C virus nonstructural protein 5A. J. Biol. Chem. 2004, 279, 40835–40843. [Google Scholar] [CrossRef] [PubMed]
- Bartenschlager, R.; Lohmann, V.; Penin, F. The molecular and structural basis of advanced antiviral therapy for hepatitis C virus infection. Nat. Rev. Microbiol. 2013, 11, 482–496. [Google Scholar] [CrossRef] [PubMed]
- Ascher, D.B.; Wielens, J.; Nero, T.L.; Doughty, L.; Morton, C.J.; Parker, M.W. Potent hepatitis C inhibitors bind directly to NS5A and reduce its affinity for RNA. Sci. Rep. 2014, 4, 4765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Targett-Adams, P.; Graham, E.J.; Middleton, J.; Palmer, A.; Shaw, S.M.; Lavender, H.; Brain, P.; Tran, T.D.; Jones, L.H.; Wakenhut, F.; et al. Small molecules targeting hepatitis C virus-encoded NS5A cause subcellular redistribution of their target: Insights into compound modes of action. J. Virol. 2011, 85, 6353–6368. [Google Scholar] [CrossRef] [PubMed]
- Barakat, K.H.; Anwar-Mohamed, A.; Tuszynski, J.A.; Robins, M.J.; Tyrrell, D.L.; Houghton, M. A Refined Model of the HCV NS5A protein bound to daclatasvir explains drug-resistant mutations and activity against divergent genotypes. J. Chem. Inf. Model. 2015, 55, 362–373. [Google Scholar] [CrossRef] [PubMed]
- Kazmierski, W.M.; Maynard, A.; Duan, M.; Baskaran, S.; Botyanszki, J.; Crosby, R.; Dickerson, S.; Tallant, M.; Grimes, R.; Hamatake, R.; et al. Novel spiroketal pyrrolidine GSK2336805 potently inhibits key hepatitis C virus genotype 1b mutants: From lead to clinical compound. J. Med. Chem. 2014, 57, 2058–2073. [Google Scholar] [CrossRef] [PubMed]
- O’Boyle Ii, D.R.; Sun, J.H.; Nower, P.T.; Lemm, J.A.; Fridell, R.A.; Wang, C.; Romine, J.L.; Belema, M.; Nguyen, V.N.; Laurent, D.R.; et al. Characterizations of HCV NS5A replication complex inhibitors. Virology 2013, 444, 343–354. [Google Scholar] [CrossRef] [PubMed]
- Lambert, S.M.; Langley, D.R.; Garnett, J.A.; Angell, R.; Hedgethorne, K.; Meanwell, N.A.; Matthews, S.J. The crystal structure of NS5A domain 1 from genotype 1a reveals new clues to the mechanism of action for dimeric HCV inhibitors. Protein Sci. 2014, 23, 723–734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nettles, J.H.; Stanton, R.A.; Broyde, J.; Amblard, F.; Zhang, H.; Zhou, L.; Shi, J.; McBrayer, T.R.; Whitaker, T.; Coats, S.J.; et al. Asymmetric binding to NS5A by daclatasvir (BMS-790052) and analogs suggests two novel modes of HCV inhibition. J. Med. Chem. 2014, 57, 10031–10043. [Google Scholar] [CrossRef] [PubMed]
- Kuiken, C.; Yusim, K.; Boykin, L.; Richardson, R. The Los Alamos HCV Sequence Database. Bioinformatics 2005, 21, 379–384. [Google Scholar] [CrossRef] [PubMed]
- Ranwez, V.; Harispe, S.; Delsuc, F.; Douzery, E.J. MACSE: Multiple Alignment of Coding SEquences accounting for frameshifts and stop codons. PLoS ONE 2011, 6, e22594. [Google Scholar] [CrossRef] [PubMed]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [PubMed]
- Brandt, B.W.; Feenstra, K.A.; Heringa, J. Multi-Harmony: Detecting functional specificity from sequence alignment. Nucleic Acids Res. 2010, 38, W35–W40. [Google Scholar] [CrossRef] [PubMed]
