Whole Exome Sequencing Identifies New Host Genomic Susceptibility Factors in Empyema Caused by Streptococcus pneumoniae in Children: A Pilot Study


 , , , , and
, , , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Design and Inclusion Criteria
2.2. Whole Exome Sequencing
2.3. Annotation of Variants and Assessment of Their Pathogenicity
2.4. Statistical Analysis of Whole Exon Sequencing Data
2.5. Statistical Analysis from Transcriptome Data
3. Results
3.1. Clinical Characteristics of Patients
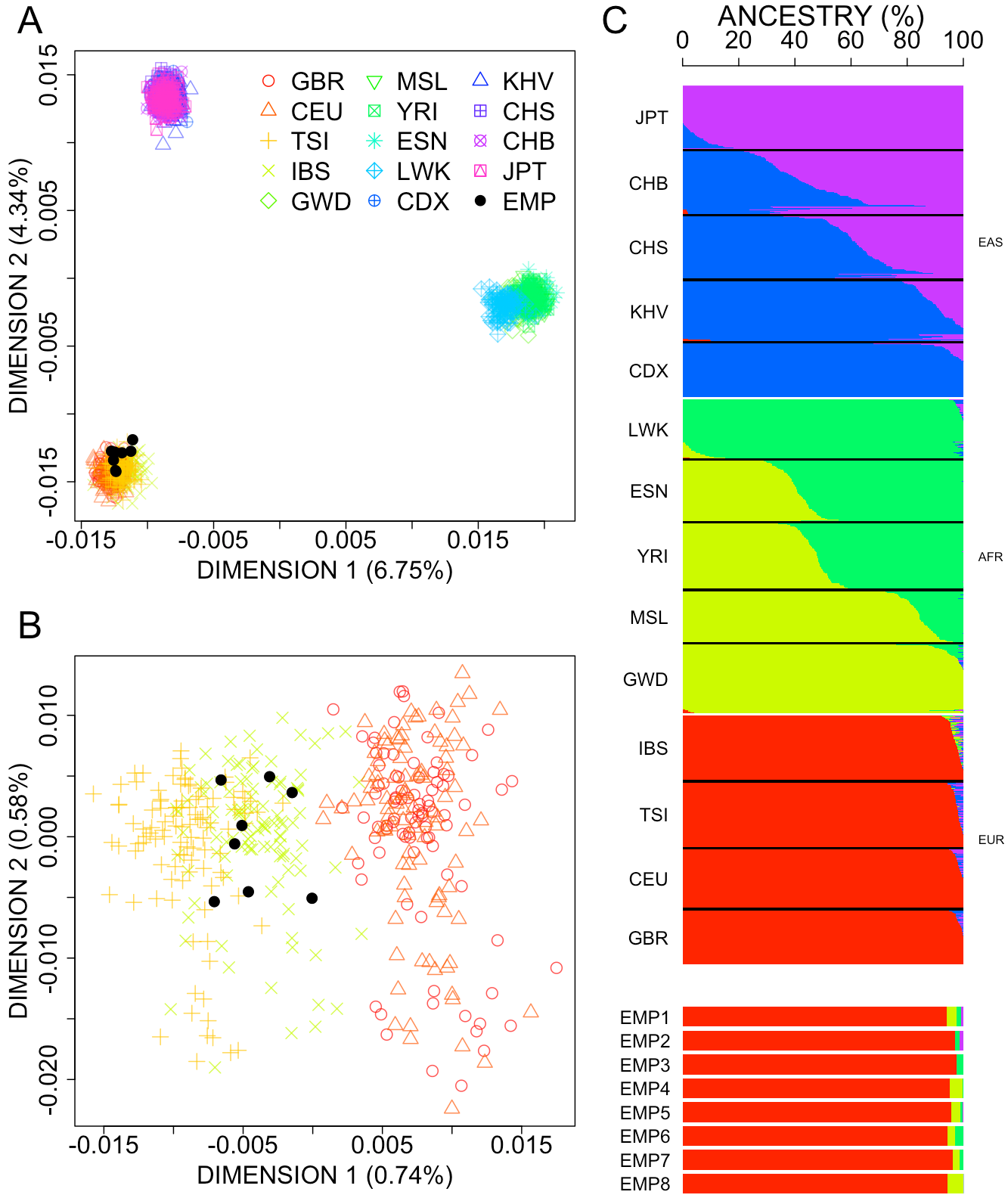
3.2. Population Genetic Characteristics of Empyema Patients
3.3. Single Nucleotide Polymorphism Association Test
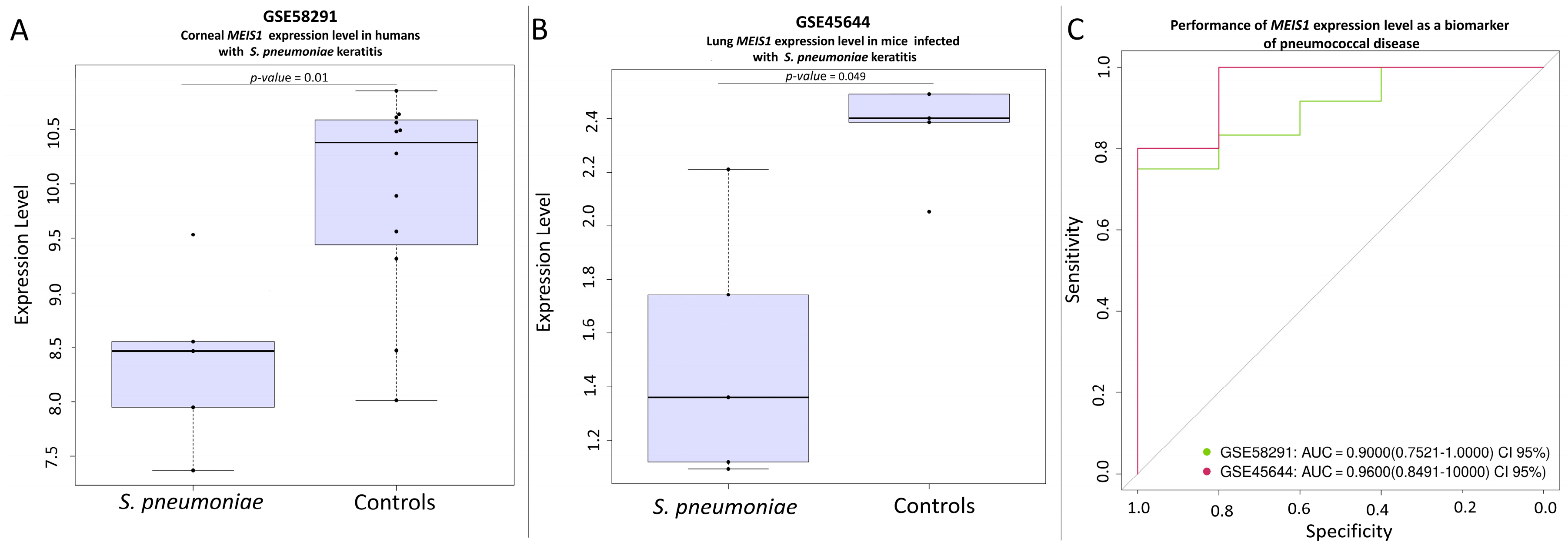
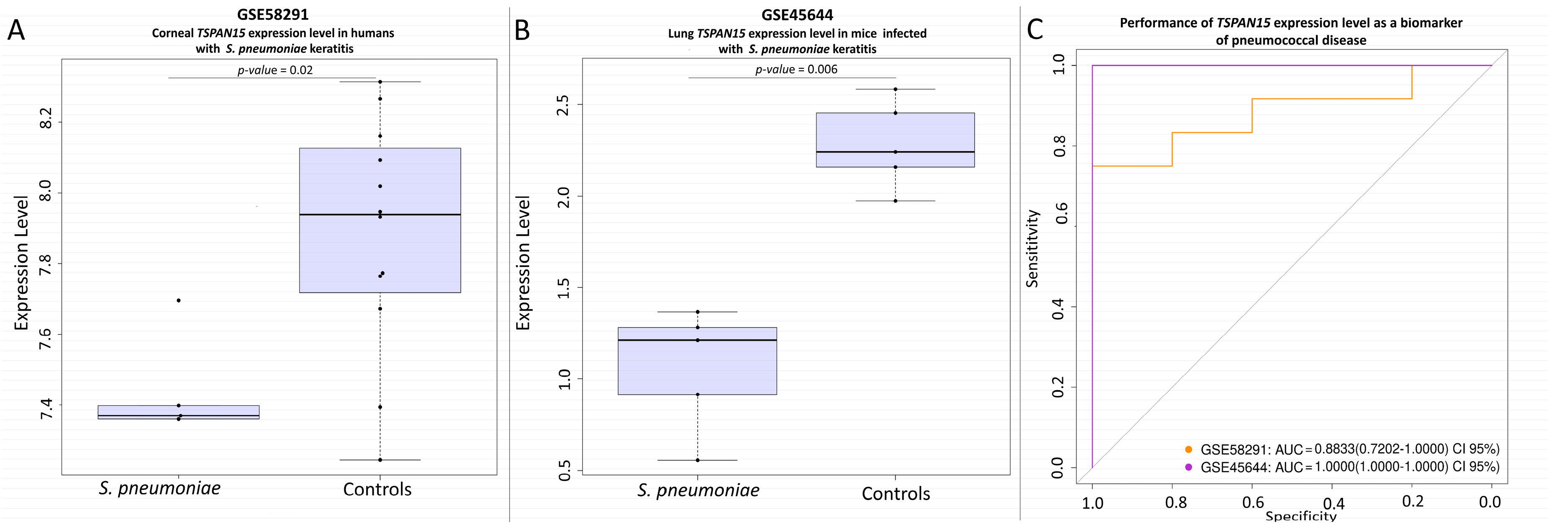
3.4. Transcription Signatures of Main Candidate Genes
4. Discussion
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- O’Brien, K.L.; Wolfson, L.J.; Watt, J.P.; Henkle, E.; Deloria-Knoll, M.; McCall, N.; Lee, E.; Mulholland, K.; Levine, O.S.; Cherian, T.; et al. Burden of disease caused by Streptococcus pneumoniae in children younger than 5 years: Global estimates. Lancet 2009, 374, 893–902. [Google Scholar] [CrossRef]
- Liu, L.; Oza, S.; Hogan, D.; Chu, Y.; Perin, J.; Zhu, J.; Lawn, J.E.; Cousens, S.; Mathers, C.; Black, R.E. Global, regional, and national causes of under-5 mortality in 2000–15: An updated systematic analysis with implications for the sustainable development goals. Lancet 2016, 388, 3027–3035. [Google Scholar] [CrossRef]
- Dion, C.F.; Ashurst, J.V. Pneumonia, Streptococcus Pneumoniae; StatPearls Publising: Treasure Island, FL, USA, 2018. [Google Scholar]
- Fletcher, M.A.; Schmitt, H.J.; Syrochkina, M.; Sylvester, G. Pneumococcal empyema and complicated pneumonias: Global trends in incidence, prevalence, and serotype epidemiology. Eur. J. Clin. Microbiol. Infect. Dis. 2014, 33, 879–910. [Google Scholar] [CrossRef] [PubMed]
- Martinón-Torres, F.; Dosil-Gallardo, S.; Pérez-del-Molino-Bernal, M.L.; Sánchez, F.P.; Tarrago, D.; Álvez, F.; Díaz, S.P.; Martinón-Torres, N.; Martinón-Sánchez, J.M. Pleural antigen assay in the diagnosis of pediatric pneumococcal empyema. J. Crit. Care 2012, 27, 321. [Google Scholar] [CrossRef] [PubMed]
