Profiling DNA Methylation Based on Next-Generation Sequencing Approaches: New Insights and Clinical Applications




Abstract
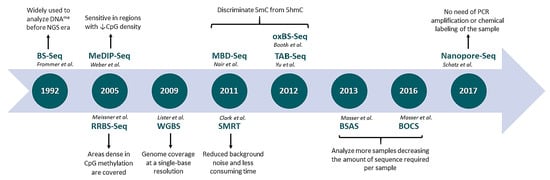
:
1. Introduction
2. DNA Methylation Profiling
2.1. Affinity Enrichment-Based Methods
2.2. Restriction Enzymes-Based Methods
2.3. Bisulfite Conversion-Based Methods
2.4. Oxidative Bisulfite Conversion-Based Methods
2.5. Capture-Based Methods
2.6. Third-Generation Sequencing
3. New Insights from Next-Generation Sequencing on Methylome Analysis: Strengths and Weaknesses
4. Non-Invasive DNA Methylation Detection Using Next-Generation Sequencing: Technical Advances and Challenges
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Esteller, M. Epigenetics in cancer. N. Engl. J. Med. 2008, 358, 1148–1159. [Google Scholar] [CrossRef] [PubMed]
- Laurent, L.; Wong, E.; Li, G.; Huynh, T.; Tsirigos, A.; Ong, C.T.; Low, H.M.; Sung, K.W.K.; Rigoutsos, I.; Loring, J. Dynamic changes in the human methylome during differentiation. Genome Res. 2010, 20, 320–331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maunakea, A.K.; Chepelev, I.; Cui, K.; Zhao, K. Intragenic DNA methylation modulates alternative splicing by recruiting MeCP2 to promote exon recognition. Cell Res. 2013, 23, 1256. [Google Scholar] [CrossRef] [PubMed]
- Smith, Z.D.; Meissner, A. DNA methylation: Roles in mammalian development. Nat. Rev. Genet. 2013, 14, 204–220. [Google Scholar] [CrossRef] [PubMed]
- Klosin, A.; Lehner, B. Mechanisms, timescales and principles of trans-generational epigenetic inheritance in animals. Curr. Opin. Genet. Dev. 2016, 36, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Issa, J.-P.J.; Kantarjian, H.M. Targeting DNA methylation. Clin. Cancer Res. 2009, 15, 3938–3946. [Google Scholar] [CrossRef] [PubMed]
- Heitzer, E.; Ulz, P.; Geigl, J.B. Circulating tumor DNA as a liquid biopsy for cancer. Clin. Chem. 2015, 61, 112–123. [Google Scholar] [CrossRef] [PubMed]
- Heyn, H.; Esteller, M. DNA methylation profiling in the clinic: Applications and challenges. Nat. Rev. Genet. 2012, 13, 679–692. [Google Scholar] [CrossRef] [PubMed]
- Soto, J.; Rodriguez-Antolin, C.; Vallespín, E.; De Castro Carpeño, J.; De Caceres, I.I. The impact of next-generation sequencing on the DNA methylation–based translational cancer research. Transl. Res. 2016, 169, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Hirst, M.; Marra, M.A. Next generation sequencing based approaches to epigenomics. Brief. Funct. Genom. 2010, 9, 455–465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hurd, P.J.; Nelson, C.J. Advantages of next-generation sequencing versus the microarray in epigenetic research. Brief. Funct. Genom. Proteom. 2009, 8, 174–183. [Google Scholar] [CrossRef] [PubMed]
- Lister, R.; Ecker, J.R. Finding the fifth base: Genome-wide sequencing of cytosine methylation. Genome Res. 2009, 19, 959–966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Behjati, S.; Tarpey, P.S. What is next generation sequencing? Arch. Dis. Child.-Educ. Pract. 2013, 98, 236–238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yong, W.-S.; F.-Hsu, M.; Chen, P.-Y. Profiling genome-wide DNA methylation. Epigenet. Chromatin 2016, 9, 26. [Google Scholar] [CrossRef] [PubMed]
