High-Throughput Approaches onto Uncover (Epi)Genomic Architecture of Type 2 Diabetes

Abstract
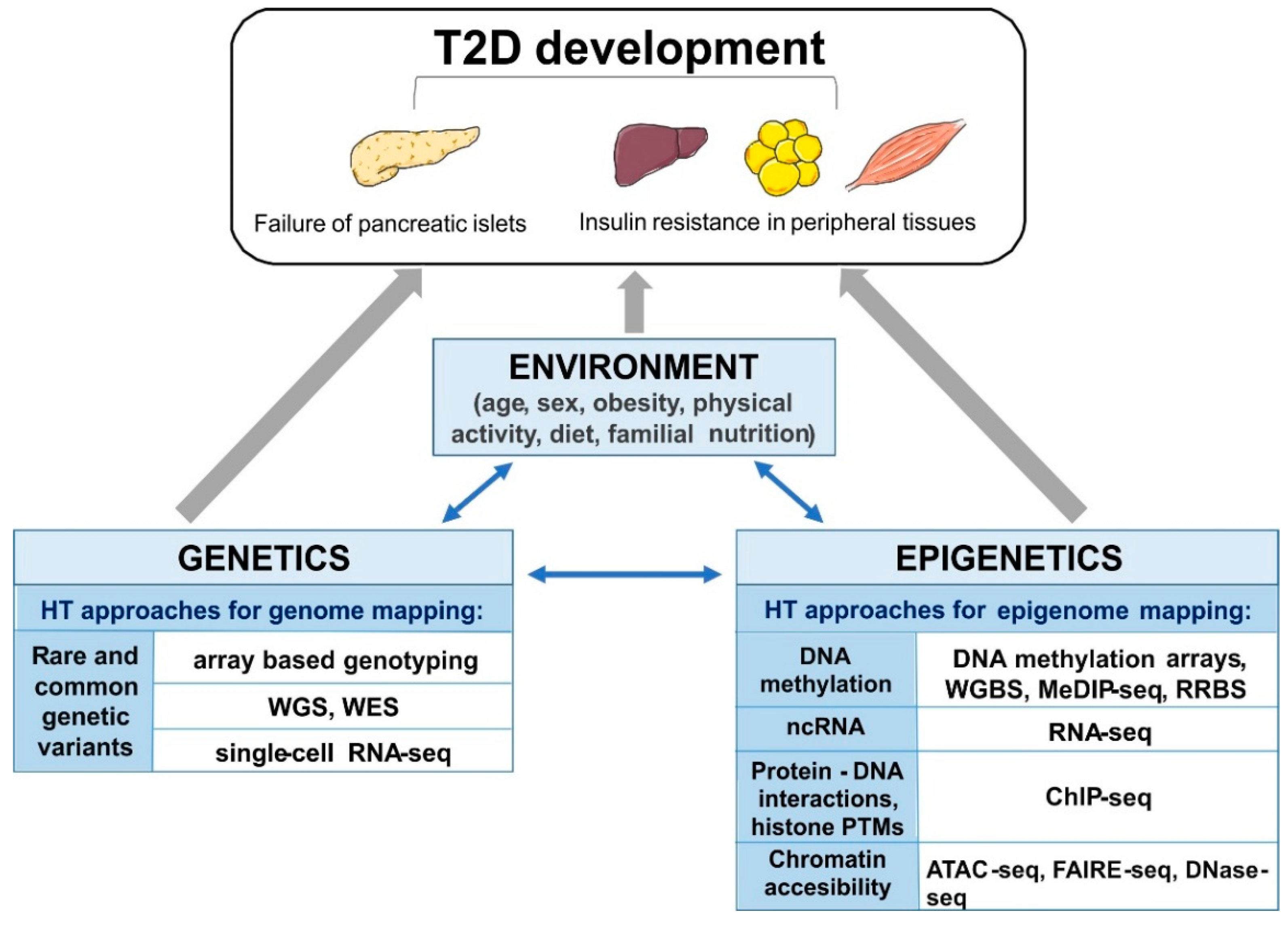
:1. Introduction
2. Genome-Wide Association Studies of Type 2 Diabetes
2.1. Discovery of Genetic Variants Associated with Type 2 Diabetes
2.2. From Genome-Wide Association Studies to Biological Function and Translational Medicine
3. Uncovering the Significance of Rare-Coding and Non-Coding Genetic Variants in the Etiology of Type 2 Diabetes
3.1. Low-Frequency Genetic Variants vs. Population Risk
3.2. Genetic Variants in Familial Studies of Type 2 Diabetes
4. Single-Cell RNA-seq as a Novel Approach in High-Throughput Type 2 Diabetes Research
5. Genome-Wide Profiling of Epigenetic Changes in Pancreatic Islets and Peripheral Tissues
5.1. DNA Methylation Signatures in Type 2 Diabetes
5.2. Chromatin Modification Profile
5.3. The Role of ncRNA in Type 2 Diabetes Pathogenesis
5.4. Epigenome-Wide Association Studies of Type 2 Diabetes
6. Interactions between Genetics and Epigenetic Control in the Pathogenesis of Type 2 Diabetes
7. Conclusions and Perspectives
Funding
Conflicts of Interest
References
- Cavan, D.; da Rocha Fernandes, J.; Makaroff, L.; Ogurtsova, K.; Webber, S. IDF Diabetes Atlas, 7th ed.; International Diabetes Federation: Brussels, Belgium, 2015; pp. 12–19. ISBN 978-2-930229-81-2. [Google Scholar]
- Prentki, M.; Nolan, C.J. Islet β cell failure in type 2 diabetes. J. Clin. Investig. 2006, 116, 1802–1812. [Google Scholar] [CrossRef] [PubMed]
- DeFronzo, R.A. Insulin resistance, lipotoxicity, type 2 diabetes and atherosclerosis: The missing links. The Claude Bernard Lecture 2009. Diabetologia 2010, 53, 1270–1287. [Google Scholar] [CrossRef] [PubMed]
- Rich, S.S. Mapping genes in diabetes: genetic epidemiological perspective. Diabetes 1990, 39, 1315–1319. [Google Scholar] [CrossRef] [PubMed]
- Mahajan, A.; Taliun, D.; Thurner, M.; Robertson, N.R.; Torres, J.M.; Rayner, N.W.; Steinthorsdottir, V.; Scott, R.A.; Grarup, N.; Cook, J.P.; et al. Fine-mapping of an expanded set of type 2 diabetes loci to single-variant resolution using high-density imputation and islet-specific epigenome maps. bioRxiv 2018, 245506. [Google Scholar] [CrossRef] [Green Version]
- Al-Goblan, A.S.; Al-Alfi, M.A.; Khan, M.Z. Mechanism linking diabetes mellitus and obesity. Diabetes Metab. Syndr. Obes. 2014, 7, 587–591. [Google Scholar] [CrossRef] [PubMed]
- Ling, C.; Groop, L. Epigenetics: A molecular link between environmental factors and type 2 diabetes. Diabetes 2009, 58, 2718–2725. [Google Scholar] [CrossRef] [PubMed]
- Hood, L.; Rowen, L. The Human Genome Project: Big science transforms biology and medicine. Genome Med. 2013, 5, 79. [Google Scholar] [CrossRef] [PubMed]
- Bonnefond, A.; Froguel, P. Rare and common genetic events in type 2 diabetes: What should biologists know? Cell Metab. 2015, 21, 357–368. [Google Scholar] [CrossRef] [PubMed]
- Billings, L.K.; Florez, J.C. The genetics of type 2 diabetes: What have we learned from GWAS? Ann. N. Y. Acad. Sci. 2010, 1212, 59–77. [Google Scholar] [CrossRef] [PubMed]
- Lawlor, N.; Khetan, S.; Ucar, D.; Stitzel, M.L. Genomics of islet (dys)function and type 2 diabetes. Trends Genet. 2017, 33, 244–255. [Google Scholar] [CrossRef] [PubMed]
- Zeggini, E.; Weedon, M.N.; Lindgren, C.M.; Frayling, T.M.; Elliott, K.S.; Lango, H.; Timpson, N.J.; Perry, J.R.; Rayner, N.W.; Freathy, R.M.; et al. Replication of genome-wide association signals in UK samples reveals risk loci for type 2 diabetes. Science 2007, 316, 1336–1341. [Google Scholar] [CrossRef] [PubMed]
- Zeggini, E.; Scott, L.J.; Saxena, R.; Voight, B.F.; Marchini, J.L.; Hu, T.; de Bakker, P.I.; Abecasis, G.R.; Almgren, P.; Andersen, G.; et al. Meta-analysis of genome-wide association data and large-scale replication identifies additional susceptibility loci for type 2 diabetes. Nat. Genet. 2008, 40, 638–645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grarup, N.; Rose, C.S.; Andersson, E.A.; Andersen, G.; Nielsen, A.L.; Albrechtsen, A.; Clausen, J.O.; Rasmussen, S.S.; Jørgensen, T.; Sandbaek, A.; et al. Studies of association of variants near the HHEX, CDKN2A/B and IGF2BP2 genes with type 2 diabetes and impaired insulin release in 10,705 Danish subjects validation and extension of genome-wide association studies. Diabetes 2007, 56, 3105–3111. [Google Scholar] [CrossRef] [PubMed]
- Omori, S.; Tanaka, Y.; Takahashi, A.; Hirose, H.; Kashiwagi, A.; Kaku, K.; Kawamori, R.; Nakamura, Y.; Maeda, S. Association of CDKAL1, IGF2BP2, CDKN2A/B, HHEX, SLC30A8, and KCNJ11 with susceptibility to type 2 diabetes in a Japanese population. Diabetes 2008, 57, 791–795. [Google Scholar] [CrossRef] [PubMed]
- Visscher, P.M.; Wray, N.R.; Zhang, Q.; Sklar, P.; McCarthy, M.I.; Brown, M.A.; Yang, J. 10 Years of GWAS discovery: Biology, function, and translation. Am. J. Hum. Genet. 2017, 101, 5–22. [Google Scholar] [CrossRef] [PubMed]
- Lyssenko, V.; Laakso, M. Genetic screening for the risk of type 2 diabetes: Worthless or valuable? Diabetes Care 2013, 36, 120–126. [Google Scholar] [CrossRef] [PubMed]
- Sladek, R.; Rocheleau, G.; Rung, J.; Dina, C.; Shen, L.; Serre, D.; Boutin, P.; Vincent, D.; Belisle, A.; Hadjadj, S.; et al. A genome-wide association study identifies novel risk loci for type 2 diabetes. Nature 2007, 445, 881–885. [Google Scholar] [CrossRef] [PubMed]
- Morris, A.P.; Voight, B.F.; Teslovich, T.M.; Ferreira, T.; Segrè, A.V.; Steinthorsdottir, V.; Strawbridge, R.J.; Khan, H.; Grallert, H.; Mahajan, A.; et al. Large-scale association analysis provides insights into the genetic architecture and pathophysiology of type 2 diabetes. Nat. Genet. 2012, 44, 981–990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scott, R.A.; Scott, L.J.; Mägi, R.; Marullo, L.; Gaulton, K.J.; Kaakinen, M.; Pervjakova, N.; Pers, T.H.; Johnson, A.D.; Eicher, J.D.; et al. An expanded genome-wide association study of type 2 diabetes in Europeans. Diabetes 2017, 66, 2888–2902. [Google Scholar] [CrossRef] [PubMed]
- Andersen, M.K.; Pedersen, C.E.; Moltke, I.; Hansen, T.; Albrechtsen, A.; Grarup, N. Genetics of type 2 diabetes: The power of isolated populations. Curr. Diabetes Rep. 2016, 16, 65. [Google Scholar] [CrossRef] [PubMed]
- Unoki, H.; Takahashi, A.; Kawaguchi, T.; Hara, K.; Horikoshi, M.; Andersen, G.; Ng, D.P.; Holmkvist, J.; Borch-Johnsen, K.; Jørgensen, T.; et al. SNPs in KCNQ1 are associated with susceptibility to type 2 diabetes in East Asian and European populations. Nat. Genet. 2008, 40, 1098–1102. [Google Scholar] [CrossRef] [PubMed]
- Fuchsberger, C.; Flannick, J.; Teslovich, T.M.; Mahajan, A.; Agarwala, V.; Gaulton, K.J.; Ma, C.; Fontanillas, P.; Moutsianas, L.; McCarthy, D.J.; et al. The genetic architecture of type 2 diabetes. Nature 2016, 536, 41–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ali, S.; Chopra, R.; Manvati, S.; Pal Singh, Y.; Kaul, N.; Behura, A.; Mahajan, A.; Sehajpal, P.; Gupta, S.; Dhar, M.K.; et al. Replication of type 2 diabetes candidate genes variations in three geographically unrelated Indian population groups. PLoS ONE 2013, 8, e58881. [Google Scholar] [CrossRef] [PubMed]
- Hanson, R.L.; Muller, Y.L.; Kobes, S.; Guo, T.; Bian, L.; Ossowski, V.; Wiedrich, K.; Sutherland, J.; Wiedrich, C.; Mahkee, D.; et al. A genome-wide association study in American Indians implicates DNER as a susceptibility locus for type 2 diabetes. Diabetes 2014, 63, 369–376. [Google Scholar] [CrossRef] [PubMed]
- Moltke, I.; Grarup, N.; Jørgensen, M.E.; Bjerregaard, P.; Treebak, J.T.; Fumagalli, M.; Korneliussen, T.S.; Andersen, M.A.; Nielsen, T.S.; Krarup, N.T.; et al. A common Greenlandic TBC1D4 variant confers muscle insulin resistance and type 2 diabetes. Nature 2014, 512, 190–193. [Google Scholar] [CrossRef] [PubMed]
- Saxena, R.; Saleheen, D.; Been, L.F.; Garavito, M.L.; Braun, T.; Bjonnes, A.; Young, R.; Ho, W.K.; Rasheed, A.; Frossard, P.; et al. Genome-wide association study identifies a novel locus contributing to type 2 diabetes susceptibility in Sikhs of Punjabi origin from India. Diabetes 2013, 62, 1746–1755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCarthy, M.I. Genetics of T2DM in 2016: Biological and translational insights from T2DM genetics. Nat. Rev. Endocrinol. 2017, 13, 71–72. [Google Scholar] [CrossRef] [PubMed]
- Grotz, A.K.; Gloyn, A.L.; Thomsen, S.K. Prioritising causal genes at type 2 diabetes risk loci. Curr. Diabetes Rep. 2017, 17, 76. [Google Scholar] [CrossRef] [PubMed]
- Ingelsson, E.; Langenberg, C.; Hivert, M.F.; Prokopenko, I.; Lyssenko, V.; Dupuis, J.; Mägi, R.; Sharp, S.; Jackson, A.U.; Assimes, T.L.; et al. Detailed physiologic characterization reveals diverse mechanisms for novel genetic loci regulating glucose and insulin metabolism in humans. Diabetes 2010, 59, 1266–1275. [Google Scholar] [CrossRef] [PubMed]
- Gaulton, K.J. Mechanisms of type 2 diabetes risk loci. Curr. Diabetes Rep. 2017, 17, 72. [Google Scholar] [CrossRef] [PubMed]
- Steinthorsdottir, V.; Thorleifsson, G.; Reynisdottir, I.; Benediktsson, R.; Jonsdottir, T.; Walters, G.B.; Styrkarsdottir, U.; Gretarsdottir, S.; Emilsson, V.; Ghosh, S.; et al. A variant in CDKAL1 influences insulin response and risk of type 2 diabetes. Nat. Genet. 2007, 39, 770–775. [Google Scholar] [CrossRef] [PubMed]
- Flannick, J.; Thorleifsson, G.; Beer, N.L.; Jacobs, S.B.; Grarup, N.; Burtt, N.P.; Mahajan, A.; Fuchsberger, C.; Atzmon, G.; Benediktsson, R.; et al. Loss-of-function mutations in SLC30A8 protect against type 2 diabetes. Nat. Genet. 2014, 46, 357–363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Florez, J.C. Pharmacogenetics in type 2 diabetes: Precision medicine or discovery tool? Diabetologia 2017, 60, 800–807. [Google Scholar] [CrossRef] [PubMed]
- Korte, A.; Farlow, A. The advantages and limitations of trait analysis with GWAS: A review. Plant Methods 2013, 9, 29. [Google Scholar] [CrossRef] [PubMed]
- Torres, J.M.; Gamazon, E.R.; Parra, E.J.; Below, J.E.; Valladares-Salgado, A.; Wacher, N.; Cruz, M.; Hanis, C.L.; Cox, N.J. Cross-tissue and tissue-specific eQTLs: Partitioning the heritability of a complex trait. Am. J. Hum. Genet. 2014, 95, 521–534. [Google Scholar] [CrossRef] [PubMed]
- Segura, V.; Vilhjalmsson, B.J.; Platt, A.; Korte, A.; Seren, U.; Long, Q.; Nordborg, M. An efficient multi-locus mixed-model approach for genome-wide association studies in structured populations. Nat. Genet. 2012, 44, 825–830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levy, S.E.; Myers, R.M. Advancements in next-generation sequencing. Annu. Rev. Genom. Hum. Genet. 2016, 17, 95–115. [Google Scholar] [CrossRef] [PubMed]
- Saint Pierre, A.; Génin, E. How important are rare variants in common disease? Brief. Funct. Genom. 2014, 13, 353–361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Estrada, K.; Aukrust, I.; Bjørkhaug, L.; Burtt, N.P.; Mercader, J.M.; García-Ortiz, H.; Huerta-Chagoya, A.; Moreno-Macías, H.; Walford, G.; Flannick, J.; et al. Association of a low-frequency variant in HNF1A with type 2 diabetes in a Latino population. JAMA 2014, 311, 2305–2314. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Li, X.; Zhang, Y.; Fan, X.; Zhang, N.; Zheng, H.; Song, Y.; Shen, C.; Shen, J.; Ren, F.; et al. Genetic variants of TPCN2 associated with type 2 diabetes risk in the Chinese population. PLoS ONE 2016, 11, e0149614. [Google Scholar] [CrossRef] [PubMed]
- Steinthorsdottir, V.; Thorleifsson, G.; Sulem, P.; Helgason, H.; Grarup, N.; Sigurdsson, A.; Helgadottir, H.T.; Johannsdottir, H.; Magnusson, O.T.; Gudjonsson, S.A.; et al. Identification of low-frequency and rare sequence variants associated with elevated or reduced risk of type 2 diabetes. Nat. Genet. 2014, 46, 294–298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prasad, R.B.; Lessmark, A.; Almgren, P.; Kovacs, G.; Hansson, O.; Oskolkov, N.; Vitai, M.; Ladenvall, C.; Kovacs, P.; Fadista, J.; et al. Excess maternal transmission of variants in the THADA gene to offspring with type 2 diabetes. Diabetologia 2016, 59, 1702–1713. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.; Nair, A.K.; Muller, Y.L.; Piaggi, P.; Bian, L.; Rosario, M.; Knowler, W.C.; Kobes, S.; Hanson, R.L.; Bogardus, C. Whole exome sequencing identifies variation in CYB5A and RNF10 associated with adiposity and type 2 diabetes. Obesity 2014, 22, 984–988. [Google Scholar] [CrossRef] [PubMed]
- SIGMA. Type 2 Diabetes Consortium. Sequence variants in SLC16A11 are a common risk factor for type 2 diabetes in Mexico. Nature 2014, 506, 97–101. [Google Scholar] [CrossRef]
- Shawl, A.I.; Park, K.; Kim, B.; Higashida, C.; Higashida, H.; Kim, U. Involvement of actin filament in the generation of Ca2+ mobilizing messengers in glucose-induced Ca2+ signaling in pancreatic β-cells. Islets 2012, 4, 145–151. [Google Scholar] [CrossRef] [PubMed]
- Park, K.H.; Kim, B.J.; Shawl, A.I.; Han, M.K.; Lee, H.C.; Kim, U.H. Autocrine/paracrine function of nicotinic acid adenine dinucleotide phosphate (NAADP) for glucose homeostasis in pancreatic beta cells and adipo-cytes. J. Biol. Chem. 2013, 288, 35548–35558. [Google Scholar] [CrossRef] [PubMed]
- Tsaih, S.W.; Holl, K.; Jia, S.; Kaldunski, M.; Tschannen, M.; He, H.; Andrae, J.W.; Li, S.H.; Stoddard, A.; Wiederhold, A.; et al. Identification of a novel gene for diabetic traits in rats, mice, and humans. Genetics 2014, 198, 17–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jun, G.; Manning, A.; Almeida, M.; Zawistowski, M.; Wood, A.R.; Teslovich, T.M.; Fuchsberger, C.; Feng, S.; Cingolani, P.; Gaulton, K.J.; et al. Evaluating the contribution of rare variants to type 2 diabetes and related traits using pedigrees. Proc. Natl. Acad. Sci. USA 2018, 115, 379–384. [Google Scholar] [CrossRef] [PubMed]
- Almgren, P.; Lehtovirta, M.; Isomaa, B.; Sarelin, L.; Taskenen, M.R.; Lyssenko, V.; Tuomi, T.; Groop, L.; Botnia Study Group. Heritability and familiality of type 2 diabetes and related quantitative traits in the Botnia Study. Diabetologia 2011, 54, 2811–2819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dobrzyn, P.; Jazurek, M.; Dobrzyn, A. Stearoyl-CoA desaturase and insulin signaling—What is the molecular switch? Biochim. Biophys. Acta 2010, 1797, 1189–1194. [Google Scholar] [CrossRef] [PubMed]
- Xin, Y.; Kim, J.; Okamoto, H.; Ni, M.; Wei, Y.; Adler, C.; Murphy, A.J.; Yancopoulos, G.D.; Lin, C.; Gromada, J. RNA sequencing of single human islet cells reveals type 2 diabetes genes. Cell Metab. 2016, 24, 608–615. [Google Scholar] [CrossRef] [PubMed]
- Weir, G.C.; Laybutt, D.R.; Kaneto, H.; Bonner-Weir, S.; Sharma, A. Beta-cell adaptation and decompensation during the progression of diabetes. Diabetes 2001, 50, S154–S159. [Google Scholar] [CrossRef] [PubMed]
- Lawlor, N.; George, J.; Bolisetty, M.; Kursawe, R.; Sun, L.; Sivakamasundari, V.; Kycia, I.; Robson, P.; Stitzel, M.L. Single-cell transcriptomes identify human islet cell signatures and reveal cell-type–specific expression changes in type 2 diabetes. Genome Res. 2017, 27, 208–222. [Google Scholar] [CrossRef] [PubMed]
- Davegårdh, C.; García-Calzón, S.; Bacos, K.; Ling, C. DNA methylation in the pathogenesis of type 2 diabetes in humans. Mol. Metab. 2018. [Google Scholar] [CrossRef] [PubMed]
- Kwak, S.H.; Park, K.S. Recent progress in genetic and epigenetic research on type 2 diabetes. Exp. Mol. Med. 2016, 48, e220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muka, T.; Nano, J.; Voortman, T.; Braun, K.V.E.; Ligthart, S.; Stranges, S.; Bramer, W.M.; Troup, J.; Chowdhury, R.; Dehghan, A.; et al. The role of global and regional DNA methylation and histone modifications in glycemic traits and type 2 diabetes: A systematic review. Nutr. Metab. Cardiovasc. Dis. 2016, 26, 553–566. [Google Scholar] [CrossRef] [PubMed]
- Robinson, S.W.; Fernandes, M.; Husi, H. Current advances in systems and integrative biology. Comput. Struct. Biotechnol. J. 2014, 11, 35–46. [Google Scholar] [CrossRef] [PubMed]
- Golson, M.L.; Kaestner, K.H. Epigenetics in formation, function, and failure of the endocrine pancreas. Mol. Metab. 2017, 6, 1066–1076. [Google Scholar] [CrossRef] [PubMed]
- Yokoi, N. Epigenetic dysregulation in pancreatic islets and pathogenesis of type 2 diabetes. J. Diabetes Investig. 2017, 9, 475–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, Y.; Ding, Y.; Liang, B.; Juanjuan Lin, J.; Kim, T.; Yu, H.; Hang, H.; Wang, K.A. Systematic study of dysregulated microRNA in type 2 diabetes mellitus. Int. J. Mol. Sci. 2017, 18, 456. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Tan, Q.; Liu, F. Differentially methylated circulating DNA: A novel biomarker to monitor beta cell death. J. Diabetes Complicat. 2018, 32, 349–353. [Google Scholar] [CrossRef] [PubMed]
- Cao, F.; Zwinderman, M.R.H.; Dekker, F.J. The process and strategy for developing selective histone deacetylase 3 inhibitors. Molecules 2018, 23, 551. [Google Scholar] [CrossRef]
- Tobi, E.W.; Lumey, L.H.; Talens, R.P.; Kremer, D.; Putter, H.; Stein, A.D.; Slagboom, P.E.; Heijmans, B.T. DNA methylation differences after exposure to prenatal famine are common and timing- and sex-specific. Hum. Mol. Genet. 2009, 18, 4046–4053. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Volkmar, M.; Dedeurwaerder, S.; Cunha, D.A.; Ndlovu, M.N.; Defrance, M.; Deplus, R.; Calonne, E.; Volkmar, U.; Igoillo-Esteve, M.; Naamane, N.; et al. DNA methylation profiling identifies epigenetic dysregulation in pancreatic islets from type 2 diabetic patients. EMBO J. 2012, 31, 1405–1426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dayeh, T.; Volkov, P.; Salö, S.; Hall, E.; Nilsson, E.; Olsson, A.H.; Kirkpatrick, C.L.; Wollheim, C.B.; Eliasson, L.; Rönn, T.; et al. Genome-wide DNA methylation analysis of human pancreatic islets from type 2 diabetic and non-diabetic donors identifies candidate genes that influence insulin secretion. PLoS Genet. 2014, 10, e1004160. [Google Scholar] [CrossRef] [PubMed]
- Volkov, P.; Bacos, K.; Ofori, J.K.; Esguerra, J.L.; Eliasson, L.; Rönn, T.; Ling, C. Whole-genome bisulfite sequencing of human pancreatic islets reveals novel differentially methylated regions in type 2 diabetes pathogenesis. Diabetes 2017, 66, 1074–1085. [Google Scholar] [CrossRef] [PubMed]
- Barres, R.; Yan, J.; Egan, B.; Treebak, J.T.; Rasmussen, M.; Fritz, T.; Caidahl, K.; Krook, A.; O’Gorman, D.J.; Zierath, J.R. Acute exercise remodels promoter methylation in human skeletal muscle. Cell Metab. 2012, 15, 405–411. [Google Scholar] [CrossRef] [PubMed]
