Speciation Theory of Carcinogenesis Explains Karyotypic Individuality and Long Latencies of Cancers

Abstract
:1. Introduction
Speciation Theory
2. Materials and Methods
2.1. Cell Cultures
2.2 Karyotyping of Rat and Human Cells by Hybridization of Metaphase-Chromosomes with Color-Coded Chromosome-Specific DNA Probes
3. Results
3.1. Initiation of Carcinogenesis by Induction of Preneoplastic Aneuploidy
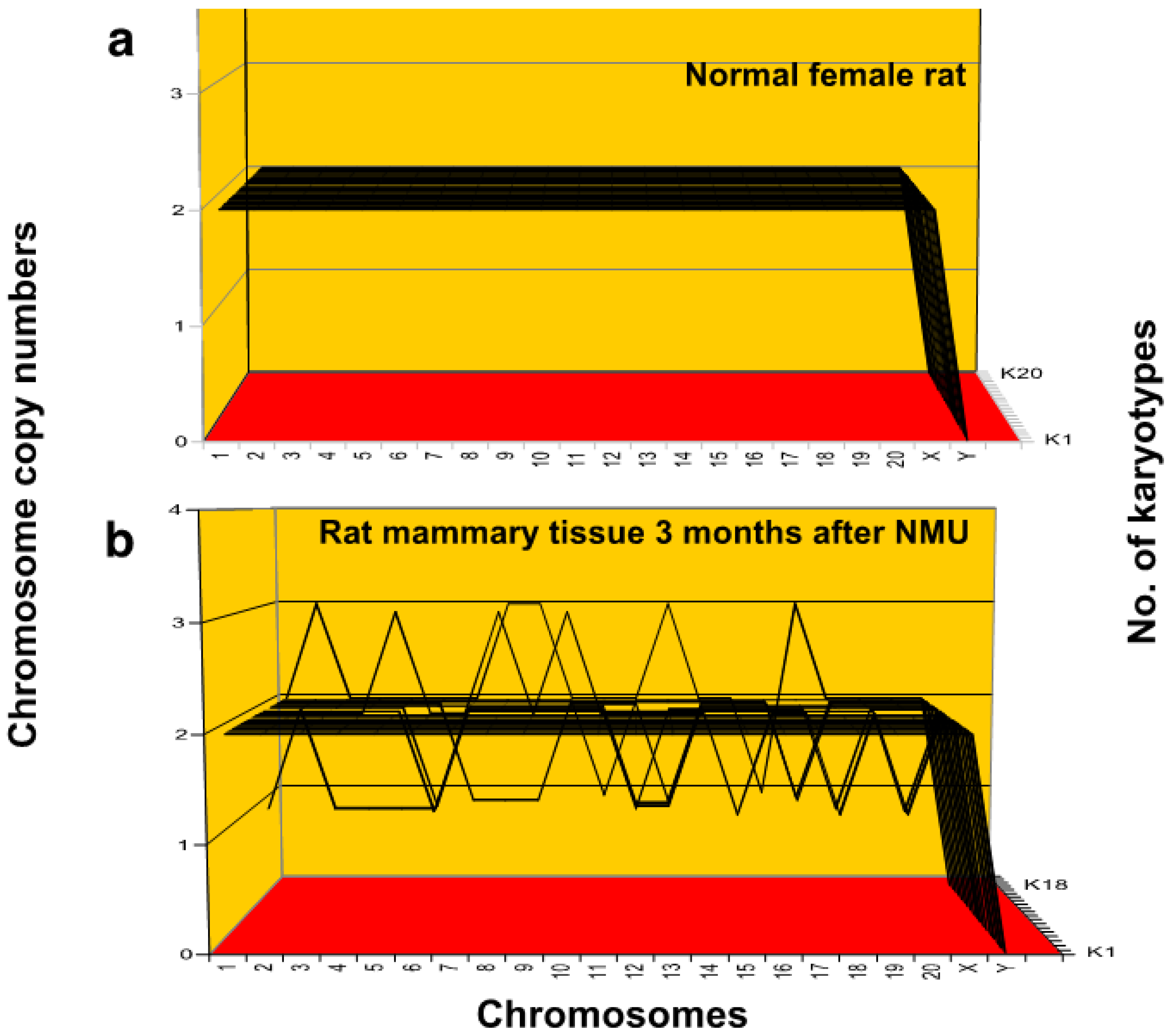
3.1.1. Preneoplastic Aneuploidy in Rats Injected with Nitrosourea
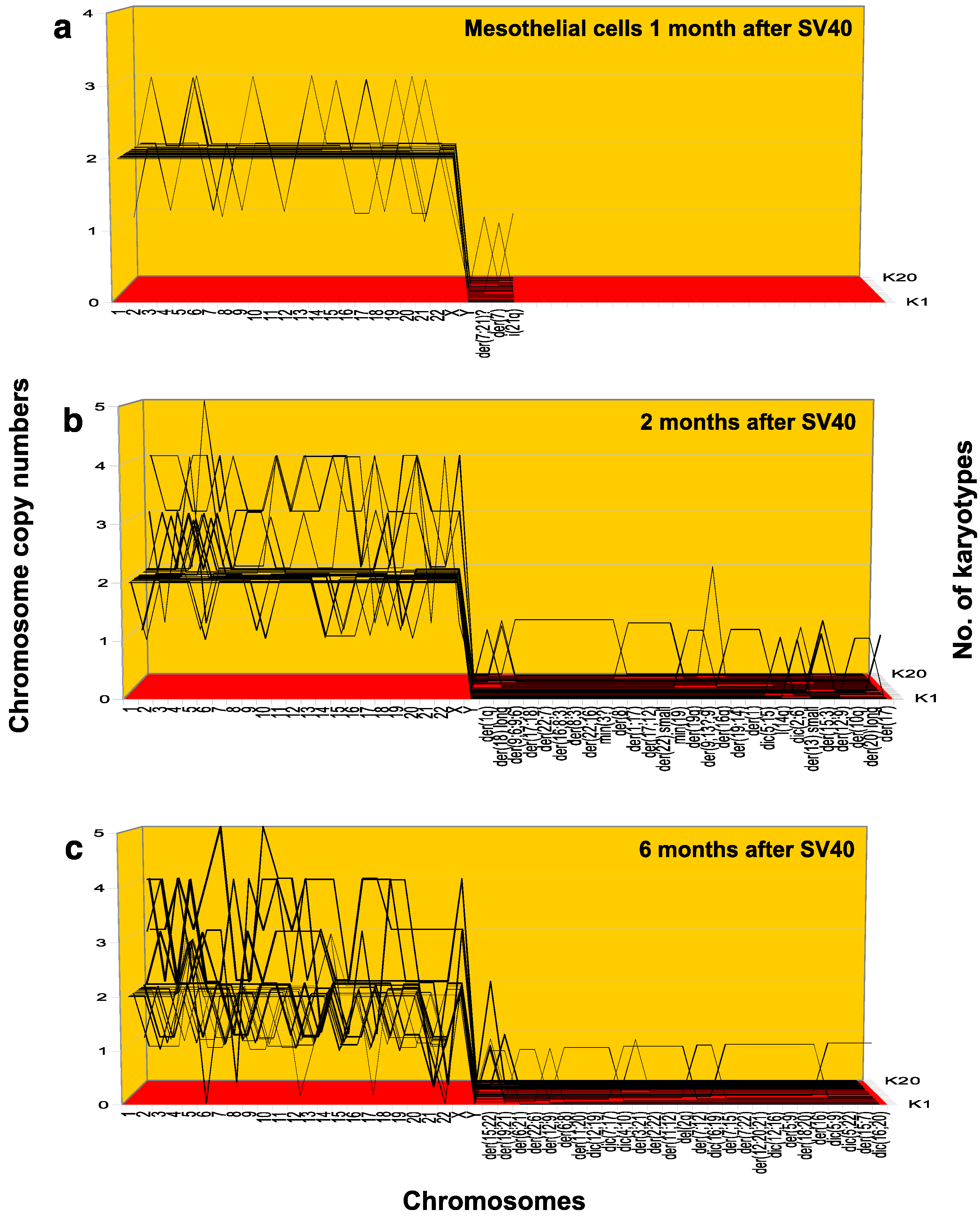
3.1.2. Preneoplastic Aneuploidy in Human Mesothelial Cells Initiated with SV40 Tumor Virus
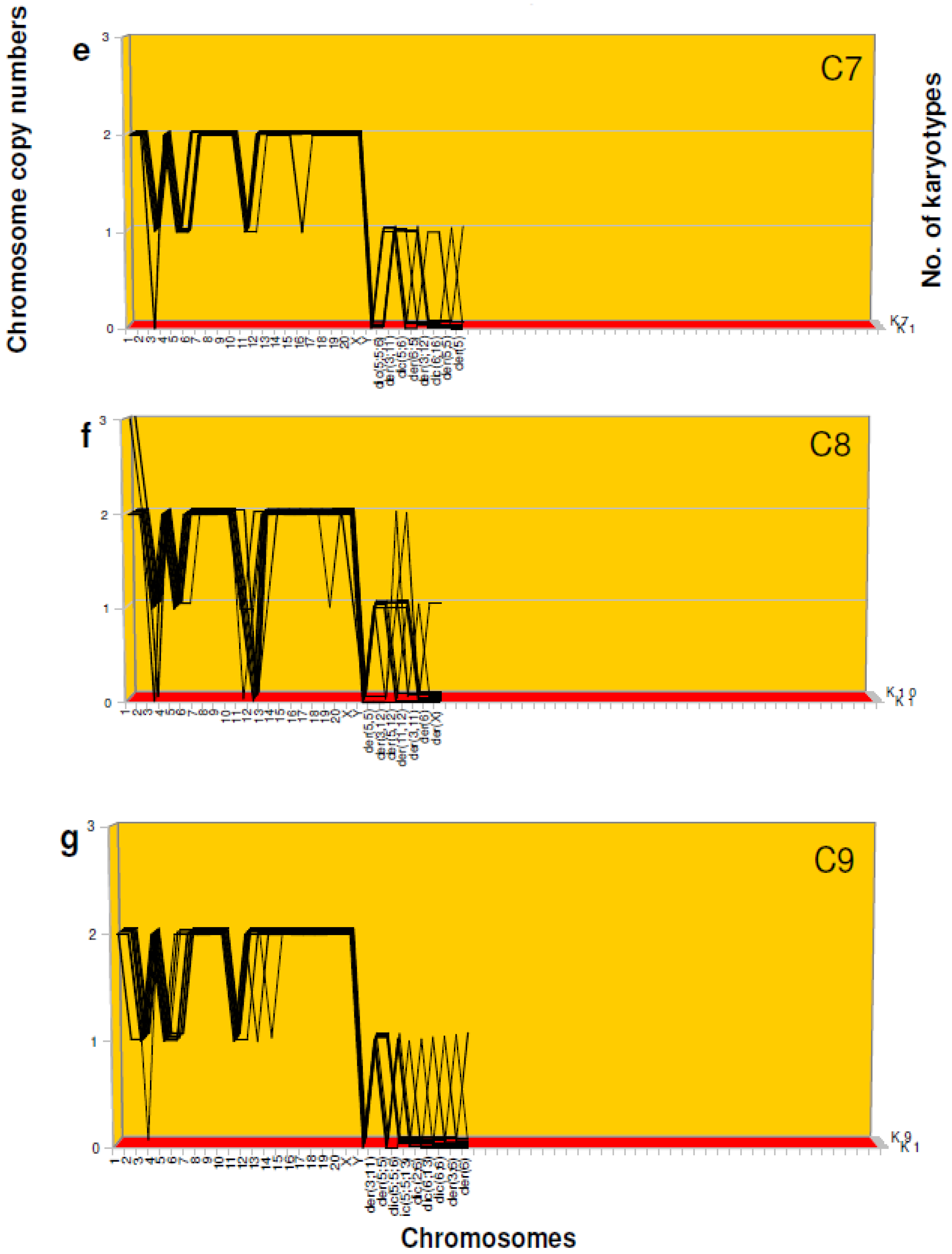
3.2. Preneoplastic Karyotypes with Multiple New Marker Chromosomes—Evidence for Saltational Origins
3.3. Individual Karyotypes of Cancers Are Heritable and Thus Segregate with All Sub-Clones
3.4. Saltational Origins of Neoplastic Karyotypes
- (1)
- (2)
- (3)
- (4)
4. Conclusions