- Laird, P.W. Principles and challenges of genome-wide DNA methylation analysis. Nat. Rev. Genet. 2010, 11, 191. [Google Scholar] [CrossRef] [PubMed]
- Anandhakumar, C.; Kizaki, S.; Bando, T.; Pandian, G.N.; Sugiyama, H. Advancing small-molecule-based chemical biology with next-generation sequencing technologies. Chembiochem 2015, 16, 20–38. [Google Scholar] [CrossRef] [PubMed]
- Serre, D.; Lee, B.H.; Ting, A.H. MBD-isolated Genome Sequencing provides a high-throughput and comprehensive survey of DNA methylation in the human genome. Nucleic Acids Res. 2009, 38, 391–399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brinkman, A.B.; Simmer, F.; Ma, K.; Kaan, A.; Zhu, J.; Stunnenberg, H.G. Whole-genome DNA methylation profiling using MethylCap-seq. Methods 2010, 52, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Cunningham, J.; Slager, S.; Kocher, J.P. Base resolution methylome profiling: Considerations in platform selection, data preprocessing and analysis. Epigenomics 2015, 7, 813–828. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Baheti, S.; Sun, Z. Statistical method evaluation for differentially methylated CpGs in base resolution next-generation DNA sequencing data. Brief. Bioinf. 2016, 19, 374–386. [Google Scholar] [CrossRef] [PubMed]
- Maunakea, A.K.; Nagarajan, R.P.; Bilenky, M.; Ballinger, T.J.; D’souza, C.; Fouse, S.D.; Johnson, B.E.; Hong, C.; Nielsen, C.; Zhao, Y.; et al. Conserved role of intragenic DNA methylation in regulating alternative promoters. Nature 2010, 466, 253. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Sun, J.; Wu, H.; Liu, S.; Wang, J.; Wu, B.; Huang, S.; Li, N.; Wang, J.; Zhang, X. Systematic assessment of reduced representation bisulfite sequencing to human blood samples: A promising method for large-sample-scale epigenomic studies. J. Biotechnol. 2012, 157, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Gu, H.; Smith, Z.D.; Bock, C.; Boyle, P.; Gnirke, A.; Meissner, A. Preparation of reduced representation bisulfite sequencing libraries for genome-scale DNA methylation profiling. Nat. Protoc. 2011, 6, 468. [Google Scholar] [CrossRef] [PubMed]
- Hayatsu, H. Discovery of bisulfite-mediated cytosine conversion to uracil, the key reaction for DNA methylation analysis—A personal account. Proc. Jpn. Acad. Ser. B 2008, 84, 321–330. [Google Scholar] [CrossRef]
- Frommer, M.; McDonald, L.E.; Millar, D.S.; Collis, C.M.; Watt, F.; Grigg, G.W.; Molloy, P.L.; Paul, C.L. A genomic sequencing protocol that yields a positive display of 5-methylcytosine residues in individual DNA strands. Proc. Natl. Acad. Sci. USA 1992, 89, 1827–1831. [Google Scholar] [CrossRef] [PubMed]
- Adusumalli, S.; Mohd Omar, M.F.; Soong, R.; Benoukraf, T. Methodological aspects of whole-genome bisulfite sequencing analysis. Brief. Bioinform. 2014, 16, 369–379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xi, Y.; Li, W. BSMAP: Whole genome bisulfite sequence MAPping program. BMC Bioinf. 2009, 10, 232. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, S.; Otto, C.; Kurtz, S.; Sharma, C.M.; Khaitovich, P.; Vogel, J.; Stadler, P.F.; Hackermüller, J. Fast mapping of short sequences with mismatches, insertions and deletions using index structures. PLoS Comput. Biol. 2009, 5, e1000502. [Google Scholar] [CrossRef] [PubMed]
- Krueger, F.; Andrews, S.R. Bismark: A flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics 2011, 27, 1571–1572. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.; Fiziev, P.; Yan, W.; Cokus, S.; Sun, X.; Zhang, M.Q.; Chen, P.Y.; Pellegrini, M. BS-Seeker2: A versatile aligning pipeline for bisulfite sequencing data. BMC Genom. 2013, 14, 774. [Google Scholar] [CrossRef] [PubMed]
- Rakyan, V.K.; Down, T.A.; Balding, D.J.; Beck, S. Epigenome-wide association studies for common human diseases. Nat. Rev. Genet. 2011, 12, 529. [Google Scholar] [CrossRef] [PubMed]
- Masser, D.R.; Berg, A.S.; Freeman, W.M. Focused, high accuracy 5-methylcytosine quantitation with base resolution by benchtop next-generation sequencing. Epigenet. Chromatin 2013, 6, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masser, D.R.; Stanford, D.R.; Freeman, W.M. Targeted DNA methylation analysis by next-generation sequencing. J. Vis. Exp. 2015. [Google Scholar] [CrossRef] [PubMed]
- Bashtrykov, P.; Jeltsch, A. DNA methylation analysis by bisulfite conversion coupled to double multiplexed amplicon-based next-generation sequencing (NGS). In Epigenome Editing; Springer Nature: Basel, Switzerland, 2018; pp. 367–382. [Google Scholar]
- Ito, S.; Shen, L.; Dai, Q.; Wu, S.C.; Collins, L.B.; Swenberg, J.A.; He, C.; Zhang, Y. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science 2011, 333, 1300–1303. [Google Scholar] [CrossRef] [PubMed]
- Shi, D.Q.; Ali, I.; Tang, J.; Yang, W.C. New insights into 5hmC DNA modification: Generation, distribution and function. Front. Genet. 2017, 8, 100. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Pastor, W.A.; Shen, Y.; Tahiliani, M.; Liu, D.R.; Rao, A. The behaviour of 5-hydroxymethylcytosine in bisulfite sequencing. PLoS ONE 2010, 5, e8888. [Google Scholar] [CrossRef] [PubMed]
- Hadad, N.; Masser, D.R.; Logan, S.; Wronowski, B.; Mangold, C.A.; Clark, N.; Otalora, L.; Unnikrishnan, A.; Ford, M.M.; Giles, C.B.; et al. Absence of genomic hypomethylation or regulation of cytosine-modifying enzymes with aging in male and female mice. Epigenet. Chromatin 2016, 9, 30. [Google Scholar] [CrossRef] [PubMed]
- Booth, M.J.; Marsico, G.; Bachman, M.; Beraldi, D.; Balasubramanian, S. Quantitative sequencing of 5-formylcytosine in DNA at single-base resolution. Nat. Chem. 2014, 6, 435. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.G.; Gross, J.A.; Lutz, P.E.; Vaillancourt, K.; Maussion, G.; Bramoulle, A.; Théroux, J.F.; Gardini, E.S.; Ehlert, U.; Bourret, G.; et al. Medium throughput bisulfite sequencing for accurate detection of 5-methylcytosine and 5-hydroxymethylcytosine. BMC Genom. 2017, 18, 96. [Google Scholar] [CrossRef] [PubMed]
