MHC-Dependent Mate Selection within 872 Spousal Pairs of European Ancestry from the Health and Retirement Study

Abstract
:1. Introduction
2. Materials and Methods
2.1. Data
2.2. Statistical Analysis
2.2.1. Major Histocompatibility Complex Region as a Whole
2.2.2. Genomic Similarity across the MHC Compared to Genomic Similarity across the Genome
2.2.3. Individual SNPs, HLA Alleles, and Amino Acids
3. Results
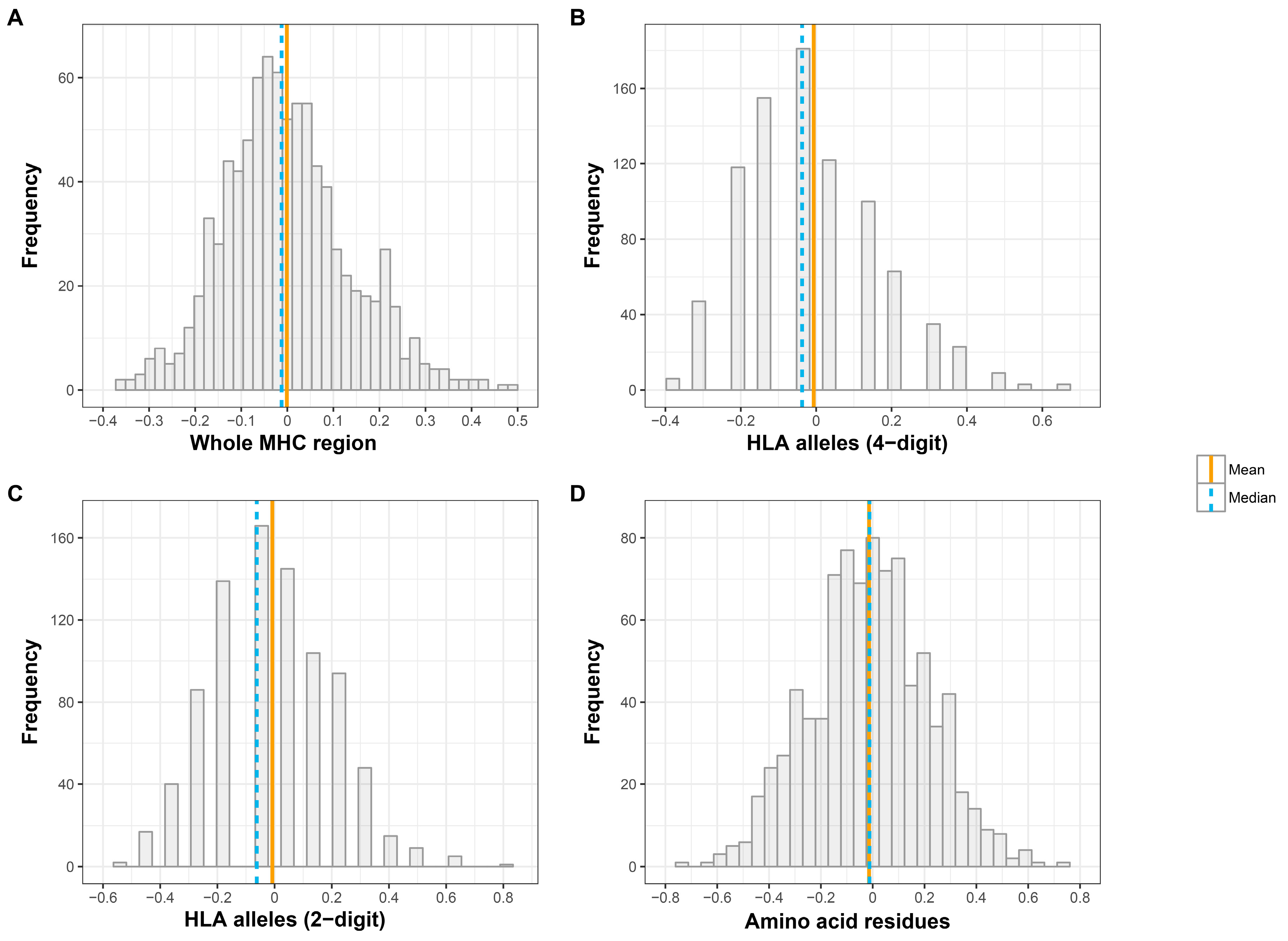
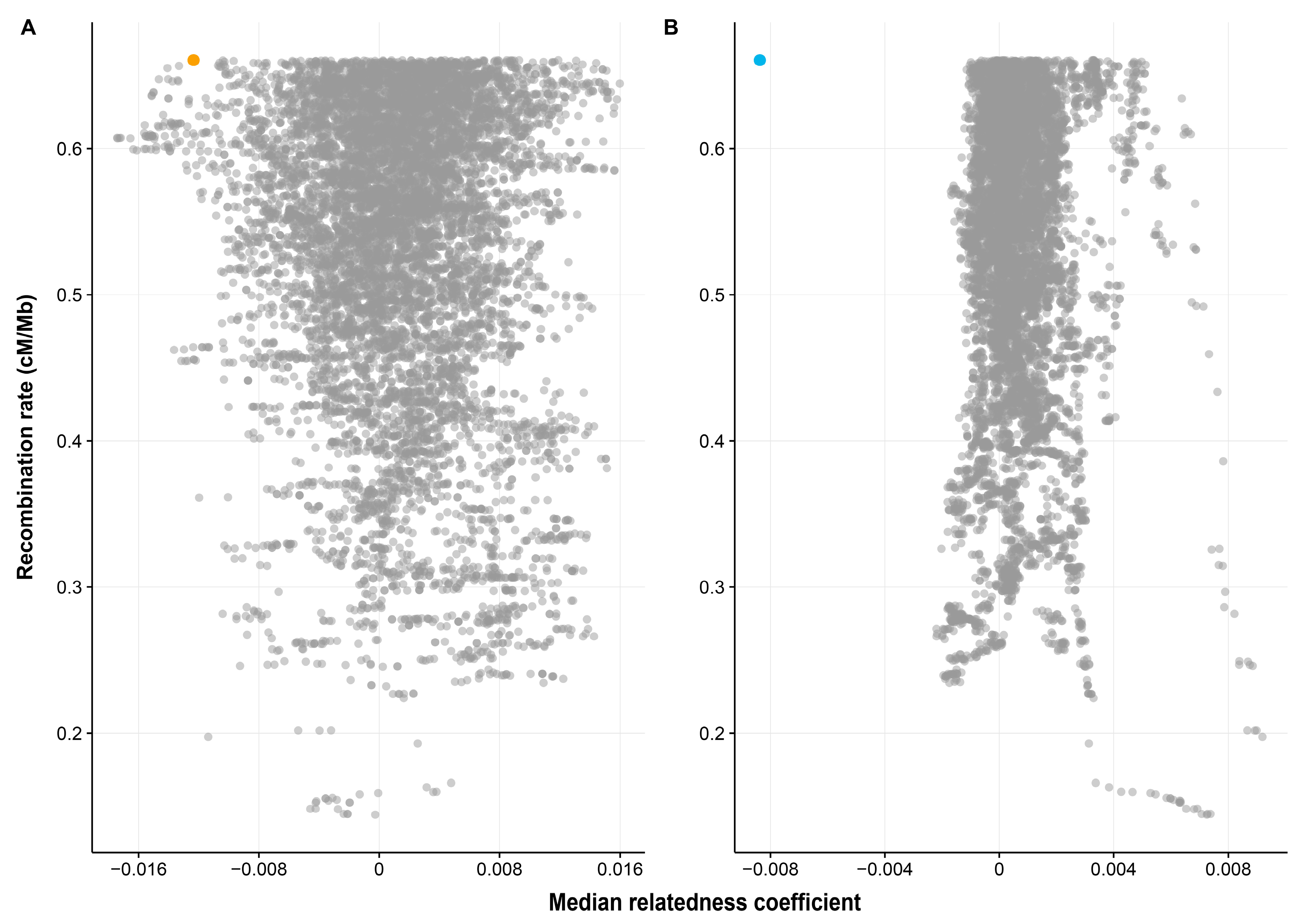
3.1. Whole MHC Region
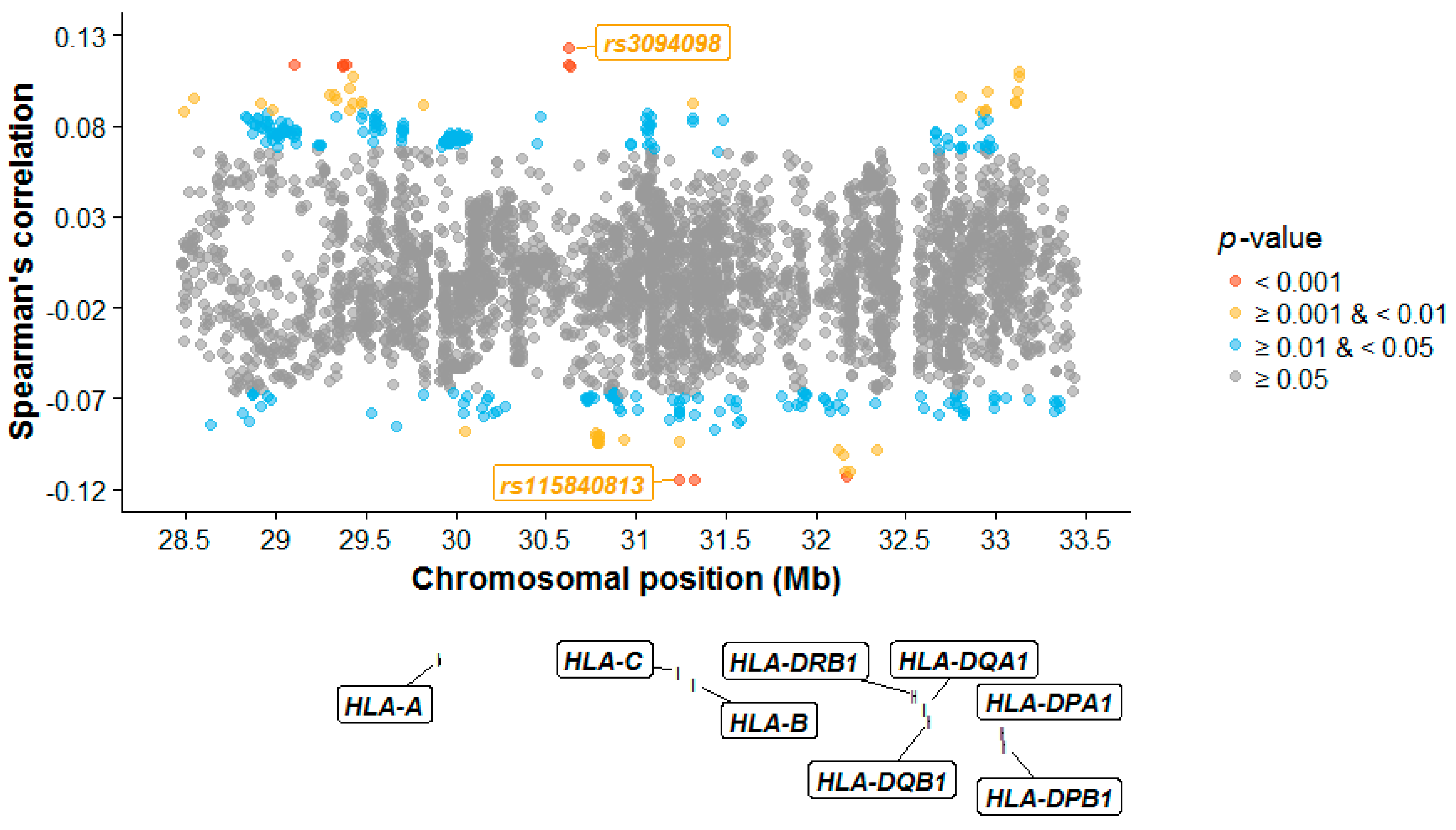
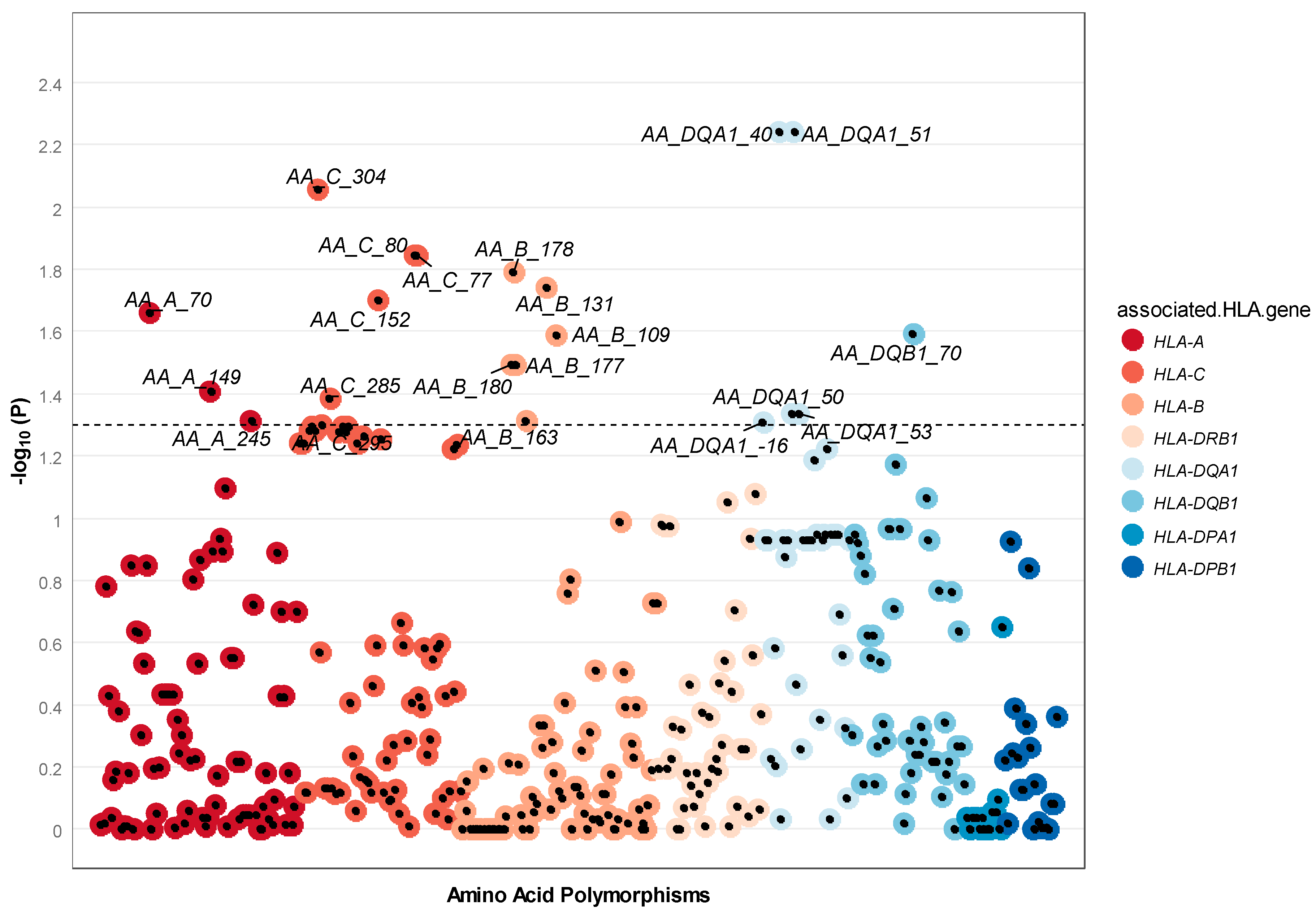
3.2. Individual SNPs, HLA Alleles, and Amino Acids
4. Discussion
4.1. Dissimilarity between Spouses across the MHC Region as a Whole
4.2. Dissimilarity between Spouses at Individual SNPs, HLA Alleles, and Amino Acids
4.3. General Discussion
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Robinson, M.R.; Kleinman, A.; Graff, M.; Vinkhuyzen, A.A.; Couper, D.; Miller, M.B.; Peyrot, W.J.; Abdellaoui, A.; Zietsch, B.P.; Nolte, I.M. Genetic evidence of assortative mating in humans. Nat. Hum. Behav. 2017, 1, 0016. [Google Scholar] [CrossRef]
- Domingue, B.W.; Fletcher, J.; Conley, D.; Boardman, J.D. Genetic and educational assortative mating among US adults. Proc. Natl. Acad. Sci. USA 2014, 111, 7996–8000. [Google Scholar] [CrossRef] [PubMed]
- Zou, J.Y.; Park, D.S.; Burchard, E.G.; Torgerson, D.G.; Pino-Yanes, M.; Song, Y.S.; Sankararaman, S.; Halperin, E.; Zaitlen, N. Genetic and socioeconomic study of mate choice in Latinos reveals novel assortment patterns. Proc. Natl. Acad. Sci. USA 2015, 112, 13621–13626. [Google Scholar] [CrossRef] [PubMed]
- Guo, G.; Wang, L.; Liu, H.; Randall, T. Genomic assortative mating in marriages in the United States. PLoS ONE 2014, 9, e112322. [Google Scholar] [CrossRef] [PubMed]
- Eaves, L.; Heath, A.; Martin, N.; Maes, H.; Neale, M.; Kendler, K.; Kirk, K.; Corey, L. Comparing the biological and cultural inheritance of personality and social attitudes in the Virginia 30,000 study of twins and their relatives. Twin Res. Hum. Genet. 1999, 2, 62–80. [Google Scholar] [CrossRef]
- Chaix, R.; Cao, C.; Donnelly, P. Is mate choice in humans MHC-dependent? PLoS Genet. 2008, 4, e1000184. [Google Scholar] [CrossRef] [PubMed]
- Havlicek, J.; Roberts, S.C. MHC-correlated mate choice in humans: A review. Psychoneuroendocrinology 2009, 34, 497–512. [Google Scholar] [CrossRef] [PubMed]
- Derti, A.; Cenik, C.; Kraft, P.; Roth, F.P. Absence of Evidence for MHC–Dependent Mate Selection within HapMap Populations. PLoS Genet. 2010, 6, e1000925. [Google Scholar] [CrossRef] [PubMed]
- Laurent, R.; Chaix, R. MHC-dependent mate choice in humans: Why genomic patterns from the HapMap European American dataset support the hypothesis. BioEssays 2012, 34, 267–271. [Google Scholar] [CrossRef] [PubMed]
- Derti, A.; Roth, F.P. Response to “MHC-dependent mate choice in humans: Why genomic patterns from the HapMap European American data set support the hypothesis”. Bioessays 2012, 34, 576–577. [Google Scholar] [CrossRef] [PubMed]
- De Bakker, P.I.; McVean, G.; Sabeti, P.C.; Miretti, M.M.; Green, T.; Marchini, J.; Ke, X.; Monsuur, A.J.; Whittaker, P.; Delgado, M. A high-resolution HLA and SNP haplotype map for disease association studies in the extended human MHC. Nat. Genet. 2006, 38, 1166–1172. [Google Scholar] [CrossRef] [PubMed]
- Roberts, S.C.; Little, A.C. Good genes, complementary genes and human mate preferences. Genetica 2008, 132, 309–321. [Google Scholar] [CrossRef] [PubMed]
- Kamiya, T.; O‘Dwyer, K.; Westerdahl, H.; Senior, A.; Nakagawa, S. A quantitative review of MHC-based mating preference: The role of diversity and dissimilarity. Mol. Ecol. 2014, 23, 5151–5163. [Google Scholar] [CrossRef] [PubMed]
- Landry, C.; Garant, D.; Duchesne, P.; Bernatchez, L. ‘Good genes as heterozygosity’: The major histocompatibility complex and mate choice in Atlantic salmon (Salmo salar). Proc. R. Soc. Lond. B: Biol. Sci. 2001, 268, 1279–1285. [Google Scholar] [CrossRef] [PubMed]
- Aeschlimann, P.; Häberli, M.; Reusch, T.; Boehm, T.; Milinski, M. Female sticklebacks Gasterosteus aculeatus use self-reference to optimize MHC allele number during mate selection. Behav. Ecol. Sociobiol. 2003, 54, 119–126. [Google Scholar]
- Olsson, M.; Madsen, T.; Nordby, J.; Wapstra, E.; Ujvari, B.; Wittsell, H. Major histocompatibility complex and mate choice in sand lizards. Proc. R. Soc. Lond. B: Biol. Sci. 2003, 270, S254–S256. [Google Scholar] [CrossRef] [PubMed]
- Richardson, D.S.; Komdeur, J.; Burke, T.; Von Schantz, T. MHC-based patterns of social and extra-pair mate choice in the Seychelles warbler. Proc. R. Soc. Lond. B: Biol. Sci. 2005, 272, 759–767. [Google Scholar] [CrossRef] [PubMed]
- Bonneaud, C.; Chastel, O.; Federici, P.; Westerdahl, H.; Sorci, G. Complex MHC-based mate choice in a wild passerine. Proc. R. Soc. Lond. B: Biol. Sci. 2006, 273, 1111–1116. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, K.; Boyse, E.; Mike, V.; Thaler, H.; Mathieson, B.; Abbott, J.; Boyse, J.; Zayas, Z.; Thomas, L. Control of mating preferences in mice by genes in the major histocompatibility complex. J. Exp. Med. 1976, 144, 1324–1335. [Google Scholar] [CrossRef] [PubMed]
- Potts, W.K.; Manning, C.J.; Wakeland, E.K. Mating patterns in seminatural populations of mice influenced by MHC genotype. Nature 1991, 352, 619–621. [Google Scholar] [CrossRef] [PubMed]
- Penn, D.J.; Potts, W.K. The evolution of mating preferences and major histocompatibility complex genes. Am. Nat. 1999, 153, 145–164. [Google Scholar] [CrossRef]
- Milinski, M. The major histocompatibility complex, sexual selection, and mate choice. Annu. Rev. Ecol. Evol. Syst. 2006, 37, 159–186. [Google Scholar] [CrossRef]
- Jacob, S.; McClintock, M.K.; Zelano, B.; Ober, C. Paternally inherited HLA alleles are associated with women‘s choice of male odor. Nat. Genet. 2002, 30, 175–179. [Google Scholar] [CrossRef] [PubMed]
- Pollack, M.S.; Wysocki, C.J.; Beauchamp, G.K.; Braun, D., Jr.; Callaway, C.; Dupont, B. Absence of HLA association or linkage for variations in sensitivity to the odor of androstenone. Immunogenetics 1982, 15, 579–589. [Google Scholar] [PubMed]
- Nordlander, C.; Hammarström, L.; Lindblom, B.; Smith, C.E. No role of HLA in mate selection. Immunogenetics 1983, 18, 429–431. [Google Scholar] [CrossRef] [PubMed]
- Sans, M.; Alvarez, I.; Callegari-Jacques, S.; Salzano, F. Genetic similarity and mate selection in Uruguay. J. Biosoc. Sci. 1994, 26, 285–289. [Google Scholar] [CrossRef] [PubMed]
- Jin, K.; Speed, T.; Thomson, G. Tests of random mating for a highly polymorphic locus: Application to HLA data. Biometrics 1995, 51, 1064–1076. [Google Scholar] [CrossRef] [PubMed]
- Hedrick, P.W.; Black, F.L. HLA and mate selection: No evidence in South Amerindians. Am. J. Hum. Genet. 1997, 61, 505–511. [Google Scholar] [CrossRef] [PubMed]
- Ihara, Y.; Aoki, K.; Tokunaga, K.; Takahashi, K.; Juji, T. HLA and Human Mate Choice. Tests on Japanese Couples. Anthropol. Sci. 2000, 108, 199–214. [Google Scholar] [CrossRef]
- Ober, C.; Weitkamp, L.R.; Cox, N.; Dytch, H.; Kostyu, D.; Elias, S. HLA and mate choice in humans. Am. J. Hum. Genet. 1997, 61, 497–504. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, L.T.; Cooperman, D.; Payn, R. HLA and mate selection. Immunogenetics 1983, 17, 89–93. [Google Scholar] [CrossRef] [PubMed]
- Khankhanian, P.; Gourraud, P.-A.; Caillier, S.J.; Santaniello, A.; Hauser, S.L.; Baranzini, S.E.; Oksenberg, J.R. Genetic variation in the odorant receptors family 13 and the MHC loci influence mate selection in a multiple sclerosis dataset. BMC Genom. 2010, 11, 626. [Google Scholar] [CrossRef] [PubMed]
- Winternitz, J.; Abbate, J.L.; Huchard, E.; Havlicek, J.; Garamszegi, L.Z. Patterns of MHC-dependent mate selection in humans and nonhuman primates: A meta-analysis. Mol. Ecol. 2017, 26, 668–688. [Google Scholar] [CrossRef] [PubMed]
- Sonnega, A.; Faul, J.D.; Ofstedal, M.B.; Langa, K.M.; Phillips, J.W.; Weir, D.R. Cohort profile: The health and retirement study (HRS). Int. J. Epidemiol. 2014, 43, 576–585. [Google Scholar] [CrossRef] [PubMed]
- Powell, J.E.; Visscher, P.M.; Goddard, M.E. Reconciling the analysis of IBD and IBS in complex trait studies. Nat. Rev. Genet. 2010, 11, 800. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Benyamin, B.; McEvoy, B.P.; Gordon, S.; Henders, A.K.; Nyholt, D.R.; Madden, P.A.; Heath, A.C.; Martin, N.G.; Montgomery, G.W. Common SNPs explain a large proportion of the heritability for human height. Nat. Genet. 2010, 42, 565–569. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Lee, S.H.; Goddard, M.E.; Visscher, P.M. GCTA: A tool for genome-wide complex trait analysis. Am. J. Hum. Genet. 2011, 88, 76–82. [Google Scholar] [CrossRef] [PubMed]
- Human Genome Region MHC. Available online: https://www.ncbi.nlm.nih.gov/grc/human/regions/MHC?asm=GRCh37 (accessed on 7 October 2017).
- Jia, X.; Han, B.; Onengut-Gumuscu, S.; Chen, W.-M.; Concannon, P.J.; Rich, S.S.; Raychaudhuri, S.; de Bakker, P.I. Imputing amino acid polymorphisms in human leukocyte antigens. PLoS ONE 2013, 8, e64683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frazer, K.A.; Ballinger, D.G.; Cox, D.R.; Hinds, D.A.; Stuve, L.L.; Gibbs, R.A.; Belmont, J.W.; Boudreau, A.; Hardenbol, P.; Leal, S.M. A second generation human haplotype map of over 3.1 million SNPs. Nature 2007, 449, 851–861. [Google Scholar] [CrossRef] [PubMed]
- Reference SNP (refSNP) Cluster Report: rs3094098. Available online: https://www.ncbi.nlm.nih.gov/projects/SNP/snp_ref.cgi?rs=3094098 (accessed on 7 October 2017).
- DHX16 DEAH-Box Helicase 16 [Homo Sapiens (Human)]. Available online: https://www.ncbi.nlm.nih.gov/gene/8449 (accessed on 7 October 2017).
- Laurent, R.; Chaix, R. HapMap European American genotypes are compatible with the hypothesis of MHC-dependent mate choice. Bioessays 2012, 34, 871–872. [Google Scholar] [CrossRef] [PubMed]
- Good, P. Permutation Tests: A Practical Guide to Resampling Methods for Testing Hypotheses; Springer Science & Business Media: Berlin, Germany, 2013. [Google Scholar]
- Laurent, R.; Toupance, B.; Chaix, R. Non-random mate choice in humans: Insights from a genome scan. Mol. Ecol. 2012, 21, 587–596. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Datasets | Parameters | Relatedness 1 | p-Value 2 |
|---|---|---|---|
| SNPs | Mean 3 | −0.001 ± 0.137 | 0.829 |
| Median 4 | −0.012 ± 0.124 | 0.218 | |
| Classical HLA genes (four-digit resolution) | Mean | −0.007 ± 0.184 | 0.258 |
| Median | −0.037 ± 0.128 | 0.500 | |
| Classical HLA genes (two-digit resolution) | Mean | −0.008 ± 0.209 | 0.211 |
| Median | −0.062 ± 0.143 | 0.419 | |
| Amino acids | Mean | −0.014 ± 0.226 | 0.040 |
| Median | −0.013 ± 0.214 | 0.235 |
| HLA Gene | Similarity Score (SC) 1 | Similarity Score (SC) 2 | Two-Sided p-Value | |
|---|---|---|---|---|
| Mean | SD | |||
| A | 411 | 421.90 | 14.37 | 0.448 |
| C | 260 | 285.20 | 13.98 | 0.071 |
| B | 191 | 207.78 | 12.31 | 0.173 |
| DRB1 | 259 | 265.61 | 13.49 | 0.624 |
| DQA1 | 488 | 497.30 | 16.47 | 0.572 |
| DQB1 | 324 | 331.61 | 14.85 | 0.608 |
| DPA1 | 1275 | 1267.73 | 11.95 | 0.543 |
| DPB1 | 585 | 586.24 | 13.72 | 0.928 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qiao, Z.; Powell, J.E.; Evans, D.M. MHC-Dependent Mate Selection within 872 Spousal Pairs of European Ancestry from the Health and Retirement Study. Genes 2018, 9, 53. https://doi.org/10.3390/genes9010053
Qiao Z, Powell JE, Evans DM. MHC-Dependent Mate Selection within 872 Spousal Pairs of European Ancestry from the Health and Retirement Study. Genes. 2018; 9(1):53. https://doi.org/10.3390/genes9010053
Chicago/Turabian StyleQiao, Zhen, Joseph E. Powell, and David M. Evans. 2018. "MHC-Dependent Mate Selection within 872 Spousal Pairs of European Ancestry from the Health and Retirement Study" Genes 9, no. 1: 53. https://doi.org/10.3390/genes9010053
APA StyleQiao, Z., Powell, J. E., & Evans, D. M. (2018). MHC-Dependent Mate Selection within 872 Spousal Pairs of European Ancestry from the Health and Retirement Study. Genes, 9(1), 53. https://doi.org/10.3390/genes9010053