- Ribel-Madsen, R.; Fraga, M.F.; Jacobsen, S.; Bork-Jensen, J.; Lara, E.; Calvanese, V.; Fernandez, A.F.; Friedrichsen, M.; Vind, B.F.; Hřjlund, K.; et al. Genome-wide analysis of DNA methylation differences in muscle and fat from monozygotic twins discordant for type 2 diabetes. PLoS ONE 2012, 7, e51302. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, S.S.; Salehzadeh, F.; Fritz, T.; Zierath, J.R.; Krook, A.; Osler, M.E. Mitochondrial regulators of fatty acid metabolism reflect metabolic dysfunction in type 2 diabetes mellitus. Metabolism 2012, 61, 175–185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaulton, K.J.; Nammo, T.; Pasquali, L.; Simon, J.M.; Giresi, P.G.; Fogarty, M.P.; Panhuis, T.M.; Mieczkowski, P.; Secchi, A.; Bosco, D.; et al. A map of open chromatin in human pancreatic islets. Nat. Genet. 2010, 42, 255–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ackermann, A.M.; Wang, Z.; Schug, J.; Naji, A.; Kaestner, K.H. Integration of ATAC-seq and RNA-seq identifies human alpha cell and beta cell signature genes. Mol. Metab. 2016, 5, 233–244. [Google Scholar] [CrossRef] [PubMed]
- Mukwevho, E.; Kohn, T.A.; Lang, D.; Nyatia, E.; Smith, J.; Ojuka, E.O. Caffeine induces hyperacetylation of histones at the MEF2 site on the Glut4 promoter and increases MEF2A binding to the site via a CaMK-dependent mechanism. Am. J. Physiol. Endocrinol. Metab. 2008, 294, 582–588. [Google Scholar] [CrossRef] [PubMed]
- Van de Bunt, M.; Gaulton, K.J.; Parts, L.; Moran, I.; Johnson, P.R.; Lindgren, C.M.; Ferrer, J.; Gloyn, A.L.; McCarthy, M.I. The miRNA profile of human pancreatic islets and beta-cells and relationship to type 2 diabetes pathogenesis. PLoS ONE 2013, 8, e55272. [Google Scholar] [CrossRef] [PubMed]
- Kameswaran, V.; Bramswig, N.C.; McKenna, L.B.; Penn, M.; Schug, J.; Hand, N.J.; Chen, Y.; Choi, I.; Vourekas, A.; Won, K.; et al. Epigenetic regulation of the MEG3-DLK1 microRNA cluster in human type 2 diabetic islets. Cell Metab. 2014, 19, 135–145. [Google Scholar] [CrossRef] [PubMed]
- Esteves, J.V.; Enguita, F.J.; Machado, U.F. MicroRNAs-mediated regulation of skeletal muscle GLUT4 expression and translocation in insulin resistance. J. Diabetes Res. 2017, 2017, 7267910. [Google Scholar] [CrossRef] [PubMed]
- Goyal, N.; Kesharwani, D.; Datta, M. Lnc-ing non-coding RNAs with metabolism and diabetes: Roles of lncRNAs. Cell. Mol. Life Sci. 2018, 75, 1827–1837. [Google Scholar] [CrossRef] [PubMed]
- Morán, I.; Akerman, I.; van de Bunt, M.; Xie, R.; Benazra, M.; Nammo, T.; Arnes, L.; Nakić, N.; García-Hurtado, J.; Rodríguez-Seguí, S.; et al. Human β cell transcriptome analysis uncovers lncRNAs that are tissue-specific, dynamically regulated, and abnormally expressed in type 2 diabetes. Cell Metab. 2012, 16, 435–448. [Google Scholar] [CrossRef] [PubMed]
- Walaszczyk, E.; Luijten, M.; Spijkerman, A.M.W.; Bonder, M.J.; Lutgers, H.L.; Snieder, H.; Wolffenbuttel, B.H.R.; van Vliet-Ostaptchouk, J.V. DNA methylation markers associated with type 2 diabetes, fasting glucose and HbA1c levels: A systematic review and replication in a case-control sample of the Lifelines study. Diabetologia 2018, 61, 354–368. [Google Scholar] [CrossRef] [PubMed]
- Chambers, J.C.; Loh, M.; Lehne, B.; Drong, A.; Kriebel, J.; Motta, V.; Wahl, S.; Elliott, H.R.; Rota, F.; Scott, W.R.; et al. Epigenome-wide association of DNA methylation markers in peripheral blood from Indian Asians and Europeans with incident type 2 diabetes: A nested case-control study. Lancet Diabetes Endocrinol. 2015, 3, 526–534. [Google Scholar] [CrossRef]
- Bacos, K.; Gillberg, L.; Volkov, P.; Olsson, A.H.; Hansen, T.; Pedersen, O.; Gjesing, A.P.; Eiberg, H.; Tuomi, T.; Almgren, P.; et al. Blood-based biomarkers of age-associated epigenetic changes in human islets associate with insulin secretion and diabetes. Nat. Commun. 2016, 7, 11089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thurner, M.; van de Bunt, M.; Torres, J.M.; Mahajan, A.; Nylander, V.; Bennett, A.J.; Gaulton, K.J.; Barrett, A.; Burrows, C.; Bell, C.G.; et al. Integration of human pancreatic islet genomic data refines regulatory mechanisms at type 2 diabetes susceptibility loci. Elife 2018, 7, e31977. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Yao, S.; Tang, J.; Liu, S.; Chen, J.; Deng, D.; Zeng, C. Integrative analysis of super enhancer SNPs for type 2 diabetes. PLoS ONE 2018, 13, e0192105. [Google Scholar] [CrossRef] [PubMed]