- Song, C.-X.; Yi, C.; He, C. Mapping recently identified nucleotide variants in the genome and transcriptome. Nat. Biotechnol. 2012, 30, 1107. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Hon, G.C.; Szulwach, K.E.; Song, C.X.; Jin, P.; Ren, B.; He, C. Tet-assisted bisulfite sequencing of 5-hydroxymethylcytosine. Nat. Protoc. 2012, 7, 2159. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Hon, G.C.; Szulwach, K.E.; Song, C.X.; Zhang, L.; Kim, A.; Li, X.; Dai, Q.; Shen, Y.; Park, B.; et al. Base-resolution analysis of 5-hydroxymethylcytosine in the mammalian genome. Cell 2012, 149, 1368–1380. [Google Scholar] [CrossRef] [PubMed]
- Booth, M.J.; Branco, M.R.; Ficz, G.; Oxley, D.; Krueger, F.; Reik, W.; Balasubramanian, S. Quantitative sequencing of 5-methylcytosine and 5-hydroxymethylcytosine at single-base resolution. Science 2012, 336, 934–937. [Google Scholar] [CrossRef] [PubMed]
- Dapprich, J.; Ferriola, D.; Mackiewicz, K.; Clark, P.M.; Rappaport, E.; D’Arcy, M.; Sasson, A.; Gai, X.; Schug, J.; Kaestner, K.H.; et al. The next generation of target capture technologies-large DNA fragment enrichment and sequencing determines regional genomic variation of high complexity. BMC Genom. 2016, 17, 486. [Google Scholar] [CrossRef] [PubMed]
- Schmidl, C.; Klug, M.; Boeld, T.J.; Andreesen, R.; Hoffmann, P.; Edinger, M.; Rehli, M. Lineage-specific DNA methylation in T cells correlates with histone methylation and enhancer activity. Genome Res. 2009. [Google Scholar] [CrossRef] [PubMed]
- Irizarry, R.A.; Ladd-Acosta, C.; Wen, B.; Wu, Z.; Montano, C.; Onyango, P.; Cui, H.; Gabo, K.; Rongione, M.; Webster, M.; et al. The human colon cancer methylome shows similar hypo-and hypermethylation at conserved tissue-specific CpG island shores. Nat. Genet. 2009, 41, 178. [Google Scholar] [CrossRef] [PubMed]
- Bäckdahl, L.; Herberth, M.; Wilson, G.; Tate, P.; Campos, L.S.; Cortese, R.; Eckhardt, F.; Beck, S. Gene body methylation of the dimethylarginine dimethylamino-hydrolase 2 (Ddah2) gene is an epigenetic biomarker for neural stem cell differentiation. Epigenetics 2009, 4, 248–254. [Google Scholar] [CrossRef] [PubMed]
- Clark, T.A.; Spittle, K.E.; Turner, S.W.; Korlach, J. Direct detection and sequencing of damaged DNA bases. Genome Integr. 2011, 2, 10. [Google Scholar] [CrossRef] [PubMed]
- Bahassi, E.M.; Stambrook, P.J. Next-generation sequencing technologies: Breaking the sound barrier of human genetics. Mutagenesis 2014, 29, 303–310. [Google Scholar] [CrossRef] [PubMed]
- Jain, M.; Koren, S.; Miga, K.H.; Quick, J.; Rand, A.C.; Sasani, T.A.; Tyson, J.R.; Beggs, A.D.; Dilthey, A.T.; Fiddes, I.T. Nanopore sequencing and assembly of a human genome with ultra-long reads. Nat. Biotechnol. 2018, 36, 338. [Google Scholar] [CrossRef] [PubMed]
- Schatz, M.C. Nanopore sequencing meets epigenetics. Nat. Methods 2017, 14, 347. [Google Scholar] [CrossRef] [PubMed]
- Hegi, M.E.; Diserens, A.C.; Gorlia, T.; Hamou, M.F.; De Tribolet, N.; Weller, M.; Kros, J.M.; Hainfellner, J.A.; Mason, W.; Mariani, L.; et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N. Engl. J. Med. 2005, 352, 997–1003. [Google Scholar] [CrossRef] [PubMed]
- Island, B.C. BRCA1 CpG island hypermethylation predicts sensitivity to poly(adenosine diphosphate)-ribose polymerase inhibitors. J. Clin. Oncol. 2010, 28, e563–e564. [Google Scholar]
- Berglund, E.C.; Kiialainen, A.; Syvänen, A.-C. Next-generation sequencing technologies and applications for human genetic history and forensics. Investig. Genet. 2011, 2, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reuter, J.A.; Spacek, D.V.; Snyder, M.P. High-throughput sequencing technologies. Mol. Cell 2015, 58, 586–597. [Google Scholar] [CrossRef] [PubMed]
- Van Dijk, E.L.; Auger, H.; Jaszczyszyn, Y.; Thermes, C. Ten years of next-generation sequencing technology. Trends Genet. 2014, 30, 418–426. [Google Scholar] [CrossRef] [PubMed]
- Eid, J.; Fehr, A.; Gray, J.; Luong, K.; Lyle, J.; Otto, G.; Peluso, P.; Rank, D.; Baybayan, P.; Bettman, B.; et al. Real-time DNA sequencing from single polymerase molecules. Science 2009, 323, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Veras, A.A.O.; de Sá, P.H.C.G.; Pinheiro, K.C.; Assis das Graças, D.; Azevedo Baraúna, R.; Cruz Schneider, M.P.; Azevedo, V.; Ramos, R.T.J.; Silva, A. Efficiency of Corynebacterium pseudotuberculosis Cp31 genome assembly with the Hi-Q enzyme on an Ion Torrent PGM sequencing platform. J. Proteom. Bioinform. 2014, 7, 374–378. [Google Scholar]
- Mardis, E.R. Next-generation sequencing platforms. Ann. Rev. Anal. Chem. 2013, 6, 287–303. [Google Scholar] [CrossRef] [PubMed]
- Jain, M.; Fiddes, I.T.; Miga, K.H.; Olsen, H.E.; Paten, B.; Akeson, M. Improved data analysis for the MinION nanopore sequencer. Nat. Methods 2015, 12, 351. [Google Scholar] [CrossRef] [PubMed]
- Miles, B.N.; Ivanov, A.P.; Wilson, K.A.; Doğan, F.; Japrung, D.; Edel, J.B. Single molecule sensing with solid-state nanopores: Novel materials, methods, and applications. Chem. Soc. Rev. 2013, 42, 15–28. [Google Scholar] [CrossRef] [PubMed]
- Van den Oord, E.J.; Bukszar, J.; Rudolf, G.; Nerella, S.; McClay, J.L.; Xie, L.Y.; Aberg, K.A. Estimation of CpG coverage in whole methylome next-generation sequencing studies. BMC Bioinform. 2013, 14, 50. [Google Scholar] [CrossRef] [PubMed]
- Hung, S.S.; Meissner, B.; Chavez, E.A.; Ben-Neriah, S.; Ennishi, D.; Jones, M.R.; Shulha, H.P.; Chan, F.C.; Boyle, M.; Kridel, R.; et al. Assessment of capture and amplicon-based approaches for the development of a targeted next-generation sequencing pipeline to personalize lymphoma management. J. Mol. Diagn. 2018, 20, 203–214. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Sun, D.; Rodriguez, B.; Zhao, Q.; Sun, H.; Zhang, Y.; Li, W. BSeQC: Quality control of bisulfite sequencing experiments. Bioinformatics 2013, 29, 3227–3229. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Noviski, A.; Yu, X. MethyQA: A pipeline for bisulfite-treated methylation sequencing quality assessment. BMC Bioinform. 2013, 14, 259. [Google Scholar] [CrossRef] [PubMed]