4.1. The Long Latent Periods and Individual Karyotypes of Cancers in the Light of the Speciation Theory
- (1)
- Long latent periods from initiating carcinogens. We adduced verifiable evidence that the low probability of creating karyotypes of new autonomous cancer species by random aneuploidizations of the karyotypes of precursors, is the reason for the long latencies of carcinogenesis.
- (2)
- Heritable individual karyotypes of cancers. Challenged by the prevailing theory that the individual karyotypes of cancers are non-clonal products of CIN mutations and thus not heritable genomes, we found instead that the karyotypes of cancers are quasi-clonal and heritable—similar to those of conventional sexual species. (Nevertheless, the karyotypes of sexual species are much less variable than those of cancers [97,110].) The experimental evidence of this finding was that the quasi-clonal karyotype of the neoplastic rat clone F10 segregated with six out of six single cell-derived sub-clones with only minor sub-clonal variations.
- (3)
- Evidence for single-step origins of cancer karyotypes. Unexpectedly, in view of the multi-step theory of carcinogenesis, we found sub-clones of F10 karyotypes with individual sets of 3–5 new marker chromosomes, but no detectable precursors carrying subsets of these new markers. We thus concluded that these unique sets of marker chromosomes were assembled in single-steps, rather than gradually by a stepwise mechanism. The suspected single-step origin of cancer karyotypes is further supported by our findings of karyotypes with 3–11 new marker chromosomes without precursors in aneuploid preneoplastic mesothelial cells (Table 2b,c).
4.2. Clinical Relevance of Heritable Individual Karyotypes and Preneoplastic Aneuploidies of Cancers
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hansemann, D. Ueber asymmetrische Zelltheilung in Epithelkrebsen und deren biologische Bedeutung. Virchows Arch. Pathol. Anat. 1890, 119, 299–326. (In German) [Google Scholar] [CrossRef]
- Winge, O. Zytologische Untersuchungen ueber die Natur maligner Tumoren. II. Teerkarzinome bei Maeusen. Z. Zellforsch. Mikrosk. Anat. 1930, 10, 683–735. (In German) [Google Scholar] [CrossRef]
- Gebhart, E.; Liehr, T. Patterns of genomic imbalances in human solid tumors. Int. J. Oncol. 2000, 16, 383–399. [Google Scholar] [CrossRef] [PubMed]
- Baudis, M. Genomic imbalances in 5918 malignant epithelial tumors: An explorative meta-analysis of chromosomal CGH data. BMC Cancer 2007, 7, 226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duesberg, P.; Mandrioli, D.; McCormack, A.; Nicholson, J.M. Is carcinogenesis a form of speciation? Cell Cycle 2011, 10, 2100–2114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitelman, F.; Johansson, B.; Mertens, F. NCI-Mitelman Database of Chromosome Aberrations and Gene Fusions in Cancer. 2017. Available online: http://cgap.nci.nih.gov/Chromosomes/Mitelman (accessed on 8 August 2018).