- Obando, I.; Camacho-Lovillo, M.S.; Porras, A.; Gandia-Gonzalez, M.A.; Molinos, A.; Vazquez-Barba, I.; Morillo-Gutierrez, B.; Neth, O.W.; Tarrago, D. Sustained high prevalence of pneumococcal serotype 1 in paediatric parapneumonic empyema in southern Spain from 2005 to 2009. Clin. Microbiol. Infect. 2012, 18, 763–768. [Google Scholar] [CrossRef] [PubMed]
- Simonsen, L.; Taylor, R.J.; Schuck-Paim, C.; Lustig, R.; Haber, M.; Klugman, K.P. Effect of 13-valent pneumococcal conjugate vaccine on admissions to hospital 2 years after its introduction in the USA: A time series analysis. Lancet Respir. Med. 2014, 2, 387–394. [Google Scholar] [CrossRef]
- Bender, J.M.; Ampofo, K.; Korgenski, K.; Daly, J.; Pavia, A.T.; Mason, E.O.; Byington, C.L. Pneumococcal necrotizing pneumonia in Utah: Does serotype matter? Clin. Infect. Dis. 2008, 46, 1346–1352. [Google Scholar] [CrossRef] [PubMed]
- Kerem, E.; Bar Ziv, Y.; Rudenski, B.; Katz, S.; Kleid, D.; Branski, D. Bacteremic necrotizing pneumococcal pneumonia in children. Am. J. Respir. Crit. Care Med. 1994, 149, 242–244. [Google Scholar] [CrossRef] [PubMed]
- Martinón-Torres, F.; Bernaola Iturbe, E.; Giménez Sánchez, F.; Baca Cots, M.; de Juan Martín, F.; Diez Domingo, J.; Garces Sánchez, M.; Gómez Campdera, J.A.; Picazo, J.J.; Pineda Solas, V. Why are pediatric empyemas on the increase in Spain? An. Pediatr. 2008, 68, 158–164. [Google Scholar] [CrossRef]
- Davila, S.; Wright, V.J.; Khor, C.C.; Sim, K.S.; Binder, A.; Breunis, W.B.; Inwald, D.; Nadel, S.; Betts, H.; Carrol, E.D.; et al. Genome-wide association study identifies variants in the cfh region associated with host susceptibility to meningococcal disease. Nat. Genet. 2010, 42, 772–776. [Google Scholar] [CrossRef] [PubMed]
- Martinon-Torres, F.; Png, E.; Khor, C.C.; Davila, S.; Wright, V.J.; Sim, K.S.; Vega, A.; Fachal, L.; Inwald, D.; Nadel, S.; et al. Natural resistance to meningococcal disease related to cfh loci: Meta-analysis of genome-wide association studies. Sci. Rep. 2016, 6, 35842. [Google Scholar] [CrossRef] [PubMed]
- Salas, A.; Pardo-Seco, J.; Cebey-Lopez, M.; Gomez-Carballa, A.; Obando-Pacheco, P.; Rivero-Calle, I.; Curras-Tuala, M.J.; Amigo, J.; Gomez-Rial, J.; Martinon-Torres, F.; et al. Whole exome sequencing reveals new candidate genes in host genomic susceptibility to respiratory syncytial virus disease. Sci. Rep. 2017, 7, 15888. [Google Scholar] [CrossRef] [PubMed]
- Chapman, S.J.; Hill, A.V. Human genetic susceptibility to infectious disease. Nat. Rev. Genet. 2012, 13, 175–188. [Google Scholar] [CrossRef] [PubMed]
- Patarčić, I.; Gelemanovic, A.; Kirin, M.; Kolčić, I.; Theodoratou, E.; Baillie, K.J.; de Jong, M.D.; Rudan, I.; Campbell, H.; Polašek, O. The role of host genetic factors in respiratory tract infectious diseases: Systematic review, meta-analyses and field synopsis. Sci. Rep. 2015, 5, 16119. [Google Scholar] [CrossRef] [PubMed]
- Smelaya, T.V.; Belopolskaya, O.B.; Smirnova, S.V.; Kuzovlev, A.N.; Moroz, V.V.; Golubev, A.M.; Pabalan, N.A.; Salnikova, L.E. Genetic dissection of host immune response in pneumonia development and progression. Sci. Rep. 2016, 6, 35021. [Google Scholar] [CrossRef] [PubMed]
- Aung, T.; Ozaki, M.; Lee, M.C.; Schlotzer-Schrehardt, U.; Thorleifsson, G.; Mizoguchi, T.; Igo, R.P., Jr.; Haripriya, A.; Williams, S.E.; Astakhov, Y.S.; et al. Genetic association study of exfoliation syndrome identifies a protective rare variant at loxl1 and five new susceptibility loci. Nat. Genet. 2017, 49, 993–1004. [Google Scholar] [CrossRef] [PubMed]
- Ng, S.B.; Turner, E.H.; Robertson, P.D.; Flygare, S.D.; Bigham, A.W.; Lee, C.; Shaffer, T.; Wong, M.; Bhattacharjee, A.; Eichler, E.E.; et al. Targeted capture and massively parallel sequencing of 12 human exomes. Nature 2009, 461, 272–276. [Google Scholar] [CrossRef] [PubMed]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef] [PubMed]
- Ng, P.C.; Henikoff, S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003, 31, 3812–3814. [Google Scholar] [CrossRef] [PubMed]
- Cooper, G.M.; Stone, E.A.; Asimenos, G.; Program, N.C.S.; Green, E.D.; Batzoglou, S.; Sidow, A. Distribution and intensity of constraint in mammalian genomic sequence. Genome Res. 2005, 15, 901–913. [Google Scholar] [CrossRef] [PubMed]
- GENDRES: Genetic, Vitamin D and Respiratory Infections Research Network. Available online: http://www.gendres.org (accessed on 1 November 2017).
- Cebey-López, M.; Herberg, J.; Pardo-Seco, J.; Gómez-Carballa, A.; Martinon-Torres, N.; Salas, A.; Martinón-Sánchez, J.M.; Gormley, S.; Sumner, E.; Fink, C.; et al. Viral co-infections in pediatric patients hospitalized with lower tract acute respiratory infections. PLoS ONE 2015, 10, e0136526. [Google Scholar] [CrossRef] [PubMed]
- Cebey-López, M.; Pardo-Seco, J.; Gómez-Carballa, A.; Martinón-Torres, N.; Rivero-Calle, I.; Justicia, A.; Redondo, L.; Martinón-Sánchez, J.M.; Martinez-Padilla, M.D.; Giménez-Sánchez, F.; et al. Vitamin D role in hospitalized children with lower tract acute respiratory infections. J. Pediatr. Gastroenterol. Nutr. 2016, 62, 479–485. [Google Scholar] [CrossRef] [PubMed]
- Hansen, J.; Black, S.; Shinefield, H.; Cherian, T.; Benson, J.; Fireman, B.; Lewis, E.; Ray, P.; Lee, J. Effectiveness of heptavalent pneumococcal conjugate vaccine in children younger than 5 years of age for prevention of pneumonia: Updated analysis using world health organization standardized interpretation of chest radiographs. Pediatr. Infect. Dis. J. 2006, 25, 779–781. [Google Scholar] [CrossRef] [PubMed]
- Korppi, M. Diagnosis and treatment of community-acquired pneumonia in children. Acta Paediatr. 2012, 101, 702–704. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with burrows-wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The genome analysis toolkit: A mapreduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed]
- Li, H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 2011, 27, 2987–2993. [Google Scholar] [CrossRef] [PubMed]
- OGT: Oxford Gene Technology. Available online: http://www.ogt.co.uk (accessed on 1 November 2017).
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef] [PubMed]
- Ensembl. Available online: http://www.ensembl.org/index.html (accessed on 1 November 2017).
- Kircher, M.; Witten, D.M.; Jain, P.; O’Roak, B.J.; Cooper, G.M.; Shendure, J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 2014, 46, 310–315. [Google Scholar] [CrossRef] [PubMed]
- Quang, D.; Chen, Y.; Xie, X. Dann: A deep learning approach for annotating the pathogenicity of genetic variants. Bioinformatics 2015, 31, 761–763. [Google Scholar] [CrossRef] [PubMed]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.; Daly, M.J.; et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [PubMed]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2012; Available online: http://www.R-project.org/ISBN 3-900051-07-0.
- The 1000 Genomes Project Consortium. An integrated map of genetic variation from 1,092 human genomes. Nature 2012, 491, 56–65. [Google Scholar] [Green Version]
- Amigo, J.; Salas, A.; Phillips, C.; Carracedo, Á. Spsmart: Adapting population based SNP genotype databases for fast and comprehensive web access. BMC Bioinform. 2008, 9, 428. [Google Scholar] [CrossRef] [PubMed]
- Amigo, J.; Salas, A.; Phillips, C. Engines: Exploring single nucleotide variation in entire human genomes. BMC Bioinform. 2011, 12, 105. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Carballa, A.; Pardo-Seco, J.; Fachal, L.; Vega, A.; Cebey, M.; Martinón-Torres, N.; Martinón-Torres, F.; Salas, A. Indian signatures in the westernmost edge of the European Romani diaspora: New insight from mitogenomes. PLoS ONE 2013, 8, e75397. [Google Scholar] [CrossRef] [PubMed]
- Dering, C.; Hemmelmann, C.; Pugh, E.; Ziegler, A. Statistical analysis of rare sequence variants: An overview of collapsing methods. Genet. Epidemiol. 2011, 35 (Suppl. 1), S12–S17. [Google Scholar] [CrossRef] [PubMed]
- Fachal, L.; Mosquera-Miguel, A.; Pastor, P.; Ortega-Cubero, S.; Lorenzo, E.; Oterino-Duran, A.; Toriello, M.; Quintans, B.; Camina-Tato, M.; Sesar, A.; et al. No evidence of association between common European mitochondrial DNA variants in Alzheimer, Parkinson, and migraine in the Spanish population. Am. J. Med. Genet. Part B 2015, 168B, 54–65. [Google Scholar] [CrossRef] [PubMed]
- Salas, A.; Fachal, L.; Marcos-Alonso, S.; Vega, A.; Martinón-Torres, F.; Grupo de Investigación ESIGEM. Investigating the role of mitochondrial haplogroups in genetic predisposition to meningococcal disease. PLoS ONE 2009, 4, e8347. [Google Scholar] [CrossRef] [PubMed]
- Dopazo, J.; Amadoz, A.; Bleda, M.; García-Alonso, L.; Alemán, A.; García-García, F.; Rodriguez, J.A.; Daub, J.T.; Muntané, G.; Rueda, A.; et al. 267 Spanish exomes reveal population-specific differences in disease-related genetic variation. Mol. Biol. Evol. 2016, 33, 1205–1218. [Google Scholar] [CrossRef] [PubMed]
- Perl. Available online: http://www.perl.org (accessed on 1 November 2017).
- Barrett, J.C.; Fry, B.; Maller, J.; Daly, M.J. Haploview: Analysis and visualization of LD and haplotype maps. Bioinformatics 2005, 21, 263–265. [Google Scholar] [CrossRef] [PubMed]
- Gabriel, S.B.; Schaffner, S.F.; Nguyen, H.; Moore, J.M.; Roy, J.; Blumenstiel, B.; Higgins, J.; DeFelice, M.; Lochner, A.; Faggart, M.; et al. The structure of haplotype blocks in the human genome. Science 2002, 296, 2225–2229. [Google Scholar] [CrossRef] [PubMed]
- GEO: Gene Expression Omnibus. Available online: https://www.ncbi.nlm.nih.gov/geo/ (accessed on 1 November 2017).
- Van Lieshout, M.H.; Scicluna, B.P.; Florquin, S.; Van der Poll, T. NLRP3 and ASC differentially affect the lung transcriptome during pneumococcal pneumonia. Am. J. Respir. Cell Mol. Biol. 2014, 50, 699–712. [Google Scholar] [CrossRef] [PubMed]
- Jonczyk, M.S.; Simon, M.; Kumar, S.; Fernandes, V.E.; Sylvius, N.; Mallon, A.M.; Denny, P.; Andrew, P.W. Genetic factors regulating lung vasculature and immune cell functions associate with resistance to pneumococcal infection. PLoS ONE 2014, 9, e89831. [Google Scholar] [CrossRef] [PubMed]
- Restori, K.H.; Kennett, M.J.; Ross, A.C. Immunization with pneumococcal polysaccharide serotype 3 and lipopolysaccharide modulates lung and liver inflammation during a virulent Streptococcus pneumoniae infection in mice. Clin. Vaccine Immunol. 2013, 20, 639–650. [Google Scholar] [CrossRef] [PubMed]
- Khaenam, P.; Rinchai, D.; Altman, M.C.; Chiche, L.; Buddhisa, S.; Kewcharoenwong, C.; Suwannasaen, D.; Mason, M.; Whalen, E.; Presnell, S.; et al. A transcriptomic reporter assay employing neutrophils to measure immunogenic activity of septic patients’ plasma. J. Transl. Med. 2014, 12, 65. [Google Scholar] [CrossRef] [PubMed]
- Ramilo, O.; Allman, W.; Chung, W.; Mejias, A.; Ardura, M.; Glaser, C.; Wittkowski, K.M.; Piqueras, B.; Banchereau, J.; Palucka, A.K.; et al. Gene expression patterns in blood leukocytes discriminate patients with acute infections. Blood 2007, 109, 2066–2077. [Google Scholar] [CrossRef] [PubMed]
- Chidambaram, J.D.; Kannambath, S.; Srikanthi, P.; Shah, M.; Lalitha, P.; Elakkiya, S.; Bauer, J.; Prajna, N.V.; Holland, M.J.; Burton, M.J. Persistence of innate immune pathways in late stage human bacterial and fungal keratitis: Results from a comparative transcriptome analysis. Front. Cell. Infect. Microbiol. 2017, 7, 193. [Google Scholar] [CrossRef] [PubMed]
- Du, P.; Kibbe, W.A.; Lin, S.M. Lumi: A pipeline for processing Illumina microarray. Bioinformatics 2008, 24, 1547–1548. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, B.S.; Irizarry, R.A. A framework for oligonucleotide microarray preprocessing. Bioinformatics 2010, 26, 2363–2367. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. Limma powers differential expression analyses for rna-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef] [PubMed]
- Alexander, D.H.; Novembre, J.; Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009, 19, 1655–1664. [Google Scholar] [CrossRef] [PubMed]
- Yildirim, I.; Shea, K.M.; Pelton, S.I. Pneumococcal disease in the era of pneumococcal conjugate vaccine. Infect. Dis. Clin. N. Am. 2015, 29, 679–697. [Google Scholar] [CrossRef] [PubMed]
- Burgos, J.; Falco, V.; Pahissa, A. The increasing incidence of empyema. Curr. Opin. Pulm. Med. 2013, 19, 350–356. [Google Scholar] [CrossRef] [PubMed]
- Byington, C.L.; Hulten, K.G.; Ampofo, K.; Sheng, X.; Pavia, A.T.; Blaschke, A.J.; Pettigrew, M.; Korgenski, K.; Daly, J.; Mason, E.O. Molecular epidemiology of pediatric pneumococcal empyema from 2001 to 2007 in Utah. J. Clin. Microbiol. 2010, 48, 520–525. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, Y.C.; Hsueh, P.R.; Lu, C.Y.; Lee, P.I.; Lee, C.Y.; Huang, L.M. Clinical manifestations and molecular epidemiology of necrotizing pneumonia and empyema caused by streptococcus pneumoniae in children in Taiwan. Clin. Infect. Dis. 2004, 38, 830–835. [Google Scholar] [CrossRef] [PubMed]
- Byington, C.L.; Korgenski, K.; Daly, J.; Ampofo, K.; Pavia, A.; Mason, E.O. Impact of the pneumococcal conjugate vaccine on pneumococcal parapneumonic empyema. Pediatr. Infect. Dis. J. 2006, 25, 250–254. [Google Scholar] [CrossRef] [PubMed]
- Rose, M.C.; Piazza, F.M.; Chen, Y.A.; Alimam, M.Z.; Bautista, M.V.; Letwin, N.; Rajput, B. Model systems for investigating mucin gene expression in airway diseases. J. Aerosol Med. 2000, 13, 245–261. [Google Scholar] [CrossRef] [PubMed]
- Linden, S.K.; Sutton, P.; Karlsson, N.G.; Korolik, V.; McGuckin, M.A. Mucins in the mucosal barrier to infection. Mucosal Immunol. 2008, 1, 183–197. [Google Scholar] [CrossRef] [PubMed]
- Weinberger, D.M.; Klugman, K.P.; Steiner, C.A.; Simonsen, L.; Viboud, C. Association between respiratory syncytial virus activity and pneumococcal disease in infants: A time series analysis of us hospitalization data. PLoS Med. 2015, 12, e1001776. [Google Scholar] [CrossRef] [PubMed]
- Cebey-López, M.; Herberg, J.; Pardo-Seco, J.; Gómez-Carballa, A.; Martinón-Torres, N.; Salas, A.; Martinón-Sánchez, J.M.; Justicia, A.; Rivero-Calle, I.; Sumner, E.; et al. Does viral co-infection influence the severity of acute respiratory infection in children? PLoS ONE 2016, 11, e0152481. [Google Scholar] [CrossRef] [PubMed]
- Germain, M.; Chasman, D.I.; de Haan, H.; Tang, W.; Lindstrom, S.; Weng, L.C.; de Andrade, M.; de Visser, M.C.; Wiggins, K.L.; Suchon, P.; et al. Meta-analysis of 65,734 individuals identifies tspan15 and slc44a2 as two susceptibility loci for venous thromboembolism. Am. J. Hum. Genet. 2015, 96, 532–542. [Google Scholar] [CrossRef] [PubMed]
- Seipold, L.; Saftig, P. The emerging role of tetraspanins in the proteolytic processing of the amyloid precursor protein. Front. Mol. Neurosci. 2016, 9, 149. [Google Scholar] [CrossRef] [PubMed]
- Charrin, S.; Jouannet, S.; Boucheix, C.; Rubinstein, E. Tetraspanins at a glance. J. Cell Sci. 2014, 127, 3641–3648. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, M.; Tachibana, I.; Takeda, Y.; He, P.; Minami, S.; Iwasaki, T.; Kida, H.; Goya, S.; Kijima, T.; Yoshida, M.; et al. Tetraspanin cd9 negatively regulates lipopolysaccharide-induced macrophage activation and lung inflammation. J. Immunol. 2009, 182, 6485–6493. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Tachibana, I.; Takeda, Y.; He, P.; Kang, S.; Suzuki, M.; Kuhara, H.; Tetsumoto, S.; Tsujino, K.; Minami, T.; et al. Statins decrease lung inflammation in mice by upregulating tetraspanin cd9 in macrophages. PLoS ONE 2013, 8, e73706. [Google Scholar] [CrossRef] [PubMed]
- Peterson, M.M.; Mack, J.L.; Hall, P.R.; Alsup, A.A.; Alexander, S.M.; Sully, E.K.; Sawires, Y.S.; Cheung, A.L.; Otto, M.; Gresham, H.D. Apolipoprotein b is an innate barrier against invasive staphylococcus aureus infection. Cell Host Microbe 2008, 4, 555–566. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variables | Pneumococcal Empyema Patients |
|---|---|
| Demographic characteristics | |
| Sex (male) a | 5/8 (62.5%) |
| Age (years) b | 5.1 (3.1) |
| Medical history | |
| Asthma a | 2/8 (25.0%) |
| Pneumococcal vaccination status a | |
| PCV | 6/8 (75.0%) |
| PCV 10 | 1/8 (12.5%) |
| PCV 13 | 1/8 (12.5%) |
| Clinical data | |
| Treatment prior to admission a | |
| Antibiotics | 3/8 (37.5%) |
| Antipyretic | 2/8 (25.0%) |
| Hospital length of stay (days) b | 21.2 (16.4) |
| PICU (days) b | 8.0 (3.8) |
| Respiratory support a | 2/8 (25.0%) |
| Oxygen a | 4/8 (50.0%) |
| Urokinase a | 5/8 (62.5%) |
| Blood test | |
| Leukocytes (c/mm3) b | 17,971.2 (5751.5) |
| Procalcitonin (ng/mL) b | 253.5 (565.4) |
| Pleural fluid test | |
| Glucose (mg/dL) b | 19.4 (25.2) |
| Proteins (g/dL) b | 5.0 (1.0) |
| Course and outcome | |
| Course a | |
| Necrotizing pneumonia | 4/8 (50.0%) |
| Sepsis | 1/8 (12.5%) |
| Sequelae a | |
| Pneumatocele | 1/8 (12.5%) |
| Exitus | 1/8 (12.5%) |
| Sequence Variation | n |
|---|---|
| Downstream | 6 |
| Exonic | 76,551 |
| Exonic/splicing | 50 |
| Intergenic | 37 |
| Intronic | 349 |
| ncRNA_exonic | 5728 |
| ncRNA_exonic; splicing | 5 |
| ncRNA_intronic | 483 |
| ncRNA_splicing | 3 |
| Splicing | 48 |
| Upstream | 17 |
| Upstream; Downstream | 2 |
| 3′-UTR | 10,687 |
| 5′-UTR | 7124 |
| 5′-UTR5/3′-UTR | 15 |
| Non-synonymous SNV | 38,911 |
| Stopgain | 368 |
| Stoploss | 40 |
| Synonymous SNV | 36,270 |
| Unknown | 1012 |
| Cohort | Statistical Values | rs201967957 (G/A) | rs576099063 (G/T) |
|---|---|---|---|
| Cases | AF | 0.9375 | 0.8125 |
| 1000G-IBS | MAF | 0.09346 | 0.1402 |
| OR | 145.50 | 26.58 | |
| p-value | 3.71 × 10−13 | 2.36 × 10−8 | |
| 1000G-CEU | MAF | 0.13920 | 0.08763 |
| OR | 92.78 | 45.12 | |
| p-value | 4.40 × 10−11 | 3.07 × 10−1 | |
| 1000G-GBR | MAF | 0.08989 | 0.15730 |
| OR | 151.90 | 23.21 | |
| p-value | 4.84 × 10−13 | 1.05 × 10−7 | |
| 1000G-TSI | MAF | 0.1557 | 0.1179 |
| OR | 81.36 | 32.41 | |
| p-value | 1.34 × 10−1 | 4.22 × 10−9 | |
| 1000G-ALL | MAF | 0.12 | 0.1262 |
| OR | 109.90 | 29.99 | |
| p-value | 6.05 × 10−13 | 1.60 × 10−9 |
| Genes | Chr. | No. SNP | p-ValueIBS | p-ValueCEU | p-ValueGBR | p-ValueTSI | p-ValueALL | p-ValueEC |
|---|---|---|---|---|---|---|---|---|
| All variants | ||||||||
| OR9G9 | 11 | 17 | 8.13 × 10−7 | 8.94 × 10−7 | 1.02 × 10−6 | 6.04 × 10−7 | 3.05 × 10−7 | − |
| MUC3A | 7 | 45 | 1.27 × 10−6 | 1.27 × 10−6 | 1.70 × 10−6 | 1.05 × 10−6 | 6.15 × 10−7 | 8.94 × 10−6 |
| MUC6 | 11 | 34 | 3.16 × 10−6 | 2.56 × 10−6 | 3.83 × 10−6 | 2.47 × 10−6 | 1.45 × 10−6 | 1.92 × 10−6 |
| Rare variants | ||||||||
| OR9G9 | 11 | 11 | 5.62 × 10−11 | 2.74 × 10−10 | 2.21 × 10−12 | 1.04 × 10−11 | 1.58 × 10−13 | − |
| MUC6 | 11 | 24 | 1.90 × 10−10 | 6.74 × 10−12 | 0 | 1.52 × 10−9 | 9.17 × 10−12 | 1.22 × 10−8 |
| MUC3A | 7 | 21 | 2.42 × 10−10 | 9.23 × 10−11 | 6.48 × 10−11 | 3.28 × 10−11 | 4.65 × 10−12 | 4.69 × 10−9 |
| APOB | 2 | 36 | 8.35 × 10−6 | 2.37 × 10−5 | 5.56 × 10−5 | 1.03 × 10−6 | 1.28 × 10−6 | 1.07 × 10−6 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Salas, A.; Pardo-Seco, J.; Barral-Arca, R.; Cebey-López, M.; Gómez-Carballa, A.; Rivero-Calle, I.; Pischedda, S.; Currás-Tuala, M.-J.; Amigo, J.; Gómez-Rial, J.; et al. Whole Exome Sequencing Identifies New Host Genomic Susceptibility Factors in Empyema Caused by Streptococcus pneumoniae in Children: A Pilot Study. Genes 2018, 9, 240. https://doi.org/10.3390/genes9050240
Salas A, Pardo-Seco J, Barral-Arca R, Cebey-López M, Gómez-Carballa A, Rivero-Calle I, Pischedda S, Currás-Tuala M-J, Amigo J, Gómez-Rial J, et al. Whole Exome Sequencing Identifies New Host Genomic Susceptibility Factors in Empyema Caused by Streptococcus pneumoniae in Children: A Pilot Study. Genes. 2018; 9(5):240. https://doi.org/10.3390/genes9050240
Chicago/Turabian StyleSalas, Antonio, Jacobo Pardo-Seco, Ruth Barral-Arca, Miriam Cebey-López, Alberto Gómez-Carballa, Irene Rivero-Calle, Sara Pischedda, María-José Currás-Tuala, Jorge Amigo, José Gómez-Rial, and et al. 2018. "Whole Exome Sequencing Identifies New Host Genomic Susceptibility Factors in Empyema Caused by Streptococcus pneumoniae in Children: A Pilot Study" Genes 9, no. 5: 240. https://doi.org/10.3390/genes9050240
APA StyleSalas, A., Pardo-Seco, J., Barral-Arca, R., Cebey-López, M., Gómez-Carballa, A., Rivero-Calle, I., Pischedda, S., Currás-Tuala, M.-J., Amigo, J., Gómez-Rial, J., Martinón-Torres, F., & On behalf of GENDRES Network. (2018). Whole Exome Sequencing Identifies New Host Genomic Susceptibility Factors in Empyema Caused by Streptococcus pneumoniae in Children: A Pilot Study. Genes, 9(5), 240. https://doi.org/10.3390/genes9050240