- Mikeska, T.; Candiloro, I.L.; Dobrovic, A. The implications of heterogeneous DNA methylation for the accurate quantification of methylation. Epigenomics 2010, 2, 561–573. [Google Scholar] [CrossRef] [PubMed]
- Macaulay, I.C.; Ponting, C.P.; Voet, T. Single-cell multiomics: Multiple measurements from single cells. Trends Genet. 2017, 33, 155–168. [Google Scholar] [CrossRef] [PubMed]
- Smallwood, S.A.; Lee, H.J.; Angermueller, C.; Krueger, F.; Saadeh, H.; Peat, J.; Andrews, S.R.; Stegle, O.; Reik, W.; Kelsey, G. Single-cell genome-wide bisulfite sequencing for assessing epigenetic heterogeneity. Nat. Methods 2014, 11, 817. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Huang, K.; An, Q.; Du, G.; Hu, G.; Xue, J.; Zhu, X.; Wang, C.Y.; Xue, Z.; Fan, G. Simultaneous profiling of transcriptome and DNA methylome from a single cell. Genome Biol. 2016, 17, 88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Angermueller, C.; Clark, S.J.; Lee, H.J.; Macaulay, I.C.; Teng, M.J.; Hu, T.X.; Krueger, F.; Smallwood, S.A.; Ponting, C.P.; Voet, T.; et al. Parallel single-cell sequencing links transcriptional and epigenetic heterogeneity. Nat. Methods 2016, 13, 229. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.; Guo, H.; Cao, C.; Li, X.; Hu, B.; Zhu, P.; Wu, X.; Wen, L.; Tang, F.; Huang, Y.; et al. Single-cell triple omics sequencing reveals genetic, epigenetic, and transcriptomic heterogeneity in hepatocellular carcinomas. Cell Res. 2016, 26, 304. [Google Scholar] [CrossRef] [PubMed]
- Tanić, M.; Beck, S. Epigenome-wide association studies for cancer biomarker discovery in circulating cell-free DNA: Technical advances and challenges. Curr. Opin. Genet. Dev. 2017, 42, 48–55. [Google Scholar] [CrossRef] [PubMed]
- Vaca-Paniagua, F.; Oliver, J.; da Costa, A.N.; Merle, P.; McKay, J.; Herceg, Z.; Holmila, R. Targeted deep DNA methylation analysis of circulating cell-free DNA in plasma using massively parallel semiconductor sequencing. Epigenomics 2015, 7, 353–362. [Google Scholar] [CrossRef] [PubMed]
- Janku, F.; Huang, H.J.; Claes, B.; Falchook, G.S.; Fu, S.; Hong, D.; Ramzanali, N.M.; Nitti, G.; Cabrilo, G.; Tsimberidou, A.M.; et al. BRAF mutation testing in cell-free DNA from the plasma of patients with advanced cancers using a rapid, automated molecular diagnostics system. Mol. Cancer Ther. 2016, 15, 1397–1404. [Google Scholar] [CrossRef] [PubMed]
- Janku, F.; Huang, H.J.; Fujii, T.; Shelton, D.N.; Madwani, K.; Fu, S.; Tsimberidou, A.M.; Piha-Paul, S.A.; Wheler, J.J.; Zinner, R.G.; et al. Multiplex KRASG12/G13 mutation testing of unamplified cell-free DNA from the plasma of patients with advanced cancers using droplet digital polymerase chain reaction. Ann. Oncol. 2017, 28, 642–650. [Google Scholar] [PubMed]