- Yamagiwa, K. Experimentelle Studie ueber die Pathogenese der Epithelialgeschwuelste. Mitteillungen Med. Fakultaet Kaiserl Univ. Tokyo 1915, 15, 295–344. (In German) [Google Scholar]
- Brown, J.R.; Thornton, J.L. Percivall Pott (1714–1788) and chimney sweepers’ cancer of the scrotum. Br. J. Ind. Med. 1957, 14, 68–70. [Google Scholar] [CrossRef] [PubMed]
- Rous, P. Surmise and fact on the nature of cancer. Nature 1959, 183, 1357–1361. [Google Scholar] [CrossRef] [PubMed]
- Shein, H.M.; Enders, J.F. Transformation induced by siminian virus 40 in human renal cell cultures, I. Morpholgy and growth characteristics. Proc. Natl. Acad. Sci. USA 1962, 48, 1164–1172. [Google Scholar] [CrossRef] [PubMed]
- Cairns, J. Cancer: Science and Society; W. H. Freeman: San Francisco, CA, USA, 1978. [Google Scholar]
- Preston, D.L.; Ron, E.; Tokuoka, S.; Funamoto, S.; Nishi, N.; Soda, M.; Mabuchi, K.; Kodama, K. Solid cancer incidence in atomic bomb survivors: 1958–1998. Radiat. Res. 2007, 168, 1–64. [Google Scholar] [CrossRef] [PubMed]
- Brash, D.; Cairns, J. The mysterious steps in carcinogenesis. Br. J. Cancer 2009, 101, 379–380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bloomfield, M.; Duesberg, P. Karyotype alteration generates the neoplastic phenotypes of SV40-infected human and rodent cells. Mol. Cytogenet. 2015, 8, 79. [Google Scholar] [CrossRef] [PubMed]
- Nordling, C.O. A new theory on cancer-inducing mechanism. Br. J. Cancer 1953, 7, 68–72. [Google Scholar] [CrossRef] [PubMed]
- Armitage, P.; Doll, R. The age distribution of cancer and a multi-stage theory of carcinogenesis. Br. J. Cancer 1954, 8, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Nowell, P.C. The clonal evolution of tumor cell populations. Science 1976, 194, 23–28. [Google Scholar] [CrossRef] [PubMed]
- Vogelstein, B.; Kinzler, K.W. The multistep nature of cancer. Trends Genet. 1993, 9, 138–141. [Google Scholar] [CrossRef]
- Bishop, J.M. Cancer: The rise of the genetic paradigm. Genes Dev. 1995, 9, 1309–1315. [Google Scholar] [CrossRef] [PubMed]
- Hahn, W.C.; Counter, C.M.; Lundberg, A.S.; Beijersbergen, R.L.; Brooks, M.W.; Weinberg, R.A. Creation of human tumour cells with defined genetic elements. Nature 1999, 400, 464–468. [Google Scholar] [CrossRef] [PubMed]
- Alberts, B.; Johnson, A.; Lewis, J.; Raff, M.; Roberts, K.; Walter, P. Molecular Biology of the Cell; Garland Publishing, Inc.: New York, NY, USA, 2014. [Google Scholar]
- Weinberg, R.A. The Biology of Cancer, 2nd ed.; Garland Science: New York, NY, USA; London, UK, 2014. [Google Scholar]
- Lengauer, C.; Kinzler, K.W.; Vogelstein, B. Genetic instability in colorectal cancers. Nature 1997, 386, 623–627. [Google Scholar] [CrossRef] [PubMed]
- Cahill, D.P.; Lengauer, C.; Yu, J.; Riggins, G.J.; Willson, J.K.V.; Markowitz, S.D.; Kinzler, K.W.; Vogelstein, B. Mutations of mitotic checkpoint genes in human cancers. Nature 1998, 392, 300–303. [Google Scholar] [CrossRef] [PubMed]
- Lengauer, C.; Kinzler, K.W.; Vogelstein, B. Genetic instabilities in human cