Unravelling the Roles of Susceptibility Loci for Autoimmune Diseases in the Post-GWAS Era



{kind=link}
{kind=link}
Abstract
:1. Introduction
2. The Genome-Wide Association Studies Era
3. Missing Heritability in Genome-Wide Association Studies
4. The Role of Complex Loci at Genetic Level
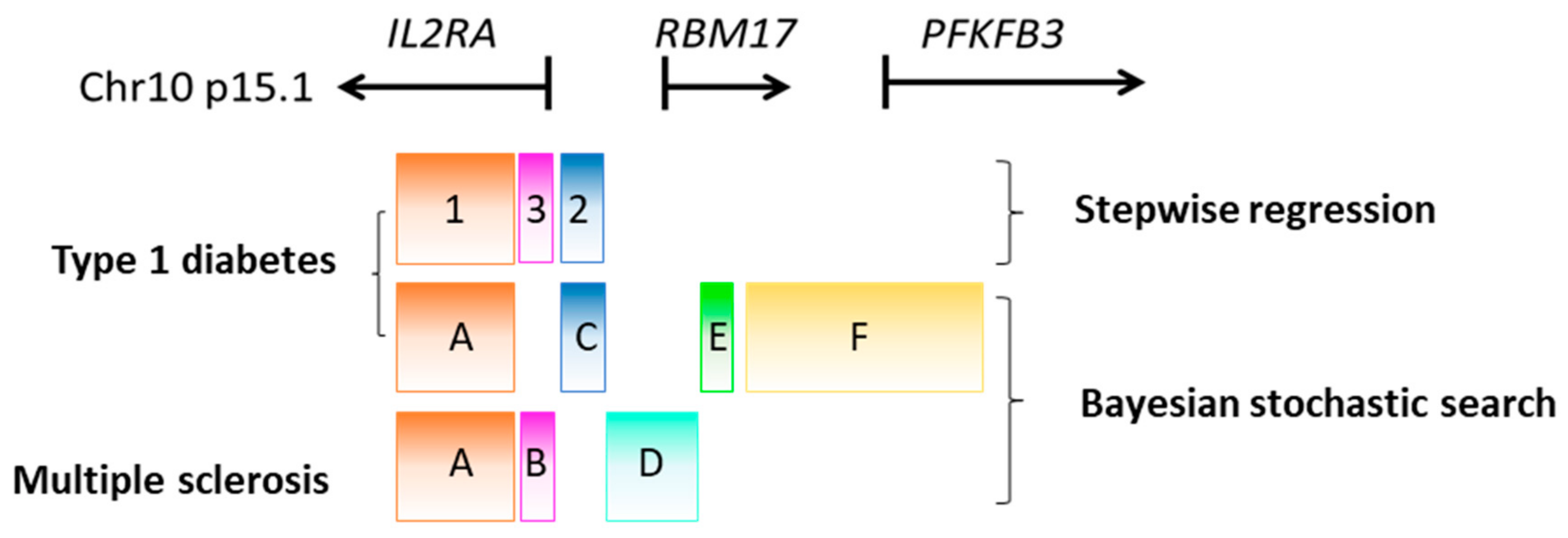
5. Identifying Candidate Causal Variants Using Fine Mapping
6. Epistasis in Autoimmune Diseases
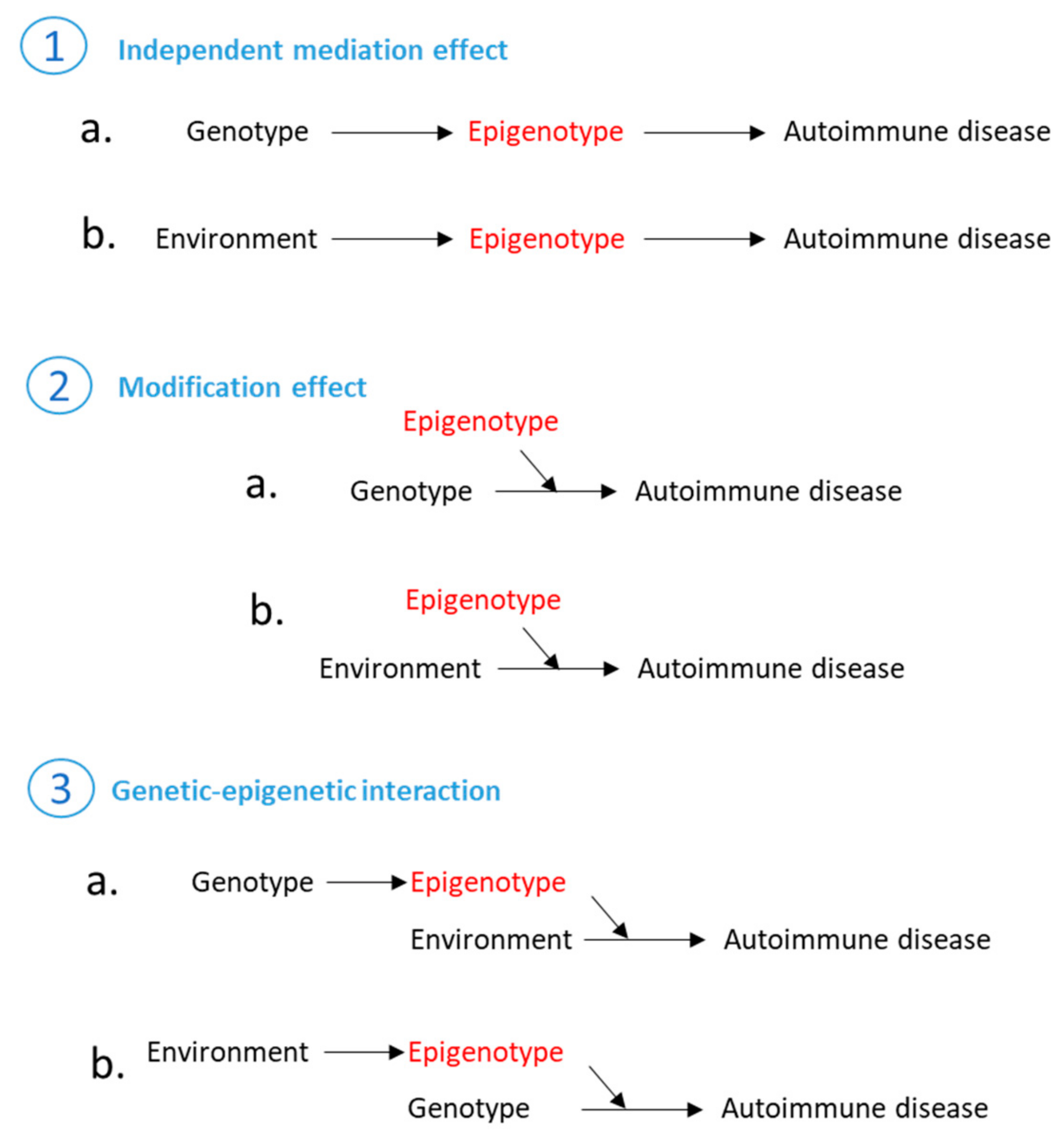
7. Epigenetic Regulation
8. Conclusions
Funding
Conflicts of Interest
References
- Campbell, A.W. Autoimmunity and the Gut. Autoimmune Dis. 2014. [Google Scholar] [CrossRef] [PubMed]
- Barrett, J.C.; Clayton, D.G.; Concannon, P.; Akolkar, B.; Cooper, J.D.; Erlich, H.A.; Julier, C.; Morahan, G.; Nerup, J.; Nierras, C.; et al. Rich, and consortium type 1 diabetes genetics. genome-wide association study and meta-analysis find that over 40 loci affect risk of type 1 diabetes. Nat. Genet. 2009, 41, 703–707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kappelman, M.D.; Moore, K.R.; Allen, J.K.; Cook, S.F. Recent trends in the prevalence of Crohn’s disease and ulcerative colitis in a commercially insured US population. Dig. Dis. Sci. 2013, 58, 519–525. [Google Scholar] [CrossRef] [PubMed]
- Benito-Leon, J. Are the prevalence and incidence of multiple sclerosis changing? Neuroepidemiology 2011, 36, 148–149. [Google Scholar] [CrossRef] [PubMed]
- Myasoedova, E.; Crowson, C.S.; Kremers, H.M.; Therneau, T.M.; Gabriel, S.E. Is the incidence of rheumatoid arthritis rising? Results from Olmsted County, Minnesota, 1955–2007. Arthritis Rheum. 2010, 62, 1576–1582. [Google Scholar] [CrossRef] [PubMed]
- Ercolini, A.M.; Miller, S.D. The role of infections in autoimmune disease. Clin. Exp. Immunol. 2009, 155, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manzel, A.; Muller, D.N.; Hafler, D.A.; Erdman, S.E.; Linker, R.A.; Kleinewietfeld, M. Role of western diet in inflammatory autoimmune diseases. Curr. Allergy Asthma Rep. 2014, 14. [Google Scholar] [CrossRef] [PubMed]
- Vieira, S.M.; Pagovich, O.E.; Kriegel, M.A. Diet, microbiota and autoimmune diseases. Lupus 2014, 23, 518–526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fronczak, C.M.; Baron, A.E.; Chase, H.P.; Ross, C.; Brady, H.L.; Hoffman, M.; Eisenbarth, G.S.; Rewers, M.; Norris, J.M. In utero dietary exposures and risk of islet autoimmunity in children. Diabetes Care 2003, 26, 3237–3242. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, C.W. Questions persist: Environmental factors in autoimmune disease. Environ. Health Perspect. 2011, 119, A249–A253. [Google Scholar] [CrossRef] [PubMed]
- Selmi, C.; Tsuneyama, K. Nutrition, geoepidemiology, and autoimmunity. Autoimmun. Rev. 2010, 9, A267–A270. [Google Scholar] [CrossRef] [PubMed]
- Stojanovich, L. Stress and Autoimmunity. Autoimmun. Rev. 2010, 9, A271–A276. [Google Scholar] [CrossRef] [PubMed]
- Knip, M.; Honkanen, J. Modulation of type 1 diabetes risk by the intestinal microbiome. Curr. Diabetes Rep. 2017, 17. [Google Scholar] [CrossRef] [PubMed]
- Watad, A.; Azrielant, S.; Bragazzi, N.L.; Sharif, K.; David, P.; Katz, I.; Aljadeff, G.; Quaresma, M.; Tanay, G.; Adawi, M.; et al. Seasonality and autoimmune diseases: The contribution of the four seasons to the mosaic of autoimmunity. J. Autoimmun. 2017, 82, 13–30. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Wong, F.S.; Wen, L. Antibiotics, gut microbiota, environment in early life and type 1 diabetes. Pharmacol. Res. 2017, 119, 219–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singal, D.P.; Blajchman, M.A. Histocompatibility (HL-A) antigens, lymphocytotoxic antibodies and tissue antibodies in patients with diabetes mellitus. Diabetes 1973, 22, 429–432. [Google Scholar] [CrossRef] [PubMed]
- Cudworth, A.G.; Woodrow, J.C. Letter: HL-A antigens and diabetes mellitus. Lancet 1974, 2. [Google Scholar] [CrossRef]
- Stastny, P. Mixed lymphocyte cultures in rheumatoid arthritis. J. Clin. Investig. 1976, 57, 1148–1157. [Google Scholar] [CrossRef] [PubMed]
- Moser, K.L.; Kelly, J.A.; Lessard, C.J.; Harley, J.B. Recent insights into the genetic basis of systemic lupus erythematosus. Genes Immun. 2009, 10, 373–379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlosstein, L.; Terasaki, P.I.; Bluestone, R.; Pearson, C.M. High association of an HL-A antigen, W27, with ankylosing spondylitis. N. Engl. J. Med. 1973, 288, 704–706. [Google Scholar] [CrossRef] [PubMed]
- Bennett, S.T.; Wilson, A.J.; Cucca, F.; Nerup, J.; Pociot, F.; McKinney, P.A.; Barnett, A.H.; Bain, S.C.; Todd, J.A. IDDM2-VNTR-encoded susceptibility to type 1 diabetes: Dominant protection and parental transmission of alleles of the insulin gene-linked minisatellite locus. J. Autoimmun. 1996, 9, 415–421. [Google Scholar] [CrossRef] [PubMed]
- Lucassen, A.M.; Julier, C.; Beressi, J.P.; Boitard, C.; Froguel, P.; Lathrop, M.; Bell, J.I. Susceptibility to insulin dependent diabetes mellitus maps to a 4.1 kb segment of DNA spanning the insulin gene and associated VNTR. Nat. Genet. 1993, 4, 305–310. [Google Scholar] [CrossRef] [PubMed]
- Bell, G.I.; Horita, S.; Karam, J.H. A polymorphic locus near the human insulin gene is associated with insulin-dependent diabetes mellitus. Diabetes 1984, 33, 176–183. [Google Scholar] [CrossRef] [PubMed]
- Rawlings, D.J.; Dai, X.; Buckner, J.H. The role of PTPN22 risk variant in the development of autoimmunity: Finding common ground between mouse and human. J. Immunol. 2015, 194, 2977–2984. [Google Scholar] [CrossRef] [PubMed]
- Gough, S.C.; Walker, L.S.; Sansom, D.M. CTLA4 gene polymorphism and autoimmunity. Immunol. Rev. 2005, 204, 102–115. [Google Scholar] [CrossRef] [PubMed]
- Martin, J.E.; Carmona, F.D.; Broen, J.C.; Simeon, C.P.; Vonk, M.C.; Carreira, P.; Rios-Fernandez, R.; Espinosa, G.; Vicente-Rabaneda, E.; Tolosa, C.; et al. The autoimmune disease-associated IL2RA locus is involved in the clinical manifestations of systemic sclerosis. Genes Immun. 2012, 13, 191–196. [Google Scholar] [CrossRef] [PubMed]
- Stahl, E.A.; Raychaudhuri, S.; Remmers, E.F.; Xie, G.; Eyre, S.; Thomson, B.P.; Li, Y.; Kurreeman, F.A.; Zhernakova, A.; Hinks, A.; et al. Genome-wide association study meta-analysis identifies seven new rheumatoid arthritis risk loci. Nat. Genet. 2010, 42, 508–514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gutierrez-Arcelus, M.; Rich, S.S.; Raychaudhuri, S. Autoimmune diseases—Connecting risk alleles with molecular traits of the immune system. Nat. Rev. Genet. 2016, 17, 160–174. [Google Scholar] [CrossRef] [PubMed]
- Wellcome Trust Case Control Consortium. Genome-wide association study of 14,000 cases of seven common diseases and 3000 shared controls. Nature 2007, 447, 661–678. [Google Scholar]
- Long, T.; Hicks, M.; Yu, H.C.; Biggs, W.H.; Kirkness, E.F.; Menni, C.; Zierer, J.; Small, K.S.; Mangino, M.; Messier, H.; et al. Whole-genome sequencing identifies common-to-rare variants associated with human blood metabolites. Nat. Genet. 2017, 49, 568–578. [Google Scholar] [CrossRef] [PubMed]
- de Vries, P.S.; Sabater-Lleal, M.; Chasman, D.I.; Trompet, S.; Ahluwalia, T.S.; Teumer, A.; Kleber, M.E.; Chen, M.H.; Wang, J.J.; Attia, J.R.; et al. Comparison of hapmap and 1000 genomes reference panels in a large-scale genome-wide association study. PLoS ONE 2017, 12. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.R.; Li, J.; Zhao, S.D.; Bradfield, J.P.; Mentch, F.D.; Maggadottir, S.M.; Hou, C.; Abrams, D.J.; Chang, D.; Gao, F.; et al. Meta-analysis of shared genetic architecture across ten pediatric autoimmune diseases. Nat. Med. 2015, 21, 1018–1027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marees, A.T.; de Kluiver, H.; Stringer, S.; Vorspan, F.; Curis, E.; Marie-Claire, C.; Derks, E.M. A Tutorial on conducting genome-wide association studies: Quality control and statistical analysis. Int. J. Methods Psychiatr. Res. 2018, 27. [Google Scholar] [CrossRef] [PubMed]
- Wray, N.R.; Yang, J.; Hayes, B.J.; Price, A.L.; Goddard, M.E.; Visscher, P.M. Pitfalls of predicting complex traits from SNPs. Nat. Rev. Genet. 2013, 14, 507–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sorosina, M.; Esposito, F.; Guaschino, C.; Clarelli, F.; Barizzone, N.; Osiceanu, A.M.; Brambilla, P.; Mascia, E.; Cavalla, P.; Gallo, P.; et al. Inverse correlation of genetic risk score with age at onset in bout-onset and progressive-onset multiple sclerosis. Mult. Scler. 2015, 21, 1463–1467. [Google Scholar] [CrossRef] [PubMed]
- Patel, K.A.; Oram, R.A.; Flanagan, S.E.; de Franco, E.; Colclough, K.; Shepherd, M.; Ellard, S.; Weedon, M.N.; Hattersley, A.T. Type 1 diabetes genetic risk score: A novel tool to discriminate monogenic and type 1 diabetes. Diabetes 2016, 65, 2094–2099. [Google Scholar] [CrossRef] [PubMed]
- Oram, R.A.; Patel, K.; Hill, A.; Shields, B.; McDonald, T.J.; Jones, A.; Hattersley, A.T.; Weedon, M.N. A type 1 diabetes genetic risk score can aid discrimination between type 1 and type 2 diabetes in young adults. Diabetes Care 2016, 39, 337–344. [Google Scholar] [CrossRef] [PubMed]
- Johnson, M.B.; Patel, K.A.; de Franco, E.; Houghton, J.A.L.; McDonald, T.J.; Ellard, S.; Flanagan, S.E.; Hattersley, A.T. A type 1 diabetes genetic risk score can discriminate monogenic autoimmunity with diabetes from early-onset clustering of polygenic autoimmunity with diabetes. Diabetologia 2018, 61, 862–869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polychronakos, C.; Li, Q. Understanding type 1 diabetes through genetics: Advances and prospects. Nat. Rev. Genet. 2011, 12, 781–792. [Google Scholar] [CrossRef] [PubMed]
- Zaitlen, N.; Kraft, A.P. Heritability in the genome-wide association era. Hum. Genet. 2012, 131, 1655–1664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.R.; Zhao, S.D.; Li, J.; Bradfield, J.P.; Mohebnasab, M.; Steel, L.; Kobie, J.; Abrams, D.J.; Mentch, F.D.; Glessner, J.T.; et al. Genetic sharing and heritability of paediatric age of onset autoimmune diseases. Nat. Commun. 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Hunt, K.A.; Mistry, V.; Bockett, N.A.; Ahmad, T.; Ban, M.; Barker, J.N.; Barrett, J.C.; Blackburn, H.; Brand, O.; Burren, O.; et al. Negligible impact of rare autoimmune-locus coding-region variants on missing heritability. Nature 2013, 498, 232–235. [Google Scholar] [CrossRef] [PubMed]
- Zuk, O.; Hechter, E.; Sunyaev, S.R.; Lander, E.S. The mystery of missing heritability: Genetic interactions create phantom heritability. Proc. Natl. Acad. Sci. USA 2012, 109, 1193–1198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kilpinen, H.; Dermitzakis, E.T. Genetic and epigenetic contribution to complex traits. Hum. Mol. Genet. 2012, 21, R24–R28. [Google Scholar] [CrossRef] [PubMed]
- Freeman, J.L.; Perry, G.H.; Feuk, L.; Redon, R.; McCarroll, S.A.; Altshuler, D.M.; Aburatani, H.; Jones, K.W.; Tyler-Smith, C.; Hurles, M.E.; et al. Copy number variation: New insights in genome diversity. Genom. Res. 2006, 16, 949–961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ricano-Ponce, I.; Zhernakova, D.V.; Deelen, P.; Luo, O.; Li, X.; Isaacs, A.; Karjalainen, J.; di Tommaso, J.; Borek, Z.A.; Zorro, M.M.; et al. Refined mapping of autoimmune disease associated genetic variants with gene expression suggests an important role for non-coding rnas. J. Autoimmun. 2016, 68, 62–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strachan, T.; Read, A.P.; Strachan, T. Human Molecular Genetics, 4th ed.; Garland Science: New York, NY, USA, 2011. [Google Scholar]
- Stead, J.D.; Buard, J.; Todd, J.A.; Jeffreys, A.J. Influence of allele lineage on the role of the insulin minisatellite in susceptibility to type 1 diabetes. Hum. Mol. Genet. 2000, 9, 2929–2935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pugliese, A.; Zeller, M.; Fernandez, A., Jr.; Zalcberg, L.J.; Bartlett, R.J.; Ricordi, C.; Pietropaolo, M.; Eisenbarth, G.S.; Bennett, S.T.; Patel, D.D. The insulin gene is transcribed in the human thymus and transcription levels correlated with allelic variation at the INS VNTR-IDDM2 susceptibility locus for type 1 diabetes. Nat. Genet. 1997, 15, 293–297. [Google Scholar] [CrossRef] [PubMed]
- Mok, C.C.; Lau, C.S. Pathogenesis of systemic lupus erythematosus. J. Clin. Pathol. 2003, 56, 481–490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.; Chung, E.K.; Wu, Y.L.; Savelli, S.L.; Nagaraja, H.N.; Zhou, B.; Hebert, M.; Jones, K.N.; Shu, Y.; Kitzmiller, K.; et al. Gene copy-number variation and associated polymorphisms of complement component C4 in human systemic lupus erythematosus (SLE): Low copy number is a risk factor for and high copy number is a protective factor against sle susceptibility in European Americans. Am. J. Hum. Genet. 2007, 80, 1037–1054. [Google Scholar] [PubMed]
- Levy, S.; Sutton, G.; Ng, P.C.; Feuk, L.; Halpern, A.L.; Walenz, B.P.; Axelrod, N.; Huang, J.; Kirkness, E.F.; Denisov, G.; et al. The diploid genome sequence of an individual human. PLoS Biol. 2007, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wheeler, D.A.; Srinivasan, M.; Egholm, M.; Shen, Y.; Chen, L.; McGuire, A.; He, W.; Chen, Y.J.; Makhijani, V.; Roth, G.T.; et al. The complete genome of an individual by massively parallel DNA sequencing. Nature 2008, 452, 872–876. [Google Scholar] [CrossRef] [PubMed]
- Wellcome Trust Case Control Consortium; Craddock, N.; Hurles, M.E.; Cardin, N.; Pearson, R.D.; Plagnol, V.; Robson, S.; Vukcevic, D.; Barnes, C.; Conrad, D.F.; et al. Genome-wide association study of CNVs in 16,000 cases of eight common diseases and 3000 shared controls. Nature 2010, 464, 713–720. [Google Scholar] [PubMed]
- Zanda, M.; Onengut-Gumuscu, S.; Walker, N.; Shtir, C.; Gallo, D.; Wallace, C.; Smyth, D.; Todd, J.A.; Hurles, M.E.; Plagnol, V.; et al. A genome-wide assessment of the role of untagged copy number variants in type 1 diabetes. PLoS Genet. 2014, 10. [Google Scholar] [CrossRef] [PubMed]
- Cooper, N.J.; Shtir, C.J.; Smyth, D.J.; Guo, H.; Swafford, A.D.; Zanda, M.; Hurles, M.E.; Walker, N.M.; Plagnol, V.; Cooper, J.D.; et al. Detection and correction of artefacts in estimation of rare copy number variants and analysis of rare deletions in type 1 diabetes. Hum. Mol. Genet. 2015, 24, 1774–1790. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, H.M.; Broderick, L. Editorial: It just takes one: Somatic mosaicism in autoinflammatory disease. Arthritis Rheumatol. 2017, 69, 253–256. [Google Scholar] [CrossRef] [PubMed]
- Bruder, C.E.; Piotrowski, A.; Gijsbers, A.A.; Andersson, R.; Erickson, S.; de Stahl, T.D.; Menzel, U.; Sandgren, J.; von Tell, D.; Poplawski, A.; et al. Phenotypically concordant and discordant monozygotic twins display different DNA copy-number-variation profiles. Am. J. Hum. Genet. 2008, 82, 763–771. [Google Scholar] [CrossRef] [PubMed]
- Selmi, C.; Cavaciocchi, F.; Lleo, A.; Cheroni, C.; de Francesco, R.; Lombardi, S.A.; de Santis, M.; Meda, F.; Raimondo, M.G.; Crotti, C.; et al. Genome-wide analysis of DNA methylation, copy number variation, and gene expression in monozygotic twins discordant for primary biliary cirrhosis. Front. Immunol. 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Rao, V.K.; Oliveira, J.B. How I treat autoimmune lymphoproliferative syndrome. Blood 2011, 118, 5741–5751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holzelova, E.; Vonarbourg, C.; Stolzenberg, M.C.; Arkwright, P.D.; Selz, F.; Prieur, A.M.; Blanche, S.; Bartunkova, J.; Vilmer, E.; Fischer, A.; et al. Autoimmune lymphoproliferative syndrome with somatic Fas mutations. N. Engl. J. Med. 2004, 351, 1409–1418. [Google Scholar] [CrossRef] [PubMed]
- Moritake, H.; Takagi, M.; Kinoshita, M.; Ohara, O.; Yamamoto, S.; Moriguchi, S.; Nunoi, H. Autoimmunity including intestinal Behçet disease bearing the KRAS mutation in lymphocytes: A case report. Pediatrics 2016, 137. [Google Scholar] [CrossRef] [PubMed]
- Mayhew, A.J.; Meyre, D. Assessing the heritability of complex traits in humans: Methodological challenges and opportunities. Curr. Genom. 2017, 18, 332–340. [Google Scholar] [CrossRef] [PubMed]
- Trerotola, M.; Relli, V.; Simeone, P.; Alberti, S. Epigenetic inheritance and the missing heritability. Hum. Genom. 2015, 9. [Google Scholar] [CrossRef] [PubMed]
- Wallace, C.; Cutler, A.J.; Pontikos, N.; Pekalski, M.L.; Burren, O.S.; Cooper, J.D.; Garcia, A.R.; Ferreira, R.C.; Guo, H.; Walker, N.M.; et al. Dissection of a complex disease susceptibility region using a bayesian stochastic search approach to fine mapping. PLoS Genet. 2015, 11. [Google Scholar] [CrossRef] [PubMed]
- International MHC and Autoimmunity Genetics Network; Rioux, J.D.; Goyette, P.; Vyse, T.J.; Hammarstrom, L.; Fernando, M.M.; Green, T.; de Jager, P.L.; Foisy, S.; et al. Mapping of multiple susceptibility variants within the MHC region for 7 immune-mediated diseases. Proc. Natl. Acad. Sci. USA 2009, 106, 18680–18685. [Google Scholar] [PubMed] [Green Version]
- International Multiple Sclerosis Genetics Consortium; Wellcome Trust Case Control Consortium 2; Sawcer, S.; Hellenthal, G.; Pirinen, M.; Spencer, C.C.; Patsopoulos, N.A.; Moutsianas, L.; Dilthey, A.; Su, Z.; et al. Genetic risk and a primary role for cell-mediated immune mechanisms in multiple sclerosis. Nature 2011, 476, 214–219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boyman, O.; Sprent, J. The role of interleukin-2 during homeostasis and activation of the immune system. Nat. Rev. Immunol. 2012, 12, 180–190. [Google Scholar] [CrossRef] [PubMed]
- Vella, A.; Cooper, J.D.; Lowe, C.E.; Walker, N.; Nutland, S.; Widmer, B.; Jones, R.; Ring, S.M.; McArdle, W.; Pembrey, M.E.; et al. Localization of a type 1 diabetes locus in the IL2RA/CD25 region by use of tag single-nucleotide polymorphisms. Am. J. Hum. Genet. 2005, 76, 773–779. [Google Scholar] [CrossRef] [PubMed]
- Lowe, C.E.; Cooper, J.D.; Brusko, T.; Walker, N.M.; Smyth, D.J.; Bailey, R.; Bourget, K.; Plagnol, V.; Field, S.; Atkinson, M.; et al. Large-scale genetic fine mapping and genotype-phenotype associations implicate polymorphism in the IL2RA region in type 1 diabetes. Nat. Genet. 2007, 39, 1074–1082. [Google Scholar] [CrossRef] [PubMed]
- Maier, L.M.; Lowe, C.E.; Cooper, J.; Downes, K.; Anderson, D.E.; Severson, C.; Clark, P.M.; Healy, B.; Walker, N.; Aubin, C.; et al. IL2RA genetic heterogeneity in multiple sclerosis and type 1 diabetes susceptibility and soluble interleukin-2 receptor production. PLoS Genet. 2009, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dendrou, C.A.; Plagnol, V.; Fung, E.; Yang, J.H.; Downes, K.; Cooper, J.D.; Nutland, S.; Coleman, G.; Himsworth, M.; Hardy, M.; et al. Cell-specific protein phenotypes for the autoimmune locus IL2RA using a genotype-selectable human bioresource. Nat. Genet. 2009, 41, 1011–1015. [Google Scholar] [CrossRef] [PubMed]
- Oksenberg, J.R.; Barcellos, L.F.; Cree, B.A.; Baranzini, S.E.; Bugawan, T.L.; Khan, O.; Lincoln, R.R.; Swerdlin, A.; Mignot, E.; Lin, L.; et al. Mapping multiple sclerosis susceptibility to the HLA-DR locus in African Americans. Am. J. Hum. Genet. 2004, 74, 160–167. [Google Scholar] [CrossRef] [PubMed]
- Noble, J.A.; Martin, A.; Valdes, A.M.; Lane, J.A.; Galgani, A.; Petrone, A.; Lorini, R.; Pozzilli, P.; Buzzetti, R.; Erlich, H.A. Type 1 diabetes risk for human leukocyte antigen (HLA)-DR3 haplotypes depends on genotypic context: Association of DPB1 and HLA class I loci among DR3- and DR4-matched Italian patients and controls. Hum. Immunol. 2008, 69, 291–300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gough, S.C.; Simmonds, M.J. The HLA region and autoimmune disease: Associations and mechanisms of action. Curr. Genom. 2007, 8, 453–465. [Google Scholar]