- Diehl, F.; Schmidt, K.; Choti, M.A.; Romans, K.; Goodman, S.; Li, M.; Thornton, K.; Agrawal, N.; Sokoll, L.; Szabo, S.A.; et al. Circulating mutant DNA to assess tumor dynamics. Nat. Med. 2008, 14, 985. [Google Scholar] [CrossRef] [PubMed]
- Hao, X.; Luo, H.; Krawczyk, M.; Wei, W.; Wang, W.; Wang, J.; Flagg, K.; Hou, J.; Zhang, H.; Yi, S.; et al. DNA methylation markers for diagnosis and prognosis of common cancers. Proc. Natl. Acad. Sci. USA 2017, 114, 7414–7419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janku, F.; Zhang, S.; Waters, J.; Liu, L.; Huang, H.J.; Subbiah, V.; Hong, D.S.; Karp, D.D.; Fu, S.; Cai, X.; et al. Development and validation of an ultradeep next-generation sequencing assay for testing of plasma cell-free DNA from patients with advanced cancer. Clin. Cancer Res. 2017, 23, 5648–5656. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, A.F.; Assenov, Y.; Martin-Subero, J.I.; Balint, B.; Siebert, R.; Taniguchi, H.; Yamamoto, H.; Hidalgo, M.; Tan, A.C.; Galm, O.; et al. A DNA methylation fingerprint of 1628 human samples. Genome Res. 2012, 22, 407–419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moran, S.; Martínez-Cardús, A.; Sayols, S.; Musulén, E.; Balañá, C.; Estival-Gonzalez, A.; Moutinho, C.; Heyn, H.; Diaz-Lagares, A.; de Moura, M.C.; et al. Epigenetic profiling to classify cancer of unknown primary: A multicentre, retrospective analysis. Lancet Oncol. 2016, 17, 1386–1395. [Google Scholar] [CrossRef]
- Chan, K.A.; Jiang, P.; Zheng, Y.W.; Liao, G.J.; Sun, H.; Wong, J.; Siu, S.S.N.; Chan, W.C.; Chan, S.L.; Chan, A.T.; et al. Cancer genome scanning in plasma: Detection of tumor-associated copy number aberrations, single-nucleotide variants, and tumoral heterogeneity by massively parallel sequencing. Clin. Chem. 2013, 59, 211–224. [Google Scholar] [CrossRef] [PubMed]
- Madic, J.; Kiialainen, A.; Bidard, F.C.; Birzele, F.; Ramey, G.; Leroy, Q.; Frio, T.R.; Vaucher, I.; Raynal, V.; Bernard, V.; et al. Circulating tumor DNA and circulating tumor cells in metastatic triple negative breast cancer patients. Int. J. Cancer 2015, 136, 2158–2165. [Google Scholar] [CrossRef] [PubMed]
- Couraud, S.; Paniagua, F.V.; Villar, S.; Oliver, J.; Schuster, T.; Blanche, H.; Girard, N.; Tredaniel, J.; Guilleminault, L.; Gervais, R.; et al. Non-invasive diagnosis of actionable mutations by deep sequencing of circulating-free DNA in non-small cell lung cancer: Findings from BioCAST/IFCT-1002. Clin. Cancer Res. 2014, 20, 4613–4624. [Google Scholar] [CrossRef] [PubMed]
- Warton, K.; Lin, V.; Navin, T.; Armstrong, N.J.; Kaplan, W.; Ying, K.; Gloss, B.; Mangs, H.; Nair, S.S.; Hacker, N.F.; et al. Methylation-capture and next-generation sequencing of free circulating DNA from human plasma. BMC Genom. 2014, 15, 476. [Google Scholar] [CrossRef] [PubMed]
- Lan, X.; Adams, C.; Landers, M.; Dudas, M.; Krissinger, D.; Marnellos, G.; Bonneville, R.; Xu, M.; Wang, J.; Huang, T.H.M.; et al. High resolution detection and analysis of CpG dinucleotides methylation using MBD-Seq technology. PLoS ONE 2011, 6, e22226. [Google Scholar] [CrossRef] [PubMed]
