Another Round of “Clue” to Uncover the Mystery of Complex Traits

Abstract
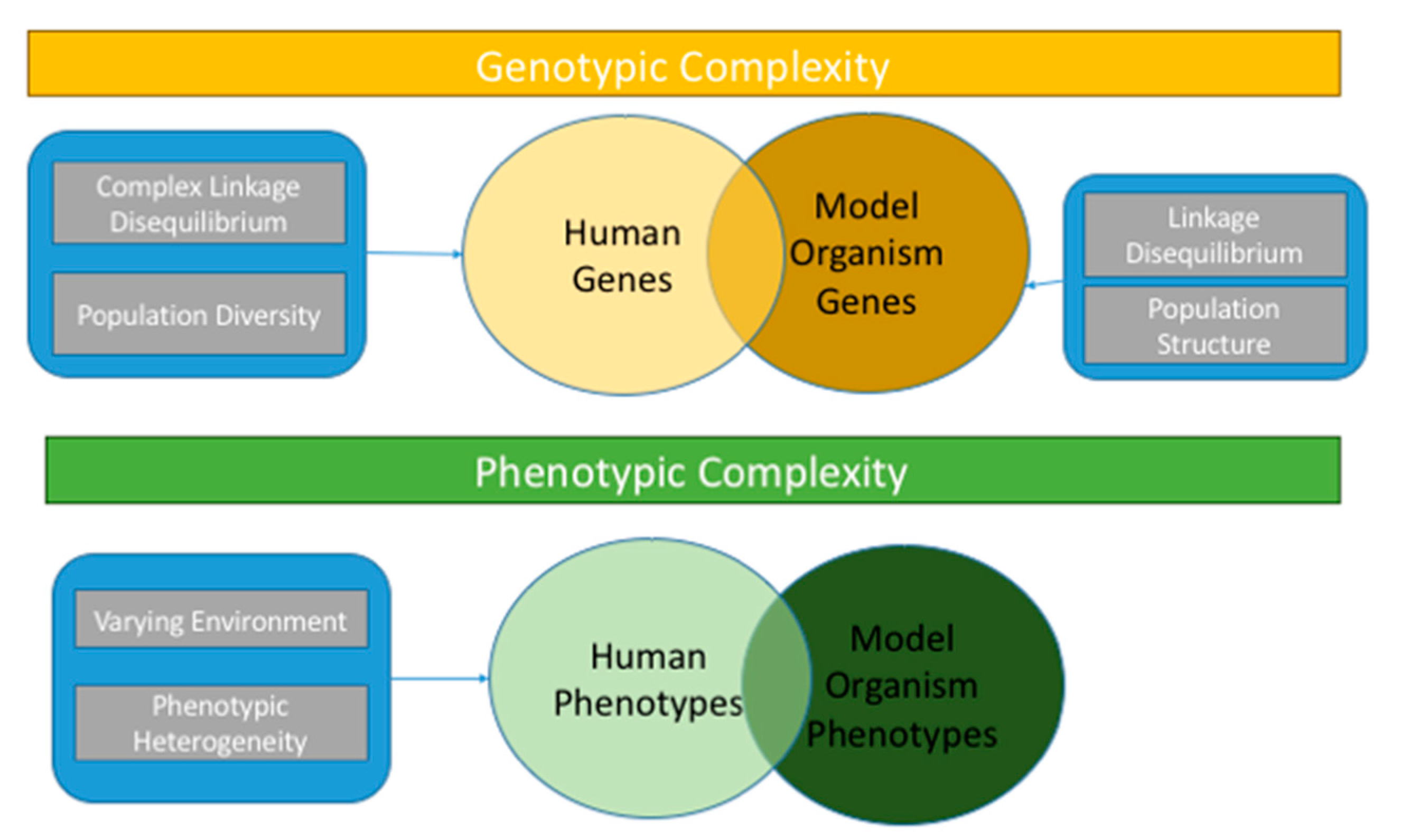
:1. Complex Diseases and the Concept of Heritability
2. Clues to Elucidating the Underlying Genetic Architecture of Complex Traits
- How much closer do we get to explaining heritability as estimated by family and twin studies by exploiting genetic variations in population based studies?
- How much of the phenotypic variance is additive (i.e., the combinatorial effect of all variations)? This is referred to as narrow sense heritability.
- How much of variance cannot be explained merely by adding all variations in a single model?
3. Suspects/Who Did It? Considering Different Types of Omics and Environmental Variability as the Suspects in the Crime (Influencing Disease Risk)
3.1. Common Variants
3.2. Rare Variants
3.3. Structural Variations
3.4. Environmental Factors
3.5. Gene Expression
3.6. Protein/Metabolites
3.7. Epigenome
4. What Is the Weapon of Choice? Which Type of Tools Can Help Elucidate the Significant Risk Factors for Complex Diseases?
5. Where? Which Tissue(s) Are Important for the Evaluation of Omics Associations?
6. Estimating Heritability (Making a Suggestion in the Game of “Clue”)
- Family and Twin studies: In family and twin studies, a set of related individuals and their phenotypic traits are analyzed to identify how heritable the phenotypic trait is in families and in sets of identical twins respectively. Family study estimates are usually lower than twin study estimates. For example, family studies for BMI estimate that BMI is 24–81% heritable whereas twin studies estimate BMI to be 47–90% heritable [34]. The estimates from studies of related individuals take the effects of the environment into consideration and, thus, generally broad sense heritability is estimated by these methods.
- Population based studies: Genomic heritability mainly refers to the proportion of trait variance that can be attributable to genetic factors such as common variants and low frequency or rare variations. Many methods and tools exist in the literature to measure heritability among a set of unrelated individuals. These include mixed model approaches (GCTA, REACTA, PLINK, etc.) [175,176], Bayesian approaches (example BGLR) [177], LD based weighted methods in mixed linear model approaches (LDAK) [178], and machine learning approaches (HERRA and MEGHA) [179,180]. All of these methods are focused towards explaining the additive variance component (i.e., narrow sense heritability). Narrow sense heritability estimates from GWAS studies and for either all variants on the genotyping chip assayed or only a subset of statistically significant variants. Locke et al. [181] determined the variance components for BMI based on statistically significant GWAS variants from their study and showed that 97 genome-wide significant loci can explain only 2.7% of the variance, whereas the overall SNP heritability (i.e., heritability from all available genotyped and imputed SNPs) is 75%, as shown by Robinson et al. [182]. Recent studies have also looked at the proportion of phenotypic variance explained by partitioning the genome. Speed et al. [183] showed how the proportion of variance explained for 19 different traits varies across the genome by chromosome and by minor allele frequency ranges. Finucane et al. [184] proposed LD score regression method using summary statistics from GWAS to partition heritability across the genome based on functional annotations.
7. The Focus of Future Studies, What to Expect?
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Cutting, G.R. Modifier genes in Mendelian disorders: The example of cystic fibrosis. Ann. N. Y. Acad. Sci. 2010, 1214, 57–69. [Google Scholar] [CrossRef] [PubMed]
- Afshari, N.A.; Igo, R.P.; Morris, N.J.; Stambolian, D.; Sharma, S.; Pulagam, V.L.; Dunn, S.; Stamler, J.F.; Truitt, B.J.; Rimmler, J.; et al. Genome-wide association study identifies three novel loci in Fuchs endothelial corneal dystrophy. Nat. Commun. 2017, 8, 14898. [Google Scholar] [CrossRef] [PubMed]
- Klein, I.; Danzi, S. Thyroid Disease and the Heart. Circulation 2007, 116, 1725–1735. [Google Scholar] [CrossRef] [PubMed]
- Fritsche, L.G.; Igl, W.; Bailey, J.N.C.; Grassmann, F.; Sengupta, S.; Bragg-Gresham, J.L.; Burdon, K.P.; Hebbring, S.J.; Wen, C.; Gorski, M.; et al. A large genome-wide association study of age-related macular degeneration highlights contributions of rare and common variants. Nat. Genet. 2016, 48, 134–143. [Google Scholar] [CrossRef] [PubMed]
- Dewey, F.E.; Gusarova, V.; O’Dushlaine, C.; Gottesman, O.; Trejos, J.; Hunt, C.; Van Hout, C.V.; Habegger, L.; Buckler, D.; Lai, K.-M.V.; et al. Inactivating Variants in ANGPTL4 and Risk of Coronary Artery Disease. N. Engl. J. Med. 2016, 374, 1123–1133. [Google Scholar] [CrossRef] [PubMed]
- Chu, X.K.; Tuo, J.; Chan, C.-C. Genetics of age-related macular degeneration: Application to drug design. Future Med. Chem. 2013, 5, 13–15. [Google Scholar] [CrossRef] [PubMed]
- Everett, B.M.; Smith, R.J.; Hiatt, W.R. Reducing LDL with PCSK9 Inhibitors—The Clinical Benefit of Lipid Drugs. N. Engl. J. Med. 2015, 373, 1588–1591. [Google Scholar] [CrossRef] [PubMed]
- McCarty, C.A.; Chisholm, R.L.; Chute, C.G.; Kullo, I.J.; Jarvik, G.P.; Larson, E.B.; Li, R.; Masys, D.R.; Ritchie, M.D.; Roden, D.M.; et al. The eMERGE Network: A consortium of biorepositories linked to electronic medical records data for conducting genomic studies. BMC Med. Genom. 2011, 4, 13. [Google Scholar] [CrossRef] [PubMed]
- Carey, D.J.; Fetterolf, S.N.; Davis, F.D.; Faucett, W.A.; Kirchner, H.L.; Mirshahi, U.; Murray, M.F.; Smelser, D.T.; Gerhard, G.S.; Ledbetter, D.H. The Geisinger MyCode community health initiative: An electronic health record-linked biobank for precision medicine research. Genet. Med. 2016, 18, 906–913. [Google Scholar] [CrossRef] [PubMed]
- Roden, D.M.; Pulley, J.M.; Basford, M.A.; Bernard, G.R.; Clayton, E.W.; Balser, J.R.; Masys, D.R. Development of a large-scale de-identified DNA biobank to enable personalized medicine. Clin. Pharmacol. Ther. 2008, 84, 362–369. [Google Scholar] [CrossRef] [PubMed]
- McQuillan, G.M.; Pan, Q.; Porter, K.S. Consent for genetic research in a general population: An update on the National Health and Nutrition Examination Survey experience. Genet. Med. 2006, 8, 354–360. [Google Scholar] [CrossRef] [PubMed]
- Speliotes, E.K.; Willer, C.J.; Berndt, S.I.; Monda, K.L.; Thorleifsson, G.; Jackson, A.U.; Lango Allen, H.; Lindgren, C.M.; Luan, J.; Mägi, R.; et al. Association analyses of 249,796 individuals reveal 18 new loci associated with body mass index. Nat. Genet. 2010, 42, 937–948. [Google Scholar] [CrossRef] [PubMed]
- Patel, C.J.; Pho, N.; McDuffie, M.; Easton-Marks, J.; Kothari, C.; Kohane, I.S.; Avillach, P. A database of human exposomes and phenomes from the US National Health and Nutrition Examination Survey. Sci. Data 2016, 3, 160096. [Google Scholar] [CrossRef] [PubMed]
- Zuk, O.; Hechter, E.; Sunyaev, S.R.; Lander, E.S. The mystery of missing heritability: Genetic interactions create phantom heritability. Proc. Natl. Acad. Sci. USA 2012, 109, 1193–1198. [Google Scholar] [CrossRef] [PubMed]
- Gibson, G. Hints of hidden heritability in GWAS. Nat. Genet. 2010, 42, 558–560. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Wray, N.R.; Goddard, M.E.; Visscher, P.M. Estimating Missing Heritability for Disease from Genome-wide Association Studies. Am. J. Hum. Genet. 2011, 88, 294–305. [Google Scholar] [CrossRef] [PubMed]
- Kerner, B.; North, K.E.; Fallin, M.D. Use of Longitudinal Data in Genetic Studies in the Genome-wide Association Studies Era: Summary of Group 14. Genet. Epidemiol. 2009, 33, S93–S98. [Google Scholar] [CrossRef] [PubMed]
- Ioannidis, J.P.A.; Loy, E.Y.; Poulton, R.; Chia, K.S. Researching genetic versus nongenetic determinants of disease: A comparison and proposed unification. Sci. Transl. Med. 2009, 1, 7ps8. [Google Scholar] [CrossRef] [PubMed]
- Cheverud, J.M.; Routman, E.J. Epistasis and its contribution to genetic variance components. Genetics 1995, 139, 1455–1461. [Google Scholar] [PubMed]
- Verma, S.S.; Cooke Bailey, J.N.; Lucas, A.; Bradford, Y.; Linneman, J.G.; Hauser, M.A.; Pasquale, L.R.; Peissig, P.L.; Brilliant, M.H.; McCarty, C.A.; et al. Epistatic Gene-Based Interaction Analyses for Glaucoma in eMERGE and NEIGHBOR Consortium. PLoS Genet. 2016, 12, e1006186. [Google Scholar] [CrossRef] [PubMed]
- Gatz, M.; Reynolds, C.A.; Fratiglioni, L.; Johansson, B.; Mortimer, J.A.; Berg, S.; Fiske, A.; Pedersen, N.L. Role of genes and environments for explaining Alzheimer disease. Arch. Gen. Psychiatry 2006, 63, 168–174. [Google Scholar] [CrossRef] [PubMed]
- Lord, J.; Lu, A.J.; Cruchaga, C. Identification of rare variants in Alzheimer’s disease. Front. Genet. 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Gu, J. Heritability of prostate cancer: A tale of rare variants and common single nucleotide polymorphisms. Ann. Transl. Med. 2016, 4. [Google Scholar] [CrossRef] [PubMed]
- Connelly, C.F.; Akey, J.M. On the Prospects of Whole-Genome Association Mapping in Saccharomyces cerevisiae. Genetics 2012, 191, 1345–1353. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, D.K.; Escott-Price, V.; Ziehm, M.; Magwire, M.M.; Mackay, T.F.C.; Partridge, L.; Thornton, J.M. Longevity GWAS Using the Drosophila Genetic Reference Panel. J. Gerontol. A Biol. Sci. Med. Sci. 2015, 70, 1470–1478. [Google Scholar] [CrossRef] [PubMed]
- Wangler, M.F.; Hu, Y.; Shulman, J.M. Drosophila and genome-wide association studies: A review and resource for the functional dissection of human complex traits. Dis. Models Mech. 2017, 10, 77–88. [Google Scholar] [CrossRef] [PubMed]
- Flint, J.; Mackay, T.F.C. Genetic architecture of quantitative traits in mice, flies, and humans. Genome Res. 2009, 19, 723–733. [Google Scholar] [CrossRef] [PubMed]
- Cox, R.D.; Church, C.D. Mouse models and the interpretation of human GWAS in type 2 diabetes and obesity. Dis. Models Mech. 2011, 4, 155–164. [Google Scholar] [CrossRef] [PubMed]
- Blake, J.A.; Richardson, J.E.; Bult, C.J.; Kadin, J.A.; Eppig, J.T. MGD: The Mouse Genome Database. Nucleic Acids Res. 2003, 31, 193–195. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.L.; Eppig, J.T. The Mammalian Phenotype Ontology: Enabling robust annotation and comparative analysis. Wiley Interdiscip. Rev. Syst. Biol. Med. 2009, 1, 390–399. [Google Scholar] [CrossRef] [PubMed]
- Queitsch, C.; Carlson, K.D.; Girirajan, S. Lessons from Model Organisms: Phenotypic Robustness and Missing Heritability in Complex Disease. PLoS Genet. 2012, 8, e1003041. [Google Scholar] [CrossRef] [PubMed]
- Silventoinen, K.; Magnusson, P.K.E.; Tynelius, P.; Kaprio, J.; Rasmussen, F. Heritability of body size and muscle strength in young adulthood: A study of one million Swedish men. Genet. Epidemiol. 2008, 32, 341–349. [Google Scholar] [CrossRef] [PubMed]
- Cross-Disorder Group of the Psychiatric Genomics Consortium; International Inflammatory Bowel Disease Genetics Consortium (IIBDGC); Lee, S.H.; Ripke, S.; Neale, B.M.; Faraone, S.V.; Purcell, S.M.; Perlis, R.H.; Mowry, B.J.; Thapar, A.; et al. Genetic relationship between five psychiatric disorders estimated from genome-wide SNPs. Nat. Genet. 2013, 45, 984–994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elks, C.E.; den Hoed, M.; Zhao, J.H.; Sharp, S.J.; Wareham, N.J.; Loos, R.J.F.; Ong, K.K. Variability in the Heritability of Body Mass Index: A Systematic Review and Meta-Regression. Front. Endocrinol. (Lausanne) 2012, 3. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Bakshi, A.; Zhu, Z.; Hemani, G.; Vinkhuyzen, A.A.E.; Lee, S.H.; Robinson, M.R.; Perry, J.R.B.; Nolte, I.M.; van Vliet-Ostaptchouk, J.V.; et al. Genetic variance estimation with imputed variants finds negligible missing heritability for human height and body mass index. Nat. Genet. 2015, 47, 1114–1120. [Google Scholar] [CrossRef] [PubMed]
- Ali, O. Genetics of type 2 diabetes. World J. Diabetes 2013, 4, 114–123. [Google Scholar] [CrossRef] [PubMed]
- Cordell, H.J. Detecting gene-gene interactions that underlie human diseases. Nat. Rev. Genet. 2009, 10, 392–404. [Google Scholar] [CrossRef] [PubMed]
- Cordell, H.J. Epistasis: What it means, what it doesn’t mean, and statistical methods to detect it in humans. Hum. Mol. Genet. 2002, 11, 2463–2468. [Google Scholar] [CrossRef] [PubMed]
- Manolio, T.A.; Collins, F.S.; Cox, N.J.; Goldstein, D.B.; Hindorff, L.A.; Hunter, D.J.; McCarthy, M.I.; Ramos, E.M.; Cardon, L.R.; Chakravarti, A.; et al. Finding the missing heritability of complex diseases. Nature 2009, 461, 747–753. [Google Scholar] [CrossRef] [PubMed]
- Gamazon, E.R.; Huang, R.S.; Dolan, M.E.; Cox, N.J.; Im, H.K. Integrative Genomics: Quantifying Significance of Phenotype-Genotype Relationships from Multiple Sources of High-Throughput Data. Front. Genet. 2013, 3. [Google Scholar] [CrossRef] [PubMed]
- Lewis, R.; Lewis, R. Like a Game of Clue, Genomics Tracks Outbreak, Revealing Evolution in Action. Available online: https://blogs.scientificamerican.com/guest-blog/like-a-game-of-clue-genomics-tracks-outbreak-revealing-evolution-in-action/ (accessed on 1 November 2017).
- Clue Emerges in Case of the Missing Heritability. Available online: https://www.genengnews.com/gen-news-highlights/clue-emerges-in-case-of-the-missing-heritability/81249819 (accessed on 1 November 2017).
- Cluedo. Available online: https://en.wikipedia.org/wiki/Cluedo (accessed on 1 November 2017).
- MacArthur, J.; Bowler, E.; Cerezo, M.; Gil, L.; Hall, P.; Hastings, E.; Junkins, H.; McMahon, A.; Milano, A.; Morales, J.; et al. The new NHGRI-EBI Catalog of published genome-wide association studies (GWAS Catalog). Nucleic Acids Res. 2017, 45, D896–D901. [Google Scholar] [CrossRef] [PubMed]
- Welter, D.; MacArthur, J.; Morales, J.; Burdett, T.; Hall, P.; Junkins, H.; Klemm, A.; Flicek, P.; Manolio, T.; Hindorff, L.; et al. The NHGRI GWAS Catalog, a curated resource of SNP-trait associations. Nucleic Acids Res. 2014, 42, D1001–D1006. [Google Scholar] [CrossRef] [PubMed]
- Hindorff, L.A.; Sethupathy, P.; Junkins, H.A.; Ramos, E.M.; Mehta, J.P.; Collins, F.S.; Manolio, T.A. Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc. Natl. Acad. Sci. USA 2009, 106, 9362–9367. [Google Scholar] [CrossRef] [PubMed]
- Hirschhorn, J.N. Genomewide Association Studies—Illuminating Biologic Pathways. N. Engl. J. Med. 2009, 360, 1699–1701. [Google Scholar] [CrossRef] [PubMed]
- Schork, N.J.; Murray, S.S.; Frazer, K.A.; Topol, E.J. Common vs. rare allele hypotheses for complex diseases. Curr. Opin. Genet. Dev. 2009, 19, 212–219. [Google Scholar] [CrossRef] [PubMed]
- Franke, A.; McGovern, D.P.B.; Barrett, J.C.; Wang, K.; Radford-Smith, G.L.; Ahmad, T.; Lees, C.W.; Balschun, T.; Lee, J.; Roberts, R.; et al. Genome-wide meta-analysis increases to 71 the number of confirmed Crohn’s disease susceptibility loci. Nat. Genet. 2010, 42, 1118–1125. [Google Scholar] [CrossRef] [PubMed]
- Fall, T.; Ingelsson, E. Genome-wide association studies of obesity and metabolic syndrome. Mol. Cell. Endocrinol. 2014, 382, 740–757. [Google Scholar] [CrossRef] [PubMed]
- Billings, L.K.; Florez, J.C. The genetics of type 2 diabetes: What have we learned from GWAS? Ann. N. Y. Acad. Sci. 2010, 1212, 59–77. [Google Scholar] [CrossRef] [PubMed]
- Bashinskaya, V.V.; Kulakova, O.G.; Boyko, A.N.; Favorov, A.V.; Favorova, O.O. A review of genome-wide association studies for multiple sclerosis: Classical and hypothesis-driven approaches. Hum. Genet. 2015, 134, 1143–1162. [Google Scholar] [CrossRef] [PubMed]
- Michailidou, K.; Beesley, J.; Lindstrom, S.; Canisius, S.; Dennis, J.; Lush, M.J.; Maranian, M.J.; Bolla, M.K.; Wang, Q.; Shah, M.; et al. Genome-wide association analysis of more than 120,000 individuals identifies 15 new susceptibility loci for breast cancer. Nat. Genet. 2015, 47, 373–380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Church, C.; Lee, S.; Bagg, E.A.L.; McTaggart, J.S.; Deacon, R.; Gerken, T.; Lee, A.; Moir, L.; Mecinović, J.; Quwailid, M.M.; et al. A mouse model for the metabolic effects of the human fat mass and obesity associated FTO gene. PLoS Genet. 2009, 5, e1000599. [Google Scholar] [CrossRef] [PubMed]
- Pennesi, M.E.; Neuringer, M.; Courtney, R.J. Animal models of age related macular degeneration. Mol. Asp. Med. 2012, 33, 487–509. [Google Scholar] [CrossRef] [PubMed]
- Culverhouse, R.; Suarez, B.K.; Lin, J.; Reich, T. A perspective on epistasis: Limits of models displaying no main effect. Am. J. Hum. Genet. 2002, 70, 461–471. [Google Scholar] [CrossRef] [PubMed]
- Moore, J.H. A global view of epistasis. Nat. Genet. 2005, 37, 13–14. [Google Scholar] [CrossRef] [PubMed]
- Moore, J.H.; Williams, S.M. Traversing the conceptual divide between biological and statistical epistasis: Systems biology and a more modern synthesis. Bioessays 2005, 27, 637–646. [Google Scholar] [CrossRef] [PubMed]
- Mackay, T.F.C. Epistasis for quantitative traits in Drosophila. Methods Mol. Biol. 2015, 1253, 47–70. [Google Scholar] [CrossRef] [PubMed]
- Mackay, T.F.C. Epistasis and Quantitative Traits: Using Model Organisms to Study Gene-Gene Interactions. Nat. Rev. Genet. 2014, 15, 22–33. [Google Scholar] [CrossRef] [PubMed]
- Reedy, J.J.; Cavalier, F.P. Epistasis in eye colors of Drosophila melanogaster. J. Hered. 1971, 62, 131–134. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Richards, S.; Carbone, M.A.; Zhu, D.; Anholt, R.R.H.; Ayroles, J.F.; Duncan, L.; Jordan, K.W.; Lawrence, F.; Magwire, M.M.; et al. Epistasis dominates the genetic architecture of Drosophila quantitative traits. Proc. Natl. Acad. Sci. USA 2012, 109, 15553–15559. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Lu, Q.; Mukherjee, S.; Mukheerjee, S.; Crane, P.K.; Elston, R.; Ritchie, M.D. Analysis pipeline for the epistasis search-statistical versus biological filtering. Front. Genet. 2014, 5, 106. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Abecasis, G.R.; Boehnke, M.; Lin, X. Rare-Variant Association Analysis: Study Designs and Statistical Tests. Am. J. Hum. Genet. 2014, 95, 5–23. [Google Scholar] [CrossRef] [PubMed]
- Moutsianas, L.; Agarwala, V.; Fuchsberger, C.; Flannick, J.; Rivas, M.A.; Gaulton, K.J.; Albers, P.K.; Consortium, G.; McVean, G.; Boehnke, M.; et al. The Power of Gene-Based Rare Variant Methods to Detect Disease-Associated Variation and Test Hypotheses About Complex Disease. PLoS Genet. 2015, 11, e1005165. [Google Scholar] [CrossRef] [PubMed]
- Moore, C.B.; Wallace, J.R.; Frase, A.T.; Pendergrass, S.A.; Ritchie, M.D. BioBin: A bioinformatics tool for automating the binning of rare variants using publicly available biological knowledge. BMC Med. Genom. 2013, 6 (Suppl. 2). [Google Scholar] [CrossRef]
- Zhu, C.S.; Pinsky, P.F.; Cramer, D.W.; Ransohoff, D.F.; Hartge, P.; Pfeiffer, R.M.; Urban, N.; Mor, G.; Bast, R.C.; Moore, L.E.; et al. A framework for evaluating biomarkers for early detection: Validation of biomarker panels for ovarian cancer. Cancer Prev. Res. (Phila.) 2011, 4, 375–383. [Google Scholar] [CrossRef] [PubMed]
- Variant Association Tools. Available online: http://varianttools.sourceforge.net (accessed on 10 November 2017).
- Zhan, X.; Hu, Y.; Li, B.; Abecasis, G.R.; Liu, D.J. RVTESTS: An efficient and comprehensive tool for rare variant association analysis using sequence data: Table 1. Bioinformatics 2016, 32, 1423–1426. [Google Scholar] [CrossRef] [PubMed]
- Nejentsev, S.; Walker, N.; Riches, D.; Egholm, M.; Todd, J.A. Rare variants of IFIH1, a gene implicated in antiviral responses, protect against type 1 diabetes. Science 2009, 324, 387–389. [Google Scholar] [CrossRef] [PubMed]
- Peterson, A.S.; Fong, L.G.; Young, S.G. PCSK9 function and physiology. J. Lipid Res. 2008, 49, 1152–1156. [Google Scholar] [CrossRef] [PubMed]
- Steinberg, D.; Witztum, J.L. Inhibition of PCSK9: A powerful weapon for achieving ideal LDL cholesterol levels. Proc. Natl. Acad. Sci. USA 2009, 106, 9546–9547. [Google Scholar] [CrossRef] [PubMed]
- Lopez, D. Inhibition of PCSK9 as a novel strategy for the treatment of hypercholesterolemia. Drug News Perspect. 2008, 21, 323–330. [Google Scholar] [CrossRef] [PubMed]
- Pettenati, M.J.; Rao, P.N.; Phelan, M.C.; Grass, F.; Rao, K.W.; Cosper, P.; Carroll, A.J.; Elder, F.; Smith, J.L.; Higgins, M.D. Paracentric inversions in humans: A review of 446 paracentric inversions with presentation of 120 new cases. Am. J. Med. Genet. 1995, 55, 171–187. [Google Scholar] [CrossRef] [PubMed]
- Mullaney, J.M.; Mills, R.E.; Pittard, W.S.; Devine, S.E. Small insertions and deletions (INDELs) in human genomes. Hum. Mol. Genet. 2010, 19, R131–R136. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Wang, W.; Ma, G.; Liang, L.; Shi, Q.; Tao, S. Patterns of Insertion and Deletion in Mammalian Genomes. Curr. Genom. 2007, 8, 370–378. [Google Scholar] [CrossRef]
- Itsara, A.; Cooper, G.M.; Baker, C.; Girirajan, S.; Li, J.; Absher, D.; Krauss, R.M.; Myers, R.M.; Ridker, P.M.; Chasman, D.I.; et al. Population analysis of large copy number variants and hotspots of human genetic disease. Am. J. Hum. Genet. 2009, 84, 148–161. [Google Scholar] [CrossRef] [PubMed]
- Wellcome Trust Case Control Consortium. Genome-wide association study of 14,000 cases of seven common diseases and 3000 shared controls. Nature 2007, 447, 661–678. [Google Scholar] [CrossRef] [Green Version]
- Chung, B.H.-Y.; Tao, V.Q.; Tso, W.W.-Y. Copy number variation and autism: New insights and clinical implications. J. Formos. Med. Assoc. 2014, 113, 400–408. [Google Scholar] [CrossRef] [PubMed]
- Patel, C.J.; Bhattacharya, J.; Butte, A.J. An Environment-Wide Association Study (EWAS) on Type 2 Diabetes Mellitus. PLoS ONE 2010, 5, e10746. [Google Scholar] [CrossRef] [PubMed]
- Hall, M.A.; Dudek, S.M.; Goodloe, R.; Crawford, D.C.; Pendergrass, S.A.; Peissig, P.; Brilliant, M.; Mccarty, C.A.; Ritchie, M.D. Environment-wide association study (EWAS) for type 2 diabetes in the Marshfield Personalized Medicine Research Project Biobank. In Proceedings of the Pacific Symposium on Biocomputing, Kohala Coast, HI, USA, 3–7 January 2014; pp. 200–211. [Google Scholar]
- Ottman, R. Gene–Environment Interaction: Definitions and Study Designs. Prev. Med. 1996, 25, 764–770. [Google Scholar] [CrossRef] [PubMed]
- Khoury, M.J. Editorial: Emergence of Gene-Environment Interaction Analysis in Epidemiologic Research. Am. J. Epidemiol. 2017, 186, 751–752. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, M.D.; Davis, J.R.; Aschard, H.; Battle, A.; Conti, D.; Du, M.; Eskin, E.; Fallin, M.D.; Hsu, L.; Kraft, P.; et al. Incorporation of Biological Knowledge Into the Study of Gene-Environment Interactions. Am. J. Epidemiol. 2017, 186, 771–777. [Google Scholar] [CrossRef] [PubMed]
- Patel, C.J.; Cullen, M.R.; Ioannidis, J.P.; Butte, A.J. Systematic evaluation of environmental factors: Persistent pollutants and nutrients correlated with serum lipid levels. Int. J. Epidemiol. 2012, 41, 828–843. [Google Scholar] [CrossRef] [PubMed]
- Thomas, D. Gene–environment-wide association studies: Emerging approaches. Nat. Rev. Genet. 2010, 11, 259–272. [Google Scholar] [CrossRef] [PubMed]
- Wright, F.A.; Sullivan, P.F.; Brooks, A.I.; Zou, F.; Sun, W.; Xia, K.; Madar, V.; Jansen, R.; Chung, W.; Zhou, Y.-H.; et al. Heritability and Genomics of Gene Expression in Peripheral Blood. Nat. Genet. 2014, 46, 430–437. [Google Scholar] [CrossRef] [PubMed]
- Moffatt, M.F.; Kabesch, M.; Liang, L.; Dixon, A.L.; Strachan, D.; Heath, S.; Depner, M.; von Berg, A.; Bufe, A.; Rietschel, E.; et al. Genetic variants regulating ORMDL3 expression contribute to the risk of childhood asthma. Nature 2007, 448, 470–473. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Zhang, F.; Hu, H.; Bakshi, A.; Robinson, M.R.; Powell, J.E.; Montgomery, G.W.; Goddard, M.E.; Wray, N.R.; Visscher, P.M.; et al. Integration of summary data from GWAS and eQTL studies predicts complex trait gene targets. Nat. Genet. 2016, 48, 481–487. [Google Scholar] [CrossRef] [PubMed]
- Barbeira, A.N.; Dickinson, S.P.; Torres, J.M.; Bonazzola, R.; Zheng, J.; Torstenson, E.S.; Wheeler, H.E.; Shah, K.P.; Edwards, T.; Garcia, T.; et al. Exploring the phenotypic consequences of tissue specific gene expression variation inferred from GWAS summary statistics. bioRxiv 2017, 045260. [Google Scholar] [CrossRef]
- Hormozdiari, F.; van de Bunt, M.; Segrè, A.V.; Li, X.; Joo, J.W.J.; Bilow, M.; Sul, J.H.; Sankararaman, S.; Pasaniuc, B.; Eskin, E. Colocalization of GWAS and eQTL Signals Detects Target Genes. Am. J. Hum. Genet. 2016, 99, 1245–1260. [Google Scholar] [CrossRef] [PubMed]
- LaCroix, B.; Gamazon, E.R.; Lenkala, D.; Im, H.K.; Geeleher, P.; Ziliak, D.; Cox, N.J.; Huang, R.S. Integrative analyses of genetic variation, epigenetic regulation, and the transcriptome to elucidate the biology of platinum sensitivity. BMC Genom. 2014, 15, 292. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Li, R.; Dudek, S.M.; Ritchie, M.D. ATHENA: Identifying interactions between different levels of genomic data associated with cancer clinical outcomes using grammatical evolution neural network. BioData Min. 2013, 6, 23. [Google Scholar] [CrossRef] [PubMed]
- Holzinger, E.R.; Dudek, S.M.; Frase, A.T.; Pendergrass, S.A.; Ritchie, M.D. ATHENA: The analysis tool for heritable and environmental network associations. Bioinformatics 2014, 30, 698–705. [Google Scholar] [CrossRef] [PubMed]
- Gusev, A.; Lee, S.H.; Trynka, G.; Finucane, H.; Vilhjálmsson, B.J.; Xu, H.; Zang, C.; Ripke, S.; Bulik-Sullivan, B.; Stahl, E.; et al. Partitioning Heritability of Regulatory and Cell-Type-Specific Variants across 11 Common Diseases. Am. J. Hum. Genet. 2014, 95, 535–552. [Google Scholar] [CrossRef] [PubMed]
- Madu, C.O.; Lu, Y. Novel diagnostic biomarkers for prostate cancer. J. Cancer 2010, 1, 150–177. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Chan, D.W. Proteomic cancer biomarkers from discovery to approval: It’s worth the effort. Expert Rev. Proteom. 2014, 11, 135–136. [Google Scholar] [CrossRef] [PubMed]
- Coticchia, C.M.; Yang, J.; Moses, M.A. Ovarian Cancer Biomarkers: Current Options and Future Promise. J. Natl. Compr. Cancer Netw. 2008, 6, 795–802. [Google Scholar] [CrossRef]
- Mai, P.L.; Wentzensen, N.; Greene, M.H. Challenges related to developing serum-based biomarkers for early ovarian cancer detection. Cancer Prev. Res. (Phila.) 2011, 4, 303–306. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.-S.; Park, J.; Kay, K.A.; Christakis, N.A.; Oltvai, Z.N.; Barabási, A.-L. The implications of human metabolic network topology for disease comorbidity. Proc. Natl. Acad. Sci. USA 2008, 105, 9880–9885. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.V.; Kardia, S.L.R. Identification of epistatic effects using a protein–protein interaction database. Hum. Mol. Genet. 2010, 19, 4345–4352. [Google Scholar] [CrossRef] [PubMed]
- Egger, G.; Liang, G.; Aparicio, A.; Jones, P.A. Epigenetics in human disease and prospects for epigenetic therapy. Nature 2004, 429, 457–463. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.A.; Baylin, S.B. The fundamental role of epigenetic events in cancer. Nat. Rev. Genet. 2002, 3, 415–428. [Google Scholar] [CrossRef] [PubMed]
- Ballestar, E.; Esteller, M. The epigenetic breakdown of cancer cells: From DNA methylation to histone modifications. Prog. Mol. Subcell. Biol. 2005, 38, 169–181. [Google Scholar] [PubMed]
- Novak, K. Epigenetics Changes in Cancer Cells. MedGenMed 2004, 6, 17. [Google Scholar] [PubMed]
- Bonasio, R. The expanding epigenetic landscape of non-model organisms. J. Exp. Biol. 2015, 218, 114–122. [Google Scholar] [CrossRef] [PubMed]
- Grunstein, M.; Gasser, S.M. Epigenetics in Saccharomyces cerevisiae. Cold Spring Harb. Perspect. Biol. 2013, 5. [Google Scholar] [CrossRef] [PubMed]
- Lyko, F.; Beisel, C.; Marhold, J.; Paro, R. Epigenetic regulation in Drosophila. Curr. Top. Microbiol. Immunol. 2006, 310, 23–44. [Google Scholar] [PubMed]
- Weissmann, F.; Muyrers-Chen, I.; Musch, T.; Stach, D.; Wiessler, M.; Paro, R.; Lyko, F. DNA Hypermethylation in Drosophila melanogaster Causes Irregular Chromosome Condensation and Dysregulation of Epigenetic Histone Modifications. Mol. Cell. Biol. 2003, 23, 2577–2586. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Mackay, T.F.; Anholt, R.R. Transcriptional and epigenetic responses to mating and aging in Drosophila melanogaster. BMC Genom. 2014, 15. [Google Scholar] [CrossRef] [PubMed]
- Mano, Y.; Kobayashi, T.J.; Nakayama, J.; Uchida, H.; Oki, M. Single Cell Visualization of Yeast Gene Expression Shows Correlation of Epigenetic Switching between Multiple Heterochromatic Regions through Multiple Generations. PLoS Biol. 2013, 11, e1001601. [Google Scholar] [CrossRef] [PubMed]
- Omic Tools. Available online: https://omictools.com (accessed on 22 January 2018).
- PLINK2. Available online: https://www.cog-genomics.org/plink2 (accessed on 22 January 2018).
- PLATO. Available online: https://ritchielab.psu.edu/software/plato-download (accessed on 22 January 2018).
- QCTOOL. Available online: http://www.well.ox.ac.uk/~gav/qctool/#overview (accessed on 22 January 2018).
- Genabel. Available online: http://www.genabel.org (accessed on 22 January 2018).
- BOLT-LMM. Available online: https://data.broadinstitute.org/alkesgroup/BOLT-LMM/ (accessed on 22 January 2018).
- FAST-LMM. Available online: https://github.com/MicrosoftGenomics/FaST-LMM (accessed on 22 January 2018).
- CNVTools. Available online: http://www.bioconductor.org/packages/release/bioc/html/CNVtools.html (accessed on 22 January 2018).
- PennCNV. Available online: http://penncnv.openbioinformatics.org/en/latest/ (accessed on 22 January 2018).
- CKAT. Available online: https://works.bepress.com/debashis_ghosh/75/ (accessed on 22 January 2018).
- ParseCNV. Available online: http://parsecnv.sourceforge.net (accessed on 22 January 2018).
- CNVassoc. Available online: https://cran.r-project.org/web/packages/CNVassoc/index.html (accessed on 22 January 2018).
- RVtests. Available online: https://genome.sph.umich.edu/wiki/Rvtests (accessed on 22 January 2018).
- PLINK/SEQ. Available online: https://atgu.mgh.harvard.edu/plinkseq/ (accessed on 22 January 2018).
- EPACTS-Genome Analysis Wiki. Available online: https://genome.sph.umich.edu/wiki/EPACTS (accessed on 29 October 2017).
- MAGMA. Available online: https://ctg.cncr.nl/software/magma (accessed on 22 January 2018).
- EMMAX. Available online: http://varianttools.sourceforge.net (accessed on 22 January 2018).
- MDR. Available online: https://sourceforge.net/projects/mdr/ (accessed on 22 January 2018).
- AntEpiSeeker. Available online: http://nce.ads.uga.edu/~romdhane/AntEpiSeeker/index.html (accessed on 22 January 2018).
- MultiSurf. Available online: https://github.com/EpistasisLab/scikit-rebate/blob/master/skrebate/multisurf.py (accessed on 22 January 2018).
- BOOST. Available online: http://bioinformatics.ust.hk/BOOST.html (accessed on 22 January 2018).
- SNPTest. Available online: https://mathgen.stats.ox.ac.uk/genetics_software/snptest/snptest.html (accessed on 22 January 2018).
- TS-GSIS. Available online: https://cran.r-project.org/web/packages/TSGSIS/index.html (accessed on 22 January 2018).
- SNPassociation. Available online: https://cran.r-project.org/web/packages/SNPassoc/index.html (accessed on 22 January 2018).
- CAPE. Available online: https://cran.r-project.org/web/packages/cape/cape.pdf (accessed on 22 January 2018).
- Colak, R.; Kim, T.; Kazan, H.; Oh, Y.; Cruz, M.; Valladares-Salgado, A.; Peralta, J.; Escobedo, J.; Parra, E.J.; Kim, P.M.; et al. JBASE: Joint Bayesian Analysis of Subphenotypes and Epistasis. Bioinformatics 2016, 32, 203–210. [Google Scholar] [CrossRef] [PubMed]
- SMR. Available online: http://cnsgenomics.com/software/smr/ (accessed on 29 October 2017).
- EWASher. Available online: https://www.microsoft.com/en-us/download/details.aspx?id=52501 (accessed on 22 January 2018).
- LiCHe. Available online: http://viq854.github.io/lichee/ (accessed on 22 January 2018).
- BUHMBOX. Available online: http://software.broadinstitute.org/mpg/buhmbox/ (accessed on 22 January 2018).
- ForestPMPlot. Available online: http://genetics.cs.ucla.edu/meta_jemdoc/ (accessed on 22 January 2018).
- NetDx. Available online: https://github.com/nrnb/GoogleSummerOfCode/issues/70 (accessed on 22 January 2018).
- BioGranat-IG. Available online: http://www.thomas-schlitt.net/Biogranat.html (accessed on 22 January 2018).
- NETAM. Available online: http://www.sailing.cs.cmu.edu/main/ (accessed on 22 January 2018).
- EINVis. Available online: http://www.robwu.net/einvis/ (accessed on 22 January 2018).
- NetDecoder. Available online: http://netdecoder.hms.harvard.edu (accessed on 22 January 2018).
- ViSEN. Available online: https://sourceforge.net/projects/visen/ (accessed on 22 January 2018).
- Cytoscape. Available online: http://www.cytoscape.org (accessed on 22 January 2018).
- PARIS. Available online: https://ritchielab.psu.edu/software/paris-download (accessed on 22 January 2018).
- SNPsea. Available online: http://pubs.broadinstitute.org/mpg/snpsea/ (accessed on 22 January 2018).
- GSEA. Available online: http://software.broadinstitute.org/gsea/index.jsp (accessed on 22 January 2018).
- Vegas2Pathway. Available online: https://vegas2.qimrberghofer.edu.au (accessed on 22 January 2018).
- MAGENTA. Available online: https://software.broadinstitute.org/mpg/magenta/ (accessed on 22 January 2018).
- ATHENA. Available online: https://ritchielab.psu.edu/software/athena-downloads (accessed on 22 January 2018).
- iCluster. Available online: https://www.mskcc.org/departments/epidemiology-biostatistics/biostatistics/icluster (accessed on 22 January 2018).
- Biofilter. Available online: https://ritchielab.psu.edu/software/biofilter-download-1 (accessed on 22 January 2018).
- SKAT. Available online: https://www.hsph.harvard.edu/skat/ (accessed on 22 January 2018).
- Biobin. Available online: https://ritchielab.psu.edu/software/biobin-download (accessed on 22 January 2018).
- GLM. Available online: https://cran.r-project.org/web/packages/glmnet/index.html (accessed on 22 January 2018).
- RANGER. Available online: https://cran.r-project.org/web/packages/ranger/ranger.pdf (accessed on 22 January 2018).
- Gradient Boosting. Available online: https://cran.r-project.org/web/packages/gbm/index.html (accessed on 22 January 2018).
- TATES. Available online: https://ctg.cncr.nl/software/tates (accessed on 22 January 2018).
- CAVIAR. Available online: http://genetics.cs.ucla.edu/caviar/ (accessed on 22 January 2018).
- PrediXcan. Available online: https://github.com/hakyimlab/PrediXcan (accessed on 22 January 2018).
- Ritchie, M.D.; Holzinger, E.R.; Li, R.; Pendergrass, S.A.; Kim, D. Methods of integrating data to uncover genotype-phenotype interactions. Nat. Rev. Genet. 2015, 16, 85–97. [Google Scholar] [CrossRef] [PubMed]
- Lonsdale, J.; Thomas, J.; Salvatore, M.; Phillips, R.; Lo, E.; Shad, S.; Hasz, R.; Walters, G.; Garcia, F.; Young, N.; et al. The Genotype-Tissue Expression (GTEx) project. Nat. Genet. 2013, 45, 580–585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braun, T.; Wagner, A.; DeLuca, A.; Casavant, T.; Scheetz, T.; Clark, A.; Mullins, R.; Stone, E. The Ocular Tissue Database. Investig. Ophthalmol. Vis. Sci. 2013, 54, 3383. [Google Scholar]
- Slowikowski, K.; Hu, X.; Raychaudhuri, S. SNPsea: An algorithm to identify cell types, tissues and pathways affected by risk loci. Bioinformatics 2014, 30, 2496–2497. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Yin, X.; Wen, L.; Yang, C.; Sheng, Y.; Lin, Y.; Zhu, Z.; Shen, C.; Shi, Y.; Zheng, Y.; et al. Several Critical Cell Types, Tissues, and Pathways Are Implicated in Genome-Wide Association Studies for Systemic Lupus Erythematosus. G3 (Bethesda) 2016, 6, 1503–1511. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Kim, H.; Stahl, E.; Plenge, R.; Daly, M.; Raychaudhuri, S. Integrating autoimmune risk loci with gene-expression data identifies specific pathogenic immune cell subsets. Am. J. Hum. Genet. 2011, 89, 496–506. [Google Scholar] [CrossRef] [PubMed]
- Fadista, J.; Vikman, P.; Laakso, E.O.; Mollet, I.G.; Esguerra, J.L.; Taneera, J.; Storm, P.; Osmark, P.; Ladenvall, C.; Prasad, R.B.; et al. Global genomic and transcriptomic analysis of human pancreatic islets reveals novel genes influencing glucose metabolism. Proc. Natl. Acad. Sci. USA 2014, 111, 13924–13929. [Google Scholar] [CrossRef] [PubMed]
- Small, K.S.; Hedman, A.K.; Grundberg, E.; Nica, A.C.; Thorleifsson, G.; Kong, A.; Thorsteindottir, U.; Shin, S.-Y.; Richards, H.B.; GIANT Consortium; et al. Identification of an imprinted master trans regulator at the KLF14 locus related to multiple metabolic phenotypes. Nat. Genet. 2011, 43, 561–564. [Google Scholar] [CrossRef] [PubMed]
- Grotz, A.K.; Gloyn, A.L.; Thomsen, S.K. Prioritising Causal Genes at Type 2 Diabetes Risk Loci. Curr. Diabetes Rep. 2017, 17. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Lee, S.H.; Goddard, M.E.; Visscher, P.M. GCTA: A Tool for Genome-wide Complex Trait Analysis. Am. J. Hum. Genet. 2011, 88, 76–82. [Google Scholar] [CrossRef] [PubMed]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.R.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.W.; Daly, M.J.; et al. PLINK: A Tool Set for Whole-Genome Association and Population-Based Linkage Analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [PubMed]
- Pérez, P.; de los Campos, G. Genome-Wide Regression and Prediction with the BGLR Statistical Package. Genetics 2014, 198, 483–495. [Google Scholar] [CrossRef] [PubMed]
- Speed, D.; Hemani, G.; Johnson, M.R.; Balding, D.J. Improved heritability estimation from genome-wide SNPs. Am. J. Hum. Genet. 2012, 91, 1011–1021. [Google Scholar] [CrossRef] [PubMed]
- Gorfine, M.; Berndt, S.I.; Chang-Claude, J.; Hoffmeister, M.; Marchand, L.L.; Potter, J.; Slattery, M.L.; Keret, N.; Peters, U.; Hsu, L. Heritability Estimation using a Regularized Regression Approach (HERRA): Applicable to continuous, dichotomous or age-at-onset outcome. PLoS ONE 2017, 12, e0181269. [Google Scholar] [CrossRef] [PubMed]
- Ge, T.; Nichols, T.E.; Lee, P.H.; Holmes, A.J.; Roffman, J.L.; Buckner, R.L.; Sabuncu, M.R.; Smoller, J.W. Massively expedited genome-wide heritability analysis (MEGHA). Proc. Natl. Acad. Sci. USA 2015, 112, 2479–2484. [Google Scholar] [CrossRef] [PubMed]
- Locke, A.E.; Kahali, B.; Berndt, S.I.; Justice, A.E.; Pers, T.H.; Day, F.R.; Powell, C.; Vedantam, S.; Buchkovich, M.L.; Yang, J.; et al. Genetic studies of body mass index yield new insights for obesity biology. Nature 2015, 518, 197–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, M.R.; English, G.; Moser, G.; Lloyd-Jones, L.R.; Triplett, M.A.; Zhu, Z.; Nolte, I.M.; van Vliet-Ostaptchouk, J.V.; Snieder, H.; The LifeLines Cohort Study; et al. Genotype-covariate interaction effects and the heritability of adult body mass index. Nat. Genet. 2017, 49, 1174–1181. [Google Scholar] [CrossRef] [PubMed]
- Speed, D.; Cai, N.; UCLEB Consortium; Johnson, M.R.; Nejentsev, S.; Balding, D.J. Reevaluation of SNP heritability in complex human traits. Nat. Genet. 2017, 49, 986–992. [Google Scholar] [CrossRef] [PubMed]
- Finucane, H.K.; Bulik-Sullivan, B.; Gusev, A.; Trynka, G.; Reshef, Y.; Loh, P.-R.; Anttila, V.; Xu, H.; Zang, C.; Farh, K.; et al. Partitioning heritability by functional annotation using genome-wide association summary statistics. Nat. Genet. 2015, 47, 1228–1235. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.J.; Leal, S.M. Estimating Genetic Effects and Quantifying Missing Heritability Explained by Identified Rare-Variant Associations. Am. J. Hum. Genet. 2012, 91, 585–596. [Google Scholar] [CrossRef] [PubMed]
- Ronnegard, L.; Shen, X. Genomic prediction and estimation of marker interaction effects. bioRxiv 2016, 038935. [Google Scholar] [CrossRef]
- Pendergrass, S.A.; Brown-Gentry, K.; Dudek, S.; Frase, A.; Torstenson, E.S.; Goodloe, R.; Ambite, J.L.; Avery, C.L.; Buyske, S.; Bůžková, P.; et al. Phenome-wide association study (PheWAS) for detection of pleiotropy within the Population Architecture using Genomics and Epidemiology (PAGE) Network. PLoS Genet. 2013, 9, e1003087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daar, E.S.; Tierney, C.; Fischl, M.A.; Sax, P.E.; Mollan, K.; Budhathoki, C.; Godfrey, C.; Jahed, N.C.; Myers, L.; Katzenstein, D.; et al. Atazanavir plus ritonavir or efavirenz as part of a 3-drug regimen for initial treatment of HIV-1. Ann. Intern. Med. 2011, 154, 445–456. [Google Scholar] [CrossRef] [PubMed]
- Denny, J.C.; Ritchie, M.D.; Basford, M.A.; Pulley, J.M.; Bastarache, L.; Brown-Gentry, K.; Wang, D.; Masys, D.R.; Roden, D.M.; Crawford, D.C. PheWAS: Demonstrating the feasibility of a phenome-wide scan to discover gene-disease associations. Bioinformatics 2010, 26, 1205–1210. [Google Scholar] [CrossRef] [PubMed]
- Verma, A.; Bradford, Y.; Verma, S.S.; Pendergrass, S.A.; Daar, E.S.; Venuto, C.; Morse, G.D.; Ritchie, M.D.; Haas, D.W. Multiphenotype association study of patients randomized to initiate antiretroviral regimens in AIDS Clinical Trials Group protocol A5202. Pharmacogenet. Genom. 2017, 27, 101–111. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Cheng, W.-Y.; Glicksberg, B.S.; Gottesman, O.; Tamler, R.; Chen, R.; Bottinger, E.P.; Dudley, J.T. Identification of type 2 diabetes subgroups through topological analysis of patient similarity. Sci. Transl. Med. 2015, 7, 311ra174. [Google Scholar] [CrossRef] [PubMed]
- Verma, A.; Leader, J.B.; Verma, S.S.; Frase, A.; Wallace, J.; Dudek, S.; Lavage, D.R.; Van Hout, C.V.; Dewey, F.E.; Penn, J.; et al. Integrating clinical laboratory measures and icd-9 code diagnoses in phenome-wide association studies. In Proceedings of the Pacific Symposium on Biocomputing, Kohala Coast, HI, USA, 4–8 January 2016; Volume 21, pp. 168–179. [Google Scholar]
- Zhao, J.; Papapetrou, P.; Asker, L.; Boström, H. Learning from heterogeneous temporal data in electronic health records. J. Biomed. Inform. 2017, 65, 105–119. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Nadkarni, G.; Gottesman, O.; Ellis, S.B.; Bottinger, E.P.; Guttag, J.V. Incorporating temporal EHR data in predictive models for risk stratification of renal function deterioration. J. Biomed. Inform. 2015, 53, 220–228. [Google Scholar] [CrossRef] [PubMed]
- Magger, O.; Waldman, Y.Y.; Ruppin, E.; Sharan, R. Enhancing the Prioritization of Disease-Causing Genes through Tissue Specific Protein Interaction Networks. PLoS Comput. Biol. 2012, 8, e1002690. [Google Scholar] [CrossRef] [PubMed]
- Rotroff, D.M.; Pijut, S.S.; Marvel, S.W.; Jack, J.R.; Havener, T.M.; Pujol, A.; Schluter, A.; Graf, G.A.; Ginsberg, H.N.; Shah, H.S.; et al. Genetic variants in HSD17B3, SMAD3, and IPO11 impact circulating lipids in response to fenofibrate in individuals with type 2 diabetes. Clin. Pharmacol. Ther. 2017. [Google Scholar] [CrossRef] [PubMed]
- Ligthart, S.; Vaez, A.; Hsu, Y.-H.; Inflammation Working Group of the CHARGE Consortium; PMI-WG-XCP; LifeLines Cohort Study; Stolk, R.; Uitterlinden, A.G.; Hofman, A.; Alizadeh, B.Z.; et al. Bivariate genome-wide association study identifies novel pleiotropic loci for lipids and inflammation. BMC Genom. 2016, 17, 443. [Google Scholar] [CrossRef]
- Surakka, I.; Horikoshi, M.; Mägi, R.; Sarin, A.-P.; Mahajan, A.; Lagou, V.; Marullo, L.; Ferreira, T.; Miraglio, B.; Timonen, S.; et al. The impact of low-frequency and rare variants on lipid levels. Nat. Genet. 2015, 47, 589–597. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Brautbar, A.; Boerwinkle, E.; Sing, C.F.; Clark, A.G.; Keinan, A. Knowledge-Driven Analysis Identifies a Gene–Gene Interaction Affecting High-Density Lipoprotein Cholesterol Levels in Multi-Ethnic Populations. PLoS Genet. 2012, 8, e1002714. [Google Scholar] [CrossRef] [PubMed]
- De, R.; Verma, S.S.; Drenos, F.; Holzinger, E.R.; Holmes, M.V.; Hall, M.A.; Crosslin, D.R.; Carrell, D.S.; Hakonarson, H.; Jarvik, G.; et al. Identifying gene-gene interactions that are highly associated with Body Mass Index using Quantitative Multifactor Dimensionality Reduction (QMDR). BioData Min. 2015, 8, 41. [Google Scholar] [CrossRef] [PubMed]
- Holzinger, E.R.; Verma, S.S.; Moore, C.B.; Hall, M.; De, R.; Gilbert-Diamond, D.; Lanktree, M.B.; Pankratz, N.; Amuzu, A.; Burt, A.; et al. Discovery and replication of SNP-SNP interactions for quantitative lipid traits in over 60,000 individuals. BioData Min. 2017, 10, 25. [Google Scholar] [CrossRef] [PubMed]
- Ordovas, J.M. Gene-diet interaction and plasma lipid responses to dietary intervention. Biochem. Soc. Trans. 2002, 30, 68–73. [Google Scholar] [CrossRef] [PubMed]
- Shungin, D.; Deng, W.Q.; Varga, T.V.; Luan, J.; Mihailov, E.; Metspalu, A.; Morris, A.P.; Forouhi, N.G.; Lindgren, C.; Magnusson, P.K.E.; et al. Ranking and characterization of established BMI and lipid associated loci as candidates for gene-environment interactions. PLoS Genet. 2017, 13. [Google Scholar] [CrossRef] [PubMed]
- Wen, X.; Pique-Regi, R.; Luca, F. Integrating molecular QTL data into genome-wide genetic association analysis: Probabilistic assessment of enrichment and colocalization. PLoS Genet. 2017, 13, e1006646. [Google Scholar] [CrossRef] [PubMed]
- Luczak, M.; Formanowicz, D.; Marczak, Ł.; Suszyńska-Zajczyk, J.; Pawliczak, E.; Wanic-Kossowska, M.; Stobiecki, M. iTRAQ-based proteomic analysis of plasma reveals abnormalities in lipid metabolism proteins in chronic kidney disease-related atherosclerosis. Sci. Rep. 2016, 6, 32511. [Google Scholar] [CrossRef] [PubMed]
- Holzinger, E.R.; Dudek, S.M.; Frase, A.T.; Krauss, R.M.; Medina, M.W.; Ritchie, M.D. ATHENA: A tool for meta-dimensional analysis applied to genotypes and gene expression data to predict HDL cholesterol levels. In Proceedings of the Pacific Symposium on Biocomputing, Kohala Coast, HI, USA, 3–7 January 2013; pp. 385–396. [Google Scholar]
- Morabia, A.; Cayanis, E.; Costanza, M.C.; Ross, B.M.; Flaherty, M.S.; Alvin, G.B.; Das, K.; Gilliam, T.C. Association of extreme blood lipid profile phenotypic variation with 11 reverse cholesterol transport genes and 10 non-genetic cardiovascular disease risk factors. Hum. Mol. Genet. 2003, 12, 2733–2743. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Weapon | Suspects | Tool Name | Reference |
|---|---|---|---|
| Additive Model | Common variations | PLINK | [113] |
| Common variations | PLATO | [114] | |
| Common variations | QCTool | [115] | |
| Common variations | GenAbel | [116] | |
| Common and Rare Variations | BOLT-LMM | [117] | |
| Common and Rare Variations | FAST-LMM | [118] | |
| Structural Variations | CNVTools | [119] | |
| Structural Variations | PennCNV | [120] | |
| Structural Variations | CKAT | [121] | |
| Structural Variations | ParseCNV | [122] | |
| SNPs and Structural Variations | CNVassoc | [123] | |
| Common and Rare Variations | RVTests | [124] | |
| Common and Rare Variations | PLINK/SEQ | [125] | |
| Rare Variations | EPACTS | [126] | |
| Common variations | MAGMA | [127] | |
| Rare Variations | EMMAX | [128] | |
| Gene–Gene Interactions Model | Common variations | MDR | [129] |
| Common variations | AntEpiSeeker | [130] | |
| Common variations | MultiSURF | [131] | |
| Common variations | BOOST | [132] | |
| Common variations | PLATO | [114] | |
| SNPs and Structural Variations | CNVassoc | [123] | |
| Common variations | SNPTEST | [133] | |
| Common variations | TS-GSIS | [134] | |
| Common variations | SNPAssociation | [135] | |
| Common variations | PLINK | [113] | |
| Common Variants and Phenotypes | CAPE | [136] | |
| Gene-Environment Interactions Model | Common variations and Environment | PLATO | [114] |
| Detecting Heterogeneity | Genetic variations and Phenotypes | JBASE | [137] |
| Gene Expression and Phenotype | SMR | [138] | |
| Gene Expression and Phenotype | FAST-LMM-EWASher | [139] | |
| Phenotype | LiCHe | [140] | |
| Genetic and phenotypic | BUHMBOX | [141] | |
| Genetic Heterogeneity | ForestPMPlot | [142] | |
| Genetic variations and Phenotypes | NetDx | [143] | |
| Genetic Heterogeneity | BioGranat-IG | [144] | |
| Network based approaches | SNPs, Phenotypes and Gene Expression | NETAM | [145] |
| Common variations | EINVis | [146] | |
| Gene Expression and Phenotype | NetDecoder | [147] | |
| Common variations | ViSEN | [148] | |
| All genetic variations | Cytoscape | [149] | |
| Pathway analyses | Common variations | PARIS | [150] |
| Genes | SNPSea | [151] | |
| Genes | GSEA | [152] | |
| Common variations | VEGAS2Pathway | [153] | |
| Common variations | MAGENTA | [154] | |
| Meta-dimensional modelling | Multi-Omic Datasets | ATHENA | [155] |
| Multi-Omic Datasets | NetDX | [143] | |
| Multi-Omic Datasets | iCluster | [156] | |
| Gene-based analyses | All genetic variations | Biofilter | [157] |
| Common and Rare Variations | SKAT | [158] | |
| Rare Variations | BioBin | [159] | |
| Rare Variations | Variant Association Tools | [68] | |
| Rare Variations | EPACTS | [126] | |
| Feature Selection/Prioritization | All genetic variations | Biofilter | [157] |
| Common variations | GLM (LASSO and Elastic-Net) | [160] | |
| Common variations | RANGER | [161] | |
| Common variations | Gradient Boosting | [162] | |
| Causal Variant Determination | Common variations | TATES | [163] |
| Common variation and eQTL | CAVIAR | [164] | |
| Common variation and eQTL | PrediXcan | [165] |
| Analysis Type | References |
|---|---|
| Common variants | Rotroff et al. [196], Ligthart et al. [197] |
| Rare Variants | Liu et al. [185], Surakka et al. [198] |
| Gene–Gene Interactions | Ma et al. [199], De et al. [200], Holzinger et al. [201] |
| Gene–Environment Interactions | Ordovas [202], Shungin et al. [203] |
| Gene Expression analysis | Wen et al. [204] |
| Proteomics | Luczak et al. [205] |
| Meta-dimensional analysis | Holzinger et al. [206] |
| Phenotype Heterogeneity | Morabia et al. [207] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Verma, S.S.; Ritchie, M.D. Another Round of “Clue” to Uncover the Mystery of Complex Traits. Genes 2018, 9, 61. https://doi.org/10.3390/genes9020061
Verma SS, Ritchie MD. Another Round of “Clue” to Uncover the Mystery of Complex Traits. Genes. 2018; 9(2):61. https://doi.org/10.3390/genes9020061
Chicago/Turabian StyleVerma, Shefali Setia, and Marylyn D. Ritchie. 2018. "Another Round of “Clue” to Uncover the Mystery of Complex Traits" Genes 9, no. 2: 61. https://doi.org/10.3390/genes9020061
APA StyleVerma, S. S., & Ritchie, M. D. (2018). Another Round of “Clue” to Uncover the Mystery of Complex Traits. Genes, 9(2), 61. https://doi.org/10.3390/genes9020061