- Nakanishi, K.; Inoko, H. Combination of HLA-A24, -DQA1*03, and -DR9 contributes to acute-onset and early complete β-cell destruction in type 1 diabetes: Longitudinal study of residual beta-cell function. Diabetes 2006, 55, 1862–1868. [Google Scholar] [CrossRef] [PubMed]
- Lipponen, K.; Gombos, Z.; Kiviniemi, M.; Siljander, H.; Lempainen, J.; Hermann, R.; Veijola, R.; Simell, O.; Knip, M.; Ilonen, J. Effect of HLA class I and class II alleles on progression from autoantibody positivity to overt type 1 diabetes in children with risk-associated class II genotypes. Diabetes 2010, 59, 3253–3256. [Google Scholar] [CrossRef] [PubMed]
- Hughes, T.; Adler, A.; Kelly, J.A.; Kaufman, K.M.; Williams, A.H.; Langefeld, C.D.; Brown, E.E.; Alarcon, G.S.; Kimberly, R.P.; Edberg, J.C.; et al. Evidence for gene-gene epistatic interactions among susceptibility loci for systemic lupus erythematosus. Arthritis Rheum. 2012, 64, 485–492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galarza-Munoz, G.; Briggs, F.B.S.; Evsyukova, I.; Schott-Lerner, G.; Kennedy, E.M.; Nyanhete, T.; Wang, L.; Bergamaschi, L.; Widen, S.G.; Tomaras, G.D.; et al. Human epistatic interaction controls IL7R splicing and increases multiple sclerosis risk. Cell 2017, 169, 72–84. [Google Scholar] [CrossRef] [PubMed]
- Visscher, P.M.; Wray, N.R.; Zhang, Q.; Sklar, P.; McCarthy, M.I.; Brown, M.A.; Yang, J. 10 years of GWAS discovery: Biology, function, and translation. Am. J. Hum. Genet. 2017, 101, 5–22. [Google Scholar] [CrossRef] [PubMed]
- Gauderman, W.J. Sample size requirements for association studies of gene-gene interaction. Am. J. Epidemiol. 2002, 155, 478–484. [Google Scholar] [CrossRef] [PubMed]
- Gaunt, T.R.; Shihab, H.A.; Hemani, G.; Min, J.L.; Woodward, G.; Lyttleton, O.; Zheng, J.; Duggirala, A.; McArdle, W.L.; Ho, K.; et al. Systematic identification of genetic influences on methylation across the human life course. Genom. Biol. 2016, 17. [Google Scholar] [CrossRef] [PubMed]
- Richardson, T.G.; Shihab, H.A.; Hemani, G.; Zheng, J.; Hannon, E.; Mill, J.; Carnero-Montoro, E.; Bell, J.T.; Lyttleton, O.; McArdle, W.L.; et al. Collapsed methylation quantitative trait loci analysis for low frequency and rare variants. Hum. Mol. Genet. 2016, 25, 4339–4349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imgenberg-Kreuz, J.; Sandling, J.K.; Almlof, J.C.; Nordlund, J.; Signer, L.; Norheim, K.B.; Omdal, R.; Ronnblom, L.; Eloranta, M.L.; Syvanen, A.C.; et al. Genome-wide DNA methylation analysis in multiple tissues in primary Sjogren’s syndrome reveals regulatory effects at interferon-induced genes. Ann. Rheum. Dis. 2016, 75, 2029–2036. [Google Scholar] [CrossRef] [PubMed]
- Lemire, M.; Zaidi, S.H.; Ban, M.; Ge, B.; Aissi, D.; Germain, M.; Kassam, I.; Wang, M.; Zanke, B.W.; Gagnon, F.; et al. Long-range epigenetic regulation is conferred by genetic variation located at thousands of independent loci. Nat. Commun. 2015, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belot, M.P.; Fradin, D.; Mai, N.; Le Fur, S.; Zelenika, D.; Kerr-Conte, J.; Pattou, F.; Lucas, B.; Bougneres, P. CpG methylation changes within the IL2RA promoter in type 1 diabetes of childhood onset. PLoS ONE 2013, 8. [Google Scholar] [CrossRef] [PubMed]
- Richardson, T.G.; Zheng, J.; Smith, G.D.; Timpson, N.J.; Gaunt, T.R.; Relton, C.L.; Hemani, G. Mendelian randomization analysis identifies CpG sites as putative mediators for genetic influences on cardiovascular disease risk. Am. J. Hum. Genet. 2017, 101, 590–602. [Google Scholar] [CrossRef] [PubMed]
- Millstein, J.; Zhang, B.; Zhu, J.; Schadt, E.E. Disentangling molecular relationships with a causal inference test. BMC Genet. 2009, 10. [Google Scholar] [CrossRef] [PubMed]
- Relton, C.L.; Smith, G.D. Two-step epigenetic mendelian randomization: A strategy for establishing the causal role of epigenetic processes in pathways to disease. Int. J. Epidemiol. 2012, 41, 161–176. [Google Scholar] [CrossRef] [PubMed]
- Relton, C.L.; Smith, G.D. Mendelian randomization: Applications and limitations in epigenetic studies. Epigenomics 2015, 7, 1239–1243. [Google Scholar] [CrossRef] [PubMed]
- Ladd-Acosta, C.; Fallin, M.D. The role of epigenetics in genetic and environmental epidemiology. Epigenomics 2016, 8, 271–283. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Aryee, M.J.; Padyukov, L.; Fallin, M.D.; Hesselberg, E.; Runarsson, A.; Reinius, L.; Acevedo, N.; Taub, M.; Ronninger, M.; et al. Epigenome-wide association data implicate DNA methylation as an intermediary of genetic risk in rheumatoid arthritis. Nat. Biotechnol. 2013, 31, 142–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olsson, A.H.; Volkov, P.; Bacos, K.; Dayeh, T.; Hall, E.; Nilsson, E.A.; Ladenvall, C.; Ronn, T.; Ling, C. Genome-wide associations between genetic and epigenetic variation influence mrna expression and insulin secretion in human pancreatic islets. PLoS Genet. 2014, 10. [Google Scholar] [CrossRef] [PubMed]
- Jody, Y.; Richardson, T.G.; McArdle, W.; Relton, C.; Gillespie, K.M.; Suderman, M.; Hemani, G. Identification of loci where DNA methylation potentially mediate genetic risk of type 1 diabetes. J. Autoimmunity 2018. [Google Scholar] [CrossRef]
- Richardson, T.G.; Haycock, P.C.; Zheng, J.; Timpson, N.J.; Gaunt, T.R.; Smith, G.D.; Relton, C.L.; Hemani, G. Systematic mendelian randomization framework elucidates hundreds of CpG sites which may mediate the influence of genetic variants on disease. Hum. Mol. Genet. 2018. [Google Scholar] [CrossRef] [PubMed]
- Richardson, T.G.; Haycock, P.C.; Zheng, J.; Timpson, N.J.; Gaunt, T.R.; Smith, G.D.; Relton, C.L.; Hemani, G. Systematic mendelian randomization framework elucidates hundreds of genetic loci which may influence disease through changes in DNA methylation levels. bioRxiv 2017. [Google Scholar] [CrossRef]
- Cucca, F.; Goy, J.V.; Kawaguchi, Y.; Esposito, L.; Merriman, M.E.; Wilson, A.J.; Cordell, H.J.; Bain, S.C.; Todd, J.A. A male-female bias in type 1 diabetes and linkage to chromosome Xp in MHC HLA-DR3-positive patients. Nat. Genet. 1998, 19, 301–302. [Google Scholar] [CrossRef] [PubMed]
- Chabchoub, G.; Uz, E.; Maalej, A.; Mustafa, C.A.; Rebai, A.; Mnif, M.; Bahloul, Z.; Farid, N.R.; Ozcelik, T.; Ayadi, H. Analysis of skewed X-chromosome inactivation in females with rheumatoid arthritis and autoimmune thyroid diseases. Arthritis Res. Ther. 2009, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simmonds, M.J.; Kavvoura, F.K.; Brand, O.J.; Newby, P.R.; Jackson, L.E.; Hargreaves, C.E.; Franklyn, J.A.; Gough, S.C. Skewed X chromosome inactivation and female preponderance in autoimmune thyroid disease: An association study and meta-analysis. J. Clin. Endocrinol. Metab. 2014, 99, E127–E131. [Google Scholar] [CrossRef] [PubMed]
- Milligan, M.J.; Lipovich, L. Pseudogene-derived lncRNAs: Emerging regulators of gene expression. Front. Genet. 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Kung, J.T.; Colognori, D.; Lee, J.T. Long noncoding RNAs: Past, present, and future. Genetics 2013, 193, 651–669. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Ding, C. Roles of lncRNAs in viral infections. Front. Cell. Infect. Microbiol. 2017, 7. [Google Scholar] [CrossRef] [PubMed]
- Spurlock, C.F.; Tossberg, J.T., 3rd; Guo, Y.; Collier, S.P.; Crooke, P.S., 3rd; Aune, T.M. Expression and functions of long noncoding RNAs during human T helper cell differentiation. Nat. Commun. 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Pearson, M.J.; Jones, S.W. Review: Long noncoding RNAs in the regulation of inflammatory pathways in rheumatoid arthritis and osteoarthritis. Arthritis Rheumatol. 2016, 68, 2575–2783. [Google Scholar] [CrossRef] [PubMed]
