Application of Whole Exome and Targeted Panel Sequencing in the Clinical Molecular Diagnosis of 319 Chinese Families with Inherited Retinal Dystrophy and Comparison Study

 ,
,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Patients
2.3. Design of Targeted Capture Panel
2.4. Whole Exome Sequencing
2.5. Next-Generation Sequencing Data Analysis
2.6. Variant Classification and Validation
2.7. Regions of Low Coverage with Next Generation Sequencing
3. Results
3.1. Patients
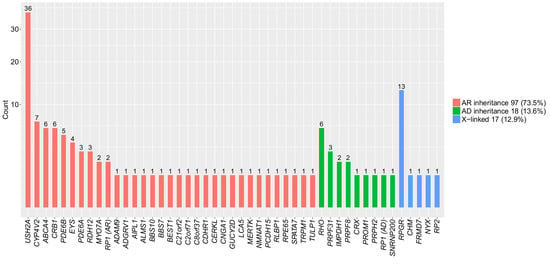
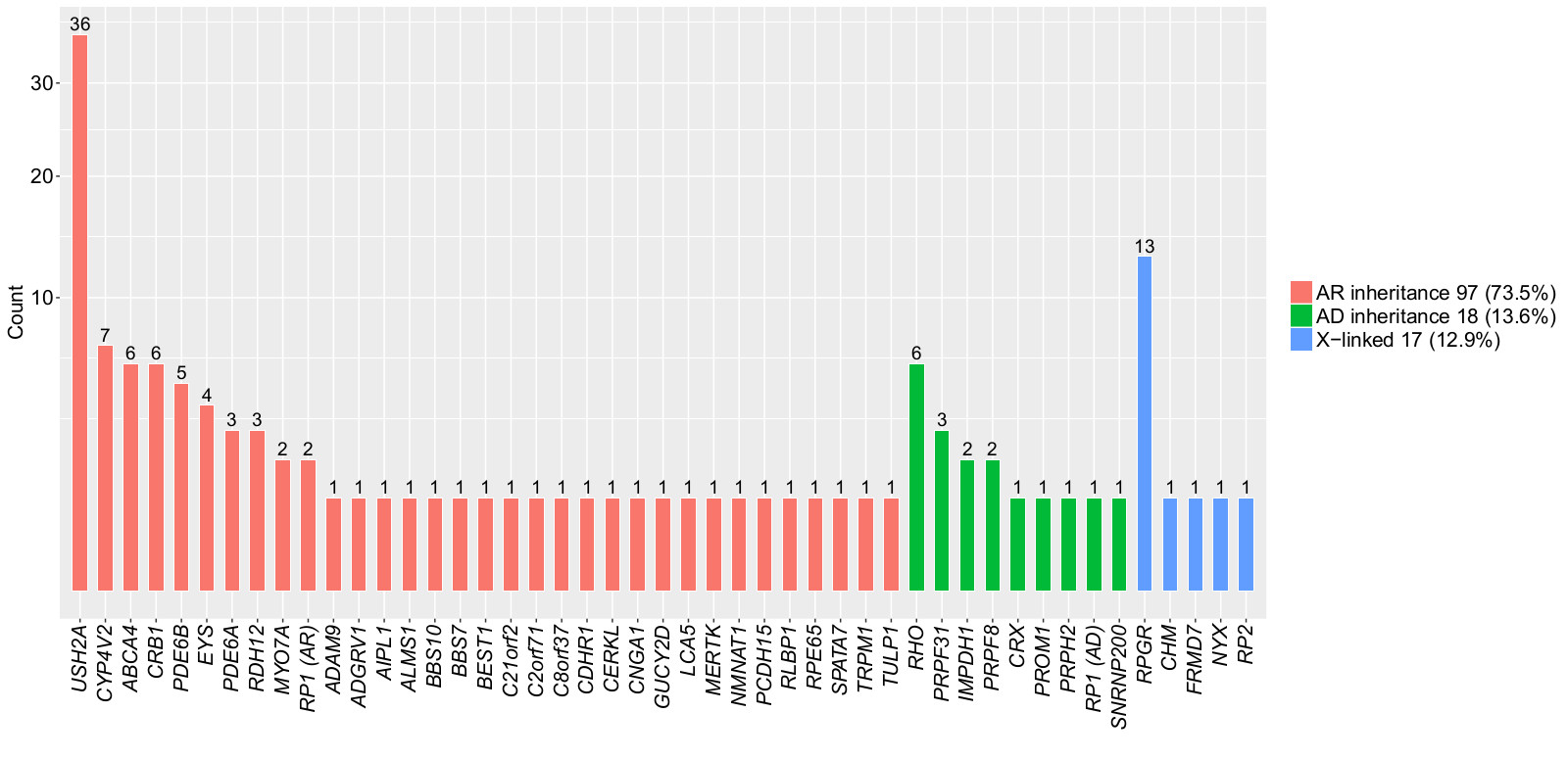
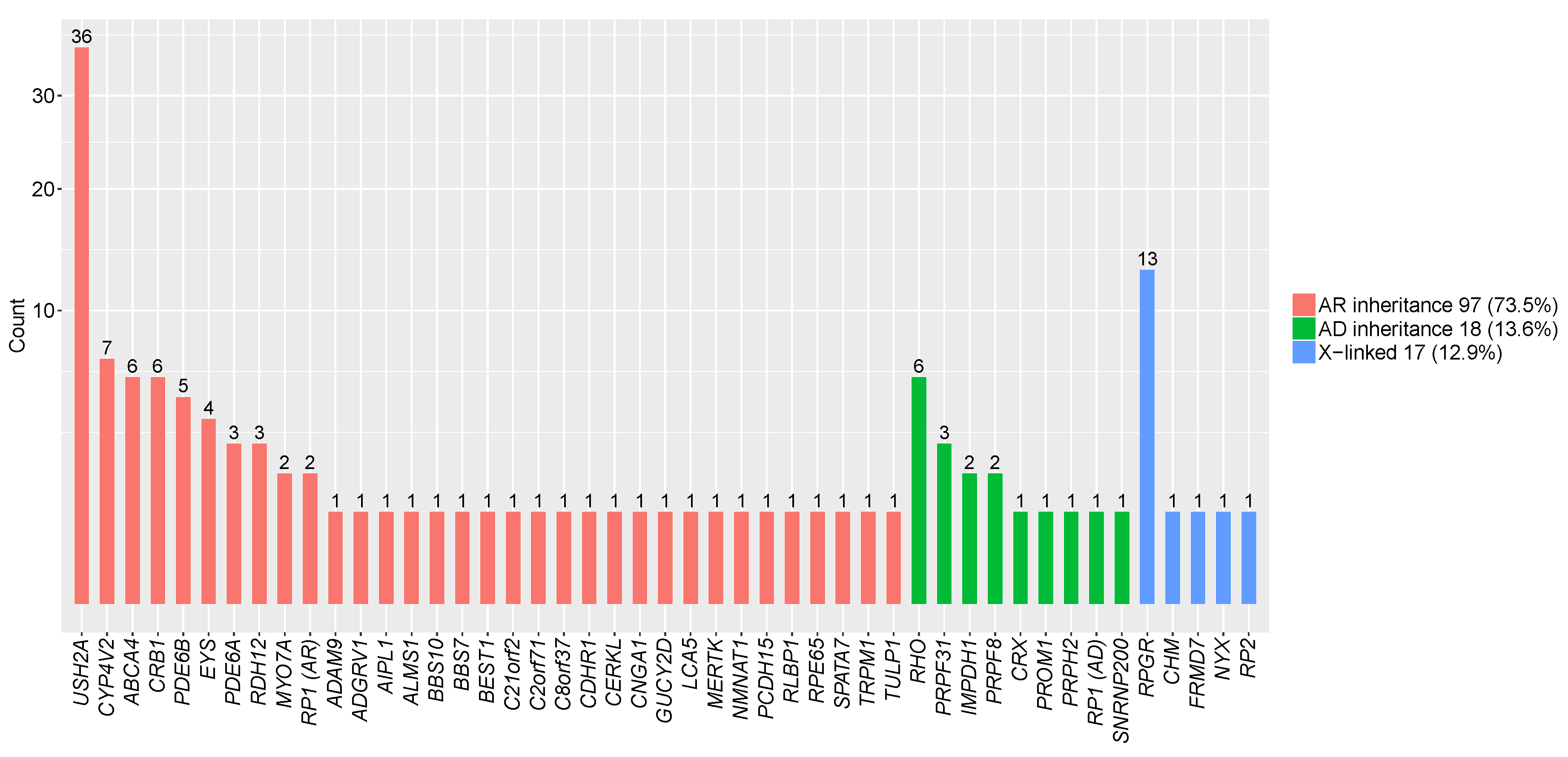
3.2. Pathogenic Mutations Identified
3.3. Revision of the Initial Clinical Diagnosis in One Patient
3.4. Observation of Mutations in ALMS1 Causing Non-Syndromic Retinal Dystrophy
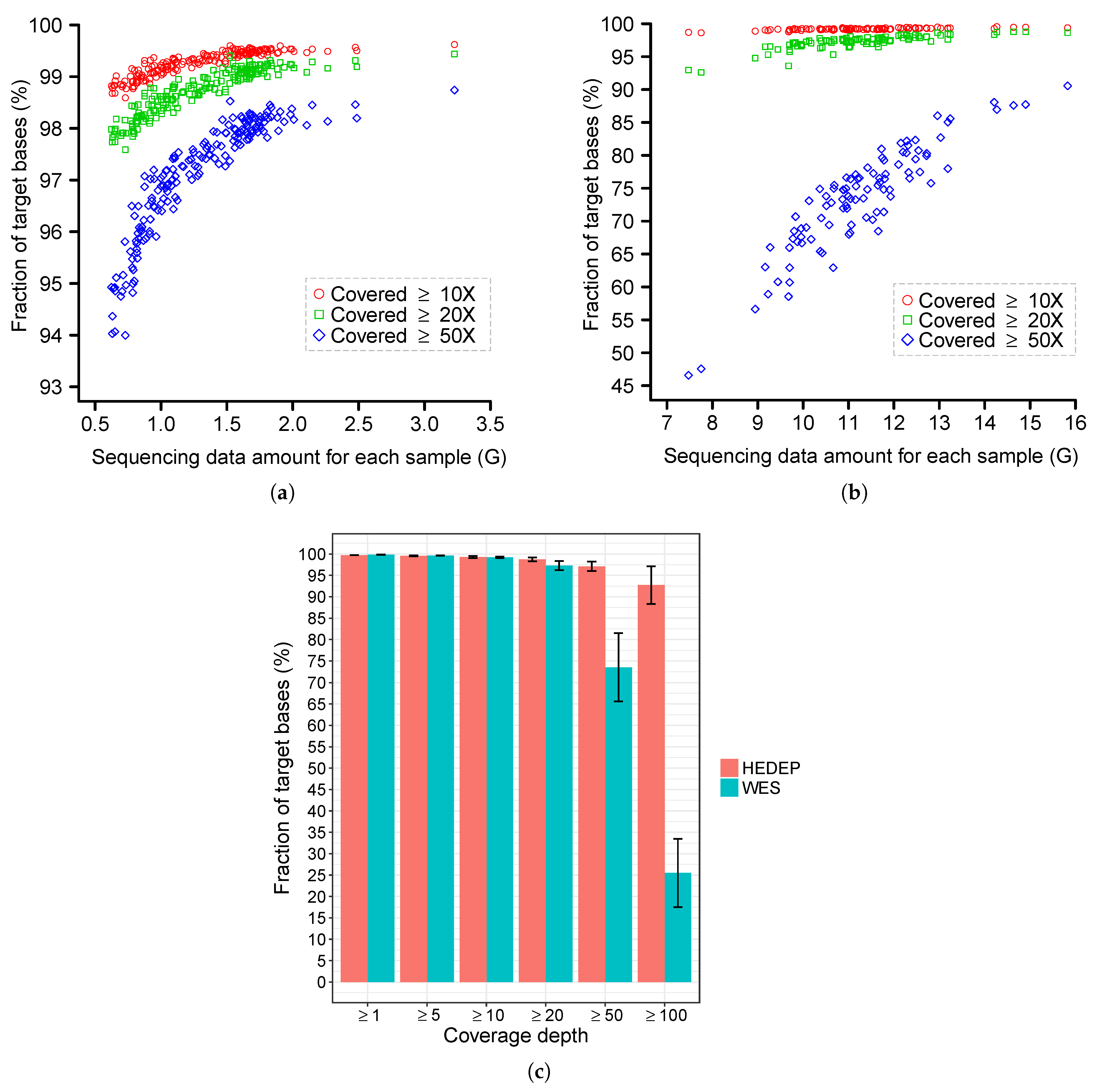
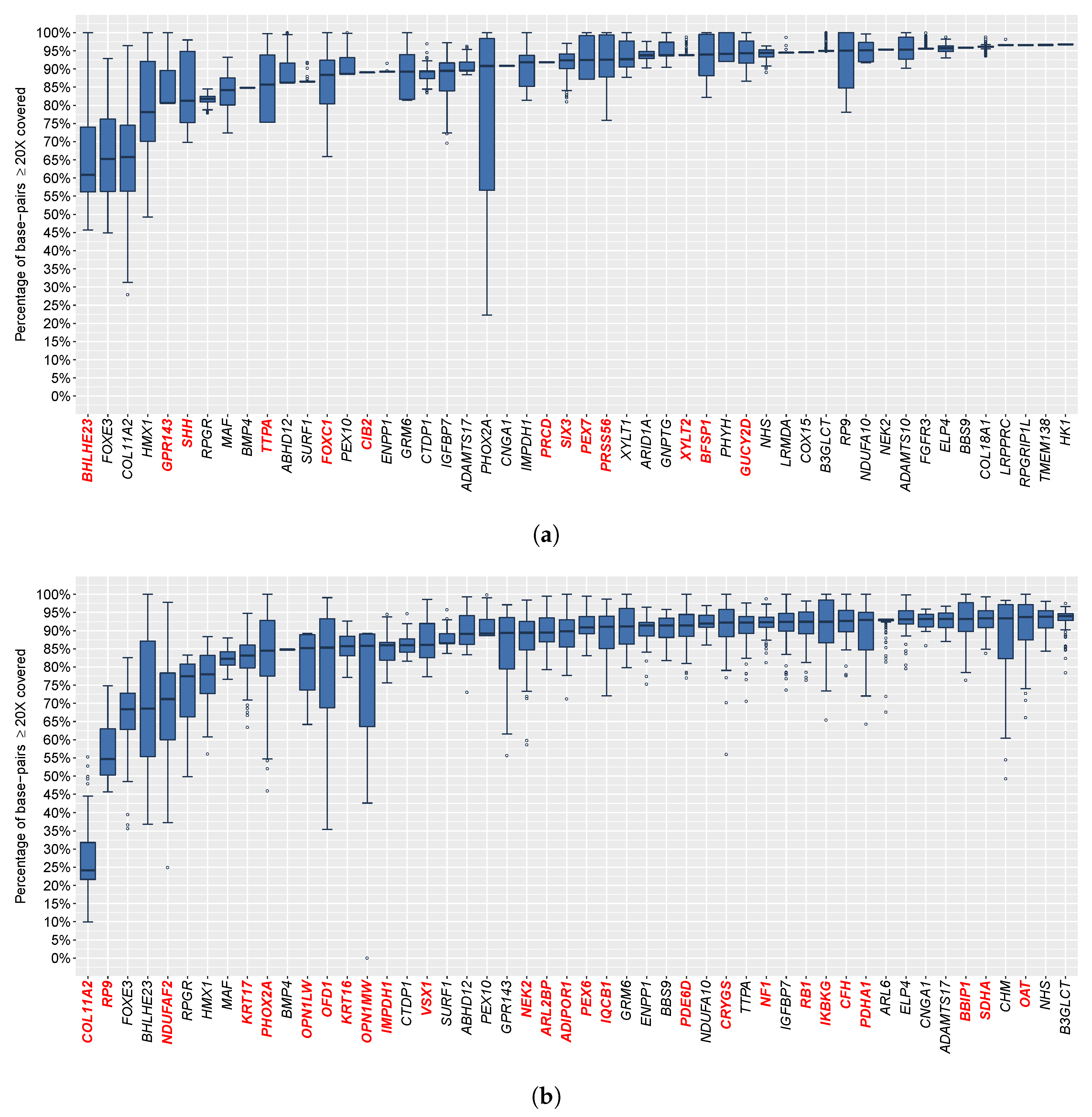
3.5. Sequence Coverage and Bioinformatics Analysis
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Berger, W.; Kloeckener-Gruissem, B.; Neidhardt, J. The molecular basis of human retinal and vitreoretinal diseases. Prog. Retin. Eye Res. 2010, 29, 335–375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Werdich, X.Q.; Place, E.M.; Pierce, E.A. Systemic diseases associated with retinal dystrophies. Semin. Ophthalmol. 2014, 29, 319–328. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Cui, H.; Yin, X.; Dou, H.; Zhao, L.; Chen, N.; Zhang, J.; Zhang, H.; Li, G.; Ma, Z. Dependable and efficient clinical molecular diagnosis of chinese RP patient with targeted exon sequencing. PLoS ONE 2015, 10, e0140684. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.F.; Huang, F.; Wu, K.C.; Wu, J.; Chen, J.; Pang, C.P.; Lu, F.; Qu, J.; Jin, Z.B. Genotype-phenotype correlation and mutation spectrum in a large cohort of patients with inherited retinal dystrophy revealed by next-generation sequencing. Genet. Med. 2015, 17, 271–278. [Google Scholar] [CrossRef] [PubMed]
- Koboldt, D.C.; Larson, D.E.; Sullivan, L.S.; Bowne, S.J.; Steinberg, K.M.; Churchill, J.D.; Buhr, A.C.; Nutter, N.; Pierce, E.A.; Blanton, S.H.; et al. Exome-based mapping and variant prioritization for inherited Mendelian disorders. Am. J. Hum. Genet. 2014, 94, 373–384. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Guan, L.; Shen, T.; Zhang, J.; Xiao, X.; Jiang, H.; Li, S.; Yang, J.; Jia, X.; Yin, Y.; et al. Mutations of 60 known causative genes in 157 families with retinitis pigmentosa based on exome sequencing. Hum. Genet. 2014, 133, 1255–1271. [Google Scholar] [CrossRef] [PubMed]
- Riera, M.; Navarro, R.; Ruiz-Nogales, S.; Méndez, P.; Burés-Jelstrup, A.; Corcóstegui, B.; Pomares, E. Whole exome sequencing using Ion Proton system enables reliable genetic diagnosis of inherited retinal dystrophies. Sci. Rep. 2017, 7, 42078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Wang, C.; Shen, Y.; Chen, N.; Wang, L.; Liang, L.; Guo, T.; Yin, X.; Ma, Z.; Zhang, B.; et al. A mutation in ADIPOR1 causes nonsyndromic autosomal dominant retinitis pigmentosa. Hum. Genet. 2016, 135, 1375–1387. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Summa, S.; Malerba, G.; Pinto, R.; Mori, A.; Mijatovic, V.; Tommasi, S. GATK hard filtering: Tunable parameters to improve variant calling for next generation sequencing targeted gene panel data. BMC Bioinform. 2017, 18, 119. [Google Scholar] [CrossRef] [PubMed]
- Li, H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 2011, 27, 2987–2993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krumm, N.; Sudmant, P.H.; Ko, A.; O’Roak, B.J.; Malig, M.; Coe, B.P.; NHLBI Exome Sequencing Project; Quinlan, A.R.; Nickerson, D.A.; Eichler, E.E. Copy number variation detection and genotyping from exome sequence data. Genome Res. 2012, 22, 1525–1532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, H.; Wang, K. Genomic variant annotation and prioritization with ANNOVAR and wANNOVAR. Nat. Protoc. 2015, 10, 1556–1566. [Google Scholar] [CrossRef] [PubMed]
- Karczewski, K.J.; Weisburd, B.; Thomas, B.; Solomonson, M.; Ruderfer, D.M.; Kavanagh, D.; Hamamsy, T.; Lek, M.; Samocha, K.E.; Cummings, B.B.; et al. The ExAC browser: Displaying reference data information from over 60,000 exomes. Nucleic Acids Res. 2017, 45, D840–D845. [Google Scholar] [CrossRef] [PubMed]
- Davydov, E.V.; Goode, D.L.; Sirota, M.; Cooper, G.M.; Sidow, A.; Batzoglou, S. Identifying a high fraction of the human genome to be under selective constraint using GERP++. PLoS Comput. Biol. 2010, 6, e1001025. [Google Scholar] [CrossRef] [PubMed]
- Adzhubei, I.; Jordan, D.M.; Sunyaev, S.R. Predicting functional effect of human missense mutations using PolyPhen-2. Curr. Protoc. Hum. Gent. 2013, 7. [Google Scholar] [CrossRef] [PubMed]
- Ng, P.C.; Henikoff, S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003, 31, 3812–3814. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, J.M.; Rödelsperger, C.; Schuelke, M.; Seelow, D. MutationTaster evaluates disease-causing potential of sequence alterations. Nat. Methods 2010, 7, 575–576. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Amendola, L.M.; Jarvik, G.P.; Leo, M.C.; McLaughlin, H.M.; Akkari, Y.; Amaral, M.D.; Berg, J.S.; Biswas, S.; Bowling, K.M.; Conlin, L.K.; et al. Performance of ACMG-AMP variant-interpretation guidelines among nine laboratories in the clinical sequencing exploratory research consortium. Am. J. Hum. Genet. 2016, 98, 1067–1076. [Google Scholar] [CrossRef] [PubMed]
- Stenson, P.D.; Ball, E.V.; Mort, M.; Phillips, A.D.; Shaw, K.; Cooper, D.N. The Human Gene Mutation Database (HGMD) and its exploitation in the fields of personalized genomics and molecular evolution. Curr. Protoc. Bioinform. 2012, 1. [Google Scholar] [CrossRef]
- Bader, I.; Brandau, O.; Achatz, H.; Apfelstedt-Sylla, E.; Hergersberg, M.; Lorenz, B.; Wissinger, B.; Wittwer, B.; Rudolph, G.; Meindl, A.; et al. X-linked retinitis pigmentosa: RPGR mutations in most families with definite X linkage and clustering of mutations in a short sequence stretch of exon ORF15. Investig. Ophthalmol. Vis. Sci. 2003, 44, 1458–1463. [Google Scholar] [CrossRef]
- Marshall, J.D.; Hinman, E.G.; Collin, G.B.; Beck, S.; Cerqueira, R.; Maffei, P.; Milan, G.; Zhang, W.; Wilson, D.I.; Hearn, T.; et al. Spectrum of ALMS1 variants and evaluation of genotype-phenotype correlations in Alström syndrome. Hum. Mutat. 2007, 28, 1114–1123. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wang, H.; Cao, M.; Li, Z.; Chen, X.; Patenia, C.; Gore, A.; Abboud, E.B.; Al-Rajhi, A.A.; Lewis, R.A.; et al. Whole-exome sequencing identifies ALMS1, IQCB1, CNGA3, and MYO7A mutations in patients with Leber congenital amaurosis. Hum. Mutat. 2011, 32, 1450–1459. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Wang, X.; Zou, X.; Xu, S.; Li, H.; Soens, Z.T.; Wang, K.; Li, Y.; Dong, F.; Chen, R.; et al. Comprehensive molecular diagnosis of a large chinese Leber congenital amaurosis cohort. Investig. Ophthalmol. Vis. Sci. 2015, 56, 3642–3655. [Google Scholar] [CrossRef] [PubMed]
- Haer-Wigman, L.; van Zelst-Stams, W.A.; Pfundt, R.; van den Born, L.I.; Klaver, C.C.; Verheij, J.B.; Hoyng, C.B.; Breuning, M.H.; Boon, C.J.; Kievit, A.J.; et al. Diagnostic exome sequencing in 266 Dutch patients with visual impairment. Eur. J. Hum. Genet. 2017, 25, 591–599. [Google Scholar] [CrossRef] [PubMed]
- Wiggs, J.L.; Pierce, E.A. Genetic testing for inherited eye disease: Who benefits? JAMA Ophthalmol. 2013, 131, 1265–1266. [Google Scholar] [CrossRef] [PubMed]
- Carss, K.J.; Arno, G.; Erwood, M.; Stephens, J.; Sanchis-Juan, A.; Hull, S.; Megy, K.; Grozeva, D.; Dewhurst, E.; Malka, S.; et al. Comprehensive rare variant analysis via Whole-Genome Sequencing to determine the molecular pathology of inherited retinal disease. Am. J. Hum. Genet. 2017, 100, 75–90. [Google Scholar] [CrossRef] [PubMed]
- Rehm, H.L. Disease-targeted sequencing: A cornerstone in the clinic. Nat. Rev. Genet. 2012, 14, 295–300. [Google Scholar] [CrossRef] [PubMed]
- Kohl, S.; Zobor, D.; Chiang, W.C.; Weisschuh, N.; Staller, J.; Gonzalez Menendez, I.; Chang, S.; Beck, S.C.; Garcia Garrido, M.; Sothilingam, V.; et al. Mutations in the unfolded protein responses regulator ATF6 causes the cone dysfunction disorder achromatopsia. Nat. Genet. 2015, 47, 757–765. [Google Scholar] [CrossRef] [PubMed]
- Angius, A.; Uva, P.; Buers, I.; Oppo, M.; Puddu, A.; Onano, S.; Persico, I.; Loi, A.; Marcia, L.; Höhne, W.; et al. Rutsch F Bi-allelic mutations in KLHL7 cause a Crisponi/CISS1-like phenotype associated with early-onset retinitis pigmentosa. Am. J. Hum. Genet. 2016, 99, 236–245. [Google Scholar] [CrossRef] [PubMed]
- Falk, M.J.; Zhang, Q.; Nakamaru-Ogiso, E.; Kannabiran, C.; Fonseca-Kelly, Z.; Chakarova, C.; Audo, I.; Mackay, D.S.; Zeitz, C.; Borman, A.D.; et al. NMNAT1 mutations cause Leber congenital amaurosis. Nat. Genet. 2012, 44, 1040–1045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiang, P.W.; Wang, J.; Chen, Y.; Fu, Q.; Zhong, J.; Chen, Y.; Yi, X.; Wu, R.; Gan, H.; Shi, Y.; et al. Exome sequencing identifies NMNAT1 mutations as a cause of Lerber congenital amarosis. Nat. Genet. 2012, 44, 972–974. [Google Scholar] [CrossRef] [PubMed]