- Mazin, P.V.; Gelfand, M.S.; Mironov, A.A.; Rakhmaninova, A.B.; Rubinov, A.R.; Russell, R.B.; Kalinina, O.V. An automated stochastic approach to the identification of the protein specificity determinants and functional subfamilies. Algorithms Mol. Biol. 2010, 5, 29. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef] [PubMed]
- Knops, E.; Kalaghatgi, P.; Neumann-Fraune, M.; Heger, E.; Schülter, E.; Lengauer, T.; Keitel, V.; Goeser, T.; Schübel, N.; von Hahn, T.; et al. Hepatitis C virus screening project of patients on current anti-HCV Therapy. J. Hepatol. 2016, 64, S402. [Google Scholar] [CrossRef]
- Kalaghatgi, P.; Sikorski, A.M.; Knops, E.; Rupp, D.; Sierra, S.; Heger, E.; Neumann-Fraune, M.; Beggel, B.; Walker, A.; Timm, J.; et al. Geno2pheno[HCV]—A Web-based interpretation system to support hepatitis C treatment decisions in the era of direct-acting antiviral agents. PLoS ONE 2016, 11, e0155869. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, P.; Schnell, G.; Tripathi, R.; Beyer, J.; Reisch, T.; Zhang, X.; Setze, C.; Rodrigues, L., Jr.; Burroughs, M.; Redman, R.; et al. Analysis of hepatitis C virus genotype 1b resistance variants in japanese patients treated with paritaprevir-ritonavir and ombitasvir. Antimicrob. Agents Chemother. 2016, 60, 1106–1113. [Google Scholar] [CrossRef] [PubMed]
- Teraoka, Y.; Uchida, T.; Imamura, M.; Osawa, M.; Tsuge, M.; Abe-Chayama, H.; Hayes, C.N.; Makokha, G.N.; Aikata, H.; Miki, D.; et al. Prevalence of NS5A resistance associated variants in NS5A inhibitor treatment failures and an effective treatment for NS5A-P32 deleted hepatitis C virus in humanized mice. Biochem. Biophys. Res. Commun. 2018, 500, 152–157. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, P.; Schnell, G.; Tripathi, R.; Beyer, J.; Reisch, T.; Dekhtyar, T.; Irvin, M.; Xie, W.; Fu, B.; Burroughs, M.; et al. Integrated Resistance Analysis of CERTAIN-1 and CERTAIN-2 studies in hepatitis C virus-infected patients receiving glecaprevir and pibrentasvir in Japan. Antimicrob. Agents Chemother. 2018, 62. [Google Scholar] [CrossRef] [PubMed]
- Macdonald, A.; Crowder, K.; Street, A.; McCormick, C.; Harris, M. The hepatitis C virus NS5A protein binds to members of the Src family of tyrosine kinases and regulates kinase activity. J. Gen. Virol. 2004, 85, 721–729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Macdonald, A.; Mazaleyrat, S.; McCormick, C.; Street, A.; Burgoyne, N.J.; Jackson, R.M.; Cazeaux, V.; Shelton, H.; Saksela, K.; Harris, M. Further studies on hepatitis C virus NS5A-SH3 domain interactions: Identification of residues critical for binding and implications for viral RNA replication and modulation of cell signalling. J. Gen. Virol. 2005, 86, 1035–1044. [Google Scholar] [CrossRef] [PubMed]
- Koharudin, L.M.; Furey, W.; Liu, H.; Liu, Y.J.; Gronenborn, A.M. The phox domain of sorting nexin 5 lacks phosphatidylinositol 3-phosphate (PtdIns(3)P) specificity and preferentially binds to phosphatidylinositol 4,5-bisphosphate (PtdIns(4,5)P2). J. Biol. Chem. 2009, 284, 23697–23707. [Google Scholar] [CrossRef] [PubMed]
- Yin, C.; Goonawardane, N.; Stewart, H.; Harris, M. A role for domain I of the hepatitis C virus NS5A protein in virus assembly. PLoS Pathog. 2018, 14, e1006834. [Google Scholar] [CrossRef] [PubMed]
- Bloom, J.D.; Gong, L.I.; Baltimore, D. Permissive secondary mutations enable the evolution of influenza oseltamivir resistance. Science 2010, 328, 1272–1275. [Google Scholar] [CrossRef] [PubMed]
- Chang, M.W.; Torbett, B.E. Accessory mutations maintain stability in drug-resistant HIV-1 protease. J. Mol. Biol. 2011, 410, 756–760. [Google Scholar] [CrossRef] [PubMed]
- Vogt, D.A.; Camus, G.; Herker, E.; Webster, B.R.; Tsou, C.L.; Greene, W.C.; Yen, T.S.; Ott, M. Lipid droplet-binding protein TIP47 regulates hepatitis C Virus RNA replication through interaction with the viral NS5A protein. PLoS Pathog. 2013, 9, e1003302. [Google Scholar] [CrossRef] [PubMed]
- Chayama, K.; Hayes, C.N. HCV drug resistance challenges in Japan: The role of pre-existing variants and emerging resistant strains in direct acting antiviral therapy. Viruses 2015, 7, 5328–5342. [Google Scholar] [CrossRef] [PubMed]
- Maisnier-Patin, S.; Andersson, D.I. Adaptation to the deleterious effects of antimicrobial drug resistance mutations by compensatory evolution. Res. Microbiol. 2004, 155, 360–369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delaney, W.E.t.; Yang, H.; Westland, C.E.; Das, K.; Arnold, E.; Gibbs, C.S.; Miller, M.D.; Xiong, S. The hepatitis B virus polymerase mutation rtV173L is selected during lamivudine therapy and enhances viral replication in vitro. J. Virol. 2003, 77, 11833–11841. [Google Scholar] [CrossRef] [PubMed]
- Levin, B.R.; Perrot, V.; Walker, N. Compensatory mutations, antibiotic resistance and the population genetics of adaptive evolution in bacteria. Genetics 2000, 154, 985–997. [Google Scholar] [PubMed]
- Verheyen, J.; Litau, E.; Sing, T.; Daumer, M.; Balduin, M.; Oette, M.; Fatkenheuer, G.; Rockstroh, J.K.; Schuldenzucker, U.; Hoffmann, D.; et al. Compensatory mutations at the HIV cleavage sites p7/p1 and p1/p6-gag in therapy-naive and therapy-experienced patients. Antivir. Ther. 2006, 11, 879–887. [Google Scholar] [PubMed]
- Nijhuis, M.; Schuurman, R.; de Jong, D.; Erickson, J.; Gustchina, E.; Albert, J.; Schipper, P.; Gulnik, S.; Boucher, C.A. Increased fitness of drug resistant HIV-1 protease as a result of acquisition of compensatory mutations during suboptimal therapy. AIDS 1999, 13, 2349–2359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez-Picado, J.; Savara, A.V.; Sutton, L.; D’Aquila, R.T. Replicative fitness of protease inhibitor-resistant mutants of human immunodeficiency virus type 1. J. Virol. 1999, 73, 3744–3752. [Google Scholar] [PubMed]
- Huigen, M.C.D.G.; Albert, J.; Lindström, A.; Ohlis, A.; Bratt, G.; de Graaf, L.; Nijhuis, M.; Boucher, C.A.B. Compensatory fixation explains long term persistence of the M41L in HIV-1 reverse transcriptase in a large transmission cluster. Antivir. Ther. 2006, 11, S113. [Google Scholar]
- McCloskey, R.M.; Liang, R.H.; Joy, J.B.; Krajden, M.; Montaner, J.S.; Harrigan, P.R.; Poon, A.F. Global origin and transmission of hepatitis C virus nonstructural protein 3 Q80K polymorphism. J. Infect. Dis. 2015, 211, 1288–1295. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
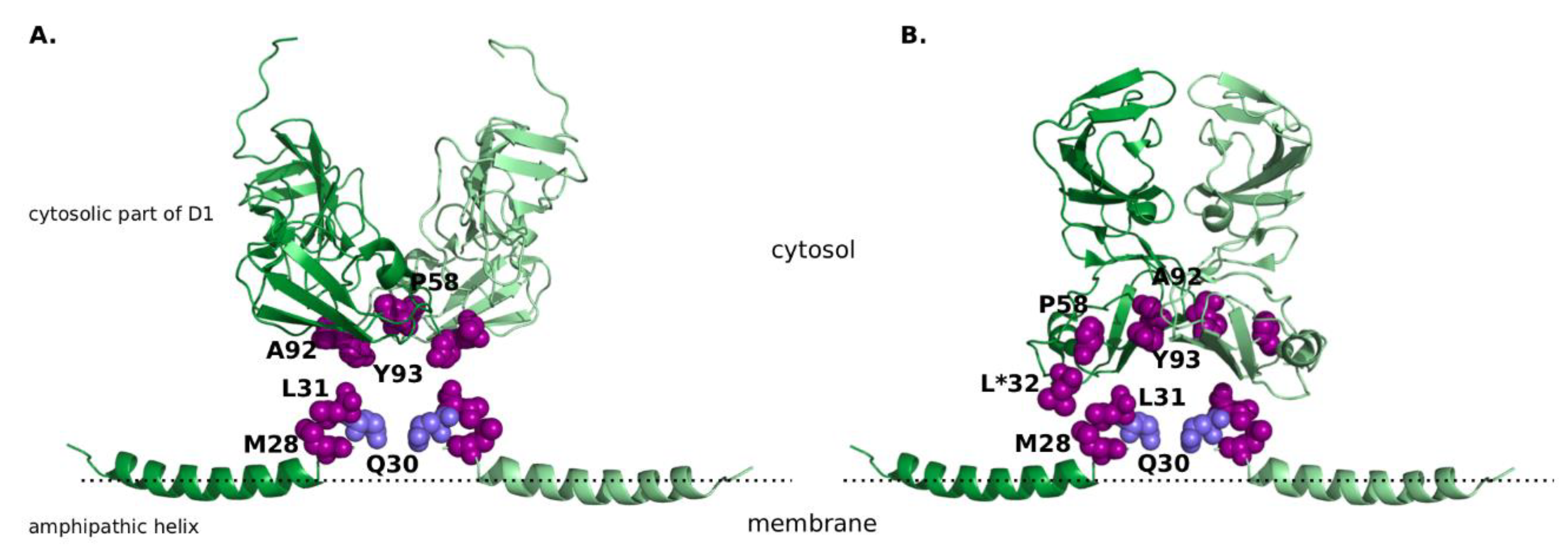
| Residue Number | Residue Identity in GT-1a | Residue Identity in GT-1b | Residue Identity in Experimentally Resolved Structures |
|---|---|---|---|
| 28 | M | L | M (1R7E) |
| 30 | Q | R | Q (1R7E) |
| 31 | L | L | L (1R7E) |
| 32 | P | P | L 1 (3FQM) |
| 58 | H | P | P (1ZH1, 3FQM) |
| 92 | A | A | A (1ZH1, 3FQM) |
| 93 | Y | Y | Y (1ZH1, 3FQM) |
| Mutation | Atypical Set | Typical Set | p-value (Fisher’s Exact Test) | Phylogeny-Aware Statistical Significance | ||
|---|---|---|---|---|---|---|
| # mutated aa | # WT aa | # mutated aa | # WT aa | |||
| V8I | 4 | 137 | 285 | 2351 | 3.977 × 10−3 | 0.962 |
| Q24K | 33 | 108 | 3 | 2633 | 1.754 × 10−121 | 0.000 |
| L28M | 5 | 136 | 13 | 2623 | 1.120 × 10-4 | 0.010 |
| V34I | 66 | 75 | 144 | 2492 | 7.108 × 10−72 | 0.000 |
| K44R | 9 | 132 | 377 | 2259 | 0.012 | 0.930 |
| Q54H | 13 | 128 | 850 | 1786 | 1.492 × 10−8 | 0.999 |
| T83M | 7 | 134 | 362 | 2274 | 4.221 × 10−3 | 0.886 |
| S107T | 1 | 140 | 128 | 2508 | 0.038 | 0.848 |
| V121I | 4 | 137 | 127 | 2509 | 0.380 | 0.595 |
| T122R | 3 | 138 | 42 | 2594 | 0.883 | 0.200 |
| V138L | 38 | 103 | 187 | 2449 | 1.455 × 10−16 | 0.002 |
| V153L | 10 | 131 | 385 | 2251 | 0.018 | 0.924 |
| D171E | 65 | 76 | 1219 | 1417 | 1.000 | 0.188 |
| N180H | 8 | 133 | 219 | 2417 | 0.340 | 0.448 |
| L183P | 60 | 81 | 426 | 2210 | 2.340 × 10−15 | 0.005 |
| Tool | Atypical GT-1b vs. Typical GT-1b | GT-1a vs. GT-1b |
|---|---|---|
| Multi-Harmony | 7, 8, 17, 24, 25, 26, 34, 37, 44, 48, 49, 52, 54, 55, 56, 62, 69, 73, 74, 75, 78, 83, 85, 101, 123, 138, 164, 174, 181, 183 | 7, 8, 14, 17, 21, 24, 25, 26, 28, 30, 34, 36, 37, 44, 46, 48, 54, 56, 58, 62, 64, 68, 71, 78, 81, 83, 85, 92, 93, 97, 101, 107, 108, 114, 116, 117, 121, 123, 124, 135, 137, 138, 146, 153, 158, 161, 164, 166, 171, 174, 176, 180, 181, 183, 222 |
| SDPfox | 24, 30, 37, 62, 164, 174, 181 | 8, 24, 25, 30, 114 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Knops, E.; Sierra, S.; Kalaghatgi, P.; Heger, E.; Kaiser, R.; Kalinina, O.V. Epistatic Interactions in NS5A of Hepatitis C Virus Suggest Drug Resistance Mechanisms. Genes 2018, 9, 343. https://doi.org/10.3390/genes9070343
Knops E, Sierra S, Kalaghatgi P, Heger E, Kaiser R, Kalinina OV. Epistatic Interactions in NS5A of Hepatitis C Virus Suggest Drug Resistance Mechanisms. Genes. 2018; 9(7):343. https://doi.org/10.3390/genes9070343
Chicago/Turabian StyleKnops, Elena, Saleta Sierra, Prabhav Kalaghatgi, Eva Heger, Rolf Kaiser, and Olga V. Kalinina. 2018. "Epistatic Interactions in NS5A of Hepatitis C Virus Suggest Drug Resistance Mechanisms" Genes 9, no. 7: 343. https://doi.org/10.3390/genes9070343
APA StyleKnops, E., Sierra, S., Kalaghatgi, P., Heger, E., Kaiser, R., & Kalinina, O. V. (2018). Epistatic Interactions in NS5A of Hepatitis C Virus Suggest Drug Resistance Mechanisms. Genes, 9(7), 343. https://doi.org/10.3390/genes9070343