- Mirza, A.H.; Kaur, S.; Brorsson, C.A.; Pociot, F. Effects of GWAS-associated genetic variants on lncRNAs within IBD and T1D candidate loci. PLoS ONE 2014, 9. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Westra, H.J.; Karjalainen, J.; Zhernakova, D.V.; Esko, T.; Hrdlickova, B.; Almeida, R.; Zhernakova, A.; Reinmaa, E.; Vosa, U.; et al. Human disease-associated genetic variation impacts large intergenic non-coding RNA expression. PLoS Genet. 2013, 9. [Google Scholar] [CrossRef] [PubMed]
- Hrdlickova, B.; Kumar, V.; Kanduri, K.; Zhernakova, D.V.; Tripathi, S.; Karjalainen, J.; Lund, R.J.; Li, Y.; Ullah, U.; Modderman, R.; et al. Expression profiles of long non-coding RNAs located in autoimmune disease-associated regions reveal immune cell-type specificity. Genom. Med. 2014, 6. [Google Scholar] [CrossRef] [PubMed]
- Pennacchio, L.A.; Bickmore, W.; Dean, A.; Nobrega, M.A.; Bejerano, G. Enhancers: Five essential questions. Nat. Rev. Genet. 2013, 14, 288–295. [Google Scholar] [CrossRef] [PubMed]
- Farh, K.K.; Marson, A.; Zhu, J.; Kleinewietfeld, M.; Housley, W.J.; Beik, S.; Shoresh, N.; Whitton, H.; Ryan, R.J.; Shishkin, A.A.; et al. Genetic and epigenetic fine mapping of causal autoimmune disease variants. Nature 2015, 518, 337–343. [Google Scholar] [CrossRef] [PubMed]
- Carella, C.; Mazziotti, G.; Amato, G.; Braverman, L.E.; Roti, E. Clinical review 169: Interferon-α-related thyroid disease: Pathophysiological, epidemiological, and clinical aspects. J. Clin. Endocrinol. Metab. 2004, 89, 3656–3661. [Google Scholar] [CrossRef] [PubMed]
- Cabrera, S.M.; Chen, Y.G.; Hagopian, W.A.; Hessner, M.J. Blood-based signatures in type 1 diabetes. Diabetologia 2016, 59, 414–425. [Google Scholar] [CrossRef] [PubMed]
- Psarras, A.; Emery, P.; Vital, E.M. Type I interferon-mediated autoimmune diseases: Pathogenesis, diagnosis and targeted therapy. Rheumatology 2017, 56, 1662–1675. [Google Scholar] [CrossRef] [PubMed]
- Stefan, M.; Wei, C.; Lombardi, A.; Li, C.W.; Concepcion, E.S.; Inabnet, W.B., 3rd; Owen, R.; Zhang, W.; Tomer, Y. Genetic-epigenetic dysregulation of thymic TSH receptor gene expression triggers thyroid autoimmunity. Proc. Natl. Acad. Sci. USA 2014, 111, 12562–12567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, J.D.; Cheng, C.W. Causal variants in autoimmune disease: A commentary on a recent published fine-mapping algorithm analysis in genome-wide association studies study. Ann. Transl. Med. 2017, 5. [Google Scholar] [CrossRef] [PubMed]
- Perez-Pinera, P.; Kocak, D.D.; Vockley, C.M.; Adler, A.F.; Kabadi, A.M.; Polstein, L.R.; Thakore, P.I.; Glass, K.A.; Ousterout, D.G.; Leong, K.W.; et al. RNA-guided gene activation by CRISPR-Cas9-based transcription factors. Nat. Methods 2013, 10, 973–976. [Google Scholar] [CrossRef] [PubMed]
- Simeonov, D.R.; Gowen, B.G.; Boontanrart, M.; Roth, T.L.; Gagnon, J.D.; Mumbach, M.R.; Satpathy, A.T.; Lee, Y.; Bray, N.L.; Chan, A.Y.; et al. Discovery of stimulation-responsive immune enhancers with CRISPR activation. Nature 2017, 549, 111–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warwick, A.; Mackay, I.R. Intolerant bodies: A short history of autoimmunity. In Johns Hopkins Biographies of Disease; Johns Hopkins University Press: Baltimore, MD, USA, 2014. [Google Scholar]


© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ye, J.; Gillespie, K.M.; Rodriguez, S. Unravelling the Roles of Susceptibility Loci for Autoimmune Diseases in the Post-GWAS Era. Genes 2018, 9, 377. https://doi.org/10.3390/genes9080377
Ye J, Gillespie KM, Rodriguez S. Unravelling the Roles of Susceptibility Loci for Autoimmune Diseases in the Post-GWAS Era. Genes. 2018; 9(8):377. https://doi.org/10.3390/genes9080377
Chicago/Turabian StyleYe, Jody, Kathleen M. Gillespie, and Santiago Rodriguez. 2018. "Unravelling the Roles of Susceptibility Loci for Autoimmune Diseases in the Post-GWAS Era" Genes 9, no. 8: 377. https://doi.org/10.3390/genes9080377
APA StyleYe, J., Gillespie, K. M., & Rodriguez, S. (2018). Unravelling the Roles of Susceptibility Loci for Autoimmune Diseases in the Post-GWAS Era. Genes, 9(8), 377. https://doi.org/10.3390/genes9080377