- Scott, L.J.; Erdos, M.R.; Huyghe, J.R.; Welch, R.P.; Beck, A.T.; Wolford, B.N.; Chines, P.S.; Didion, J.P.; Narisu, N.; Stringham, H.M.; et al. The genetic regulatory signature of type 2 diabetes in human skeletal muscle. Nat. Commun. 2016, 7, 11764. [Google Scholar] [CrossRef] [PubMed]
- Bagnato, P.; Barone, V.; Giacomello, E.; Rossi, D.; Sorrentino, V. Binding of an ankyrin-1 isoform to obscurin suggests a molecular link between the sarcoplasmic reticulum and myofibrils in striated muscles. J. Cell Biol. 2003, 160, 245–253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caruso, M.; Ma, D.; Msallaty, Z.; Lewis, M.; Seyoum, B.; Al-janabi, W.; Diamond, M.; Abou-Samra, A.B.; Højlund, K.; Tagett, R.; et al. Increased interaction with insulin receptor substrate 1, a novel abnormality in insulin resistance and type 2 diabetes. Diabetes 2014, 63, 1933–1947. [Google Scholar] [CrossRef] [PubMed]
- Wahl, S.; Drong, A.; Lehne, B.; Loh, M.; Scott, W.R.; Kunze, S.; Tsai, P.C.; Ried, J.S.; Zhang, W.; Yang, Y.; et al. Epigenome-wide association study of body mass index, and the adverse outcomes of adiposity. Nature 2017, 541, 81–86. [Google Scholar] [CrossRef] [PubMed]
- Laker, R.C.; Garde, C.; Camera, D.M.; Smiles, W.J.; Zierath, J.R.; Hawley, J.A.; Barrès, R. Transcriptomic and epigenetic responses to short-term nutrient-exercise stress in humans. Sci. Rep. 2017, 7, e15134. [Google Scholar] [CrossRef] [PubMed]
- Donkin, I.; Versteyhe, S.; Ingerslev, L.R.; Qian, K.; Mechta, M.; Nordkap, L.; Mortensen, B.; Appel, E.V.; Jørgensen, N.; Kristiansen, V.B.; et al. Obesity and bariatric surgery drive epigenetic variation of spermatozoa in humans. Cell Metab. 2016, 23, 369–378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dayeh, T.A.; Olsson, A.H.; Volkov, P.; Almgren, P.; Rönn, T.; Ling, C. Identification of CpG-SNPs associated with type 2 diabetes and differential DNA methylation in human pancreatic islets. Diabetologia 2013, 56, 1036–1046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olsson, A.H.; Volkov, P.; Bacos, K.; Dayeh, T.; Hall, E.; Nilsson, E.A.; Ladenvall, C.; Rönn, T.; Ling, C. Genome-ide associations between genetic and epigenetic variation influence mRNA expression and insulin secretion in human pancreatic islets. PLoS Genet. 2014, 10, e1004735. [Google Scholar] [CrossRef] [PubMed]
- Keller, M.; Hopp, L.; Liu, X.; Wohland, T.; Rohde, K.; Cancello, R.; Klös, M.; Bacos, K.; Kern, M.; Eichelmann, E.; et al. Genome-wide DNA promoter methylation and transcriptome analysis in human adipose tissue unravels novel candidate genes for obesity. Mol. Metab. 2017, 6, 86–100. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Meng, R.W.; Kunutsor, S.K.; Chowdhury, R.; Yuan, J.M.; Koh, W.P.; Pan, A. Plasma adiponectin levels and type 2 diabetes risk: A nested case-control study in a Chinese population and an updated meta-analysis. Sci. Rep. 2018, 8, 406. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Gene | Chr. | Variant | Type/Location | Risk allele/aa Change | Ethnicity | Pathogenicity | Reference |
|---|---|---|---|---|---|---|---|
| HNF1A | 12 | chr12:121437091 | missense | E508K | US Latino | higher | [40] |
| SLC16A11 | 17 | rs75493593 rs75418188 rs13342692 rs117767867 | missense | P443T G40S D127G V113I | European | higher | [40] |
| TPCN2 | 11 | rs1551305 | intronic | G | Chinese | higher | [41] |
| SLC30A8 | 8 | 8q24.11 | missense | R138X | Northern European | reduced | [33] |
| CCND2 | 12 | rs76895963 | intronic | G | Icelandic Danish | reduced | [42] |
| PDX1 | 13 | chr13:27396636delT | frameshift | G218Afs*12 | higher | ||
| PAM | 5 | rs35658696 rs78408340 | missense | D563G S539W | higher | ||