- Koenekoop, R.K.; Wang, H.; Majewski, J.; Wang, X.; Lopez, I.; Ren, H.; Chen, Y.; Li, Y.; Fishman, G.A.; Genead, M.; et al. Mutations in NMNAT1 cause leber congenital amaurosis and identify a new disease pathway for retinal degeneration. Nat. Genet. 2012, 44, 1035–1039. [Google Scholar] [CrossRef] [PubMed]
- ACMG Board of Directors. Points to consider in the clinical application of genomic sequencing. Genet. Med. 2012, 14, 759–761. [Google Scholar] [CrossRef]
- Broadgate, S.; Yu, J.; Downes, S.M.; Halford, S. Unravelling the genetics of inherited retinal dystrophies: Past, present and future. Prog. Retin. Eye Res. 2017, 59, 53–96. [Google Scholar] [CrossRef] [PubMed]
- Bonnet, C.; Riahi, Z.; Chantot-Bastaraud, S.; Smagghe, L.; Letexier, M.; Marcaillou, C.; Lefèvre, G.M.; Hardelin, J.P.; El-Amraoui, A.; Singh-Estivalet, A.; et al. Aninnovative strategy for the molecular diagnosis of Usher syndrome identifies causal biallelic mutations in 93% of European patients. Eur. J. Hum. Genet. 2016, 24, 1730–1738. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell-Luria, A.H.; Miller, D.T. A Clinician’s perspective on clinical exome sequencing. Hum. Genet. 2016, 135, 643–654. [Google Scholar] [CrossRef] [PubMed]