- Nair, S.S.; Coolen, M.W.; Stirzaker, C.; Song, J.Z.; Statham, A.L.; Strbenac, D.; Robinson, M.D.; Clark, S.J. Comparison of methyl-DNA immunoprecipitation (MeDIP) and methyl-CpG binding domain (MBD) protein capture for genome-wide DNA methylation analysis reveal CpG sequence coverage bias. Epigenetics 2011, 6, 34–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]


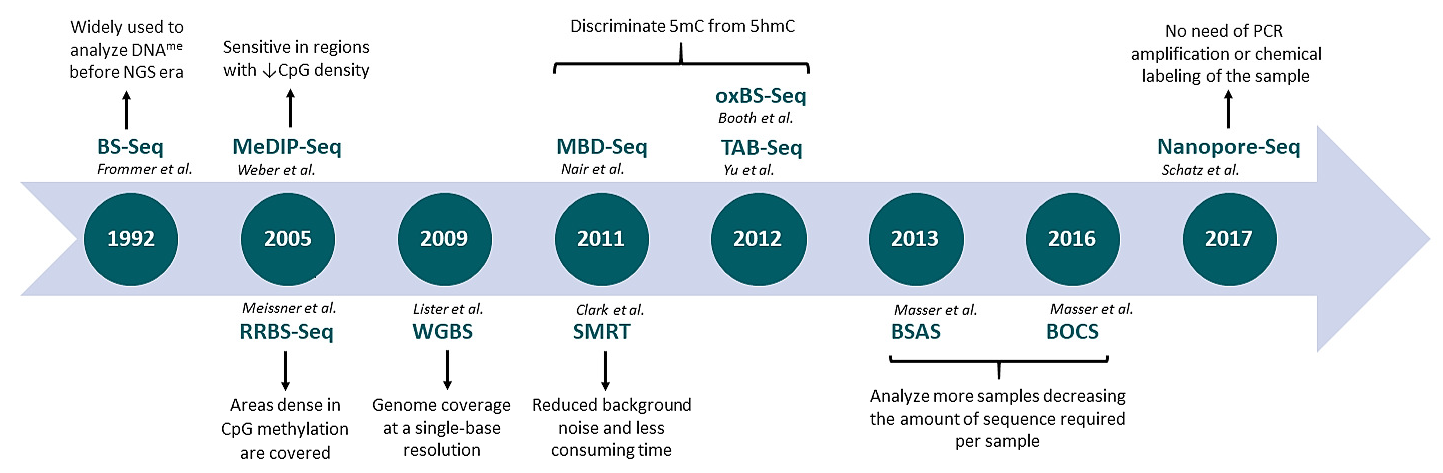{kind=link}
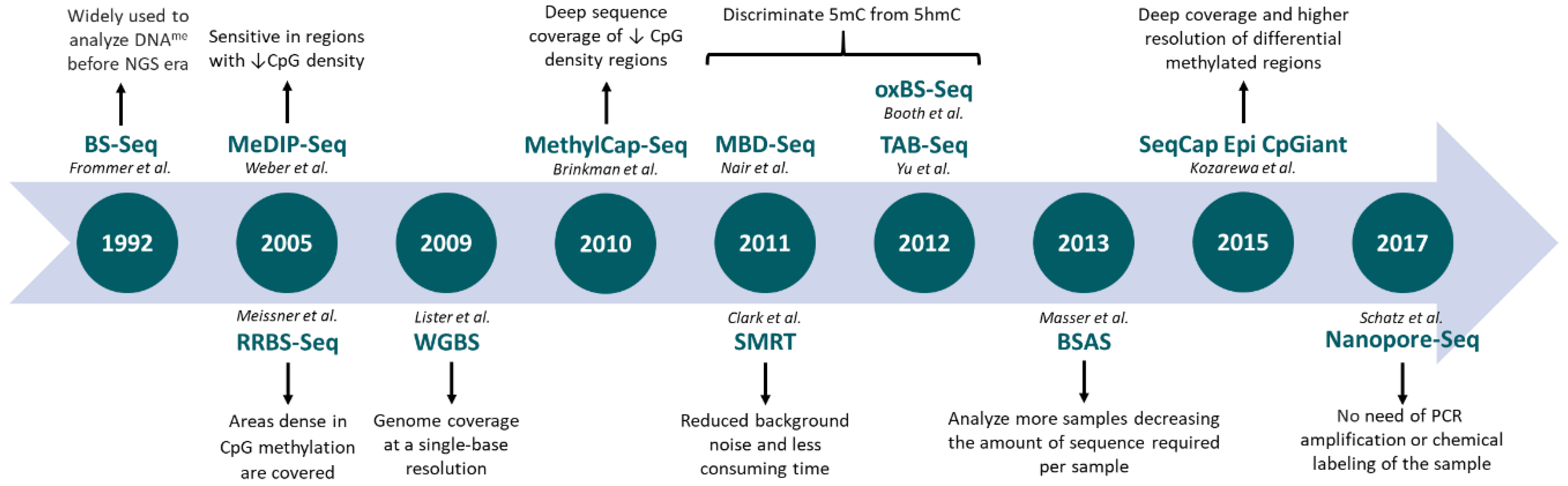{kind=link}
| Affinity Enrichment-Based Methods | Restriction Enzymes-Based Methods | Bisulfite Conversion-Based Methods | |
|---|---|---|---|
| Resolution | ~150 bp | Single-base | Single-base |
| Reads/sample | ~30–50 million reads | ~10 million reads | >500 million reads |
| CpGs covered | ~23 million CpGs | ~2 million CpGs | >28 million CpGs |
| Pros | Cost-effective method No mutations introduced | High sensitivity with lower costs | Evaluate methylation status of every CpG site |
| Cons | Biased toward hypermethylated regions Inability to predict absolute methylation level | CpGs in regions without the enzyme restriction site are not covered | Higher costs Requires high DNA input Substantial DNA degradation after bisulfite treatment |
| Application | Suitable for rapid, large scale and low-resolution studies | Suitable for site-specific/targeted studies | Suitable for high resolution studies |
| Sequencing Platform Developers | Sequencing Principle | Key Features | Limitations | Reference |
|---|---|---|---|---|
| Illumina | Sequencing by synthesis | High throughput | Higher cost per read | [55,56,57] |
| Life Technologies Ion Torrent | Polymerization | Simple detection method | Low read number per run | [58,59] |
| Pacific Biosciences PacBio | Single molecule real time ligation | Single molecule detection and long read length | High error rates (13%) and low read number per run | [60] |
| Oxford Nanopore | Nanopore sensing | Single molecule and label-free detection with reduced costs | High error rates (38.2%) | [61,62] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barros-Silva, D.; Marques, C.J.; Henrique, R.; Jerónimo, C. Profiling DNA Methylation Based on Next-Generation Sequencing Approaches: New Insights and Clinical Applications. Genes 2018, 9, 429. https://doi.org/10.3390/genes9090429
Barros-Silva D, Marques CJ, Henrique R, Jerónimo C. Profiling DNA Methylation Based on Next-Generation Sequencing Approaches: New Insights and Clinical Applications. Genes. 2018; 9(9):429. https://doi.org/10.3390/genes9090429
Chicago/Turabian StyleBarros-Silva, Daniela, C. Joana Marques, Rui Henrique, and Carmen Jerónimo. 2018. "Profiling DNA Methylation Based on Next-Generation Sequencing Approaches: New Insights and Clinical Applications" Genes 9, no. 9: 429. https://doi.org/10.3390/genes9090429
APA StyleBarros-Silva, D., Marques, C. J., Henrique, R., & Jerónimo, C. (2018). Profiling DNA Methylation Based on Next-Generation Sequencing Approaches: New Insights and Clinical Applications. Genes, 9(9), 429. https://doi.org/10.3390/genes9090429