| THADA | 2 | rs7578597 | intronic | T | European | higher | [43] |
| KCNQ1 | 11 | rs163184 | intronic | G | higher | ||
| TCF7L2 | 10 | rs7903146 | intronic | T | higher | ||
| ADRA2A | 10 | rs10885122 | intronic | G | higher | ||
| CYB5A | 18 | rs7238987 | missense | P96P | Pima Indians | higher | [44] |
| RNF10 | 12 | chr12:120990399 | missense | R151H | higher |
| T2D loci | Effector Transcript | Epigenetic Signature | Tissue | Approach | Reference Study |
|---|---|---|---|---|---|
| 8q24.11 | SLC30A8 | DMR, open chromatin regions | skeletal muscle, subcutaneous adipose tissue pancreatic islets | DNA methylation array, FAIRE-seq, RRBS | [69,71,90] |
| 13q12.2 | PDX1 | open chromatin regions | pancreatic islets | FAIRE-seq | [71] |
| 4q21.23 | NKX6.1 | ||||
| 10q25.2-q25.3 | TCF7L2 | DMR | pancreatic islets | WGBS, RRBS | [67,90] |
| 9p24.2 | GLIS3 | DMR | pancreatic islets | WGBS | [67] |
| 2p21 | THADA | ||||
| 3p25.2 | PPARG | DMR | pancreatic islets | RRBS | [90] |
| 6p22.3 | CDKAL1 | ||||
| 17p13.3 | SRR | ||||
| 11p15.5 | DUSP8 | ||||
| 12q14.3 | HMGA2 | ||||
| 11p15.5-p15.4 | KCNQ1 | DMR | skeletal muscle, subcutaneous adipose tissue pancreatic islets | DNA methylation array, WGBS, RRBS | [69] [67] [90] |
| 10q23.33 | HHEX | DMR | skeletal muscle pancreatic islets | DNA methylation array, RRBS | [69] [90] |
| 3q21.1 | ADCY5 | DMR | subcutaneous adipose tissue pancreatic islets | DNA methylation array, RRBS | [69] [90] |
| Xq28 | DUSP9 | DMR | skeletal muscle, subcutaneous adipose tissuepancreatic islets | DNA methylation array, RRBS | [69,90] |
| 9p21.3 | CDKN2A | ||||
| 12q21.1 | TSPAN8 | DMR | subcutaneous adipose tissue pancreatic islets | DNA methylation array, RRBS | [69,90] |
| 4p16.1 | WFS1 | ||||
| 2q36.3 | IRS1 | ||||
| 16q12.2 | FTO | DMR | gametes | RRBS | [89] |
| 16p11.2 | SH2B1 | ||||
| 19q13.11 | CHST8 | ||||
| 1q21.1 | TXNIP | DMR | whole blood, liver, pancreatic islets, skeletal muscle | EWAS, WGBS | [80,81] |
| 21q22.3 | ABCG1 | DMR | whole blood | EWAS | [80,87] |
| 17q25.3 | SOCS3 | DMR | whole blood, liver | EWAS, WGBS | [80,81,87] |
| 17p11.2 | SREBF1 | whole blood, pancreatic islets | |||
| 7q32.2 | KLF14 | DMR | whole blood | WGBS | [81] |
| 2q12.2 | FHL2 | ||||
| 14q22.1 | GNPNAT1 | ||||
| 11p15.5 | IGF2 | DMR | whole blood | WGBS | [64] |
| 20q13.32 | GNASAS | ||||
| 7q32.1 | LEP | ||||
| 1q32.1 | IL10 | ||||
| 9q31.1 | ABCA1 | ||||
| 11p15.5 | INS-IGF2 | ||||
| 14q32.2 | MEG3 | ||||
| 3q26.2 | SLC2A2 | ||||
| 9q31.1 | NR4A3 | ||||
| 6q26 | PARK2 | ||||
| 12q21.3 | SOCS2 | ||||
| 2q36.3 | PID1 | ||||
| 4p15.2 | PPARGC1A | DMR | skeletal muscle | DNA methylation array | [68,69] |
| 4q13.3 | IL8 | [69] | |||
| 2p25.1 | KLF11 | ||||
| 20q13.12 | HNF4A | DMR | skeletal muscle, subcutaneous adipose tissue | DNA methylation array | [69] |
| 7q31.2 | CAV1 | subcutaneous adipose tissue | |||
| 3p25.3 | CIDEC | ||||
| 9p21.3 | CDKN2B | ||||
| 10q23.33 | IDE | ||||
| 11q14.3 | MTNR1B |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dziewulska, A.; Dobosz, A.M.; Dobrzyn, A. High-Throughput Approaches onto Uncover (Epi)Genomic Architecture of Type 2 Diabetes. Genes 2018, 9, 374. https://doi.org/10.3390/genes9080374
Dziewulska A, Dobosz AM, Dobrzyn A. High-Throughput Approaches onto Uncover (Epi)Genomic Architecture of Type 2 Diabetes. Genes. 2018; 9(8):374. https://doi.org/10.3390/genes9080374
Chicago/Turabian StyleDziewulska, Anna, Aneta M. Dobosz, and Agnieszka Dobrzyn. 2018. "High-Throughput Approaches onto Uncover (Epi)Genomic Architecture of Type 2 Diabetes" Genes 9, no. 8: 374. https://doi.org/10.3390/genes9080374
APA StyleDziewulska, A., Dobosz, A. M., & Dobrzyn, A. (2018). High-Throughput Approaches onto Uncover (Epi)Genomic Architecture of Type 2 Diabetes. Genes, 9(8), 374. https://doi.org/10.3390/genes9080374