- Carrigan, M.; Duignan, E.; Malone, C.P.; Stephenson, K.; Saad, T.; McDermott, C.; Green, A.; Keegan, D.; Humphries, P.; Kenna, P.F.; et al. Panel-based population next-generation sequencing for inherited retinal degenerations. Sci. Rep. 2016, 6, 33248. [Google Scholar] [CrossRef] [PubMed]



© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, L.; Zhang, J.; Chen, N.; Wang, L.; Zhang, F.; Ma, Z.; Li, G.; Yang, L. Application of Whole Exome and Targeted Panel Sequencing in the Clinical Molecular Diagnosis of 319 Chinese Families with Inherited Retinal Dystrophy and Comparison Study. Genes 2018, 9, 360. https://doi.org/10.3390/genes9070360
Wang L, Zhang J, Chen N, Wang L, Zhang F, Ma Z, Li G, Yang L. Application of Whole Exome and Targeted Panel Sequencing in the Clinical Molecular Diagnosis of 319 Chinese Families with Inherited Retinal Dystrophy and Comparison Study. Genes. 2018; 9(7):360. https://doi.org/10.3390/genes9070360
Chicago/Turabian StyleWang, Likun, Jinlu Zhang, Ningning Chen, Lei Wang, Fengsheng Zhang, Zhizhong Ma, Genlin Li, and Liping Yang. 2018. "Application of Whole Exome and Targeted Panel Sequencing in the Clinical Molecular Diagnosis of 319 Chinese Families with Inherited Retinal Dystrophy and Comparison Study" Genes 9, no. 7: 360. https://doi.org/10.3390/genes9070360
APA StyleWang, L., Zhang, J., Chen, N., Wang, L., Zhang, F., Ma, Z., Li, G., & Yang, L. (2018). Application of Whole Exome and Targeted Panel Sequencing in the Clinical Molecular Diagnosis of 319 Chinese Families with Inherited Retinal Dystrophy and Comparison Study. Genes, 9(7), 360. https://doi.org/10.3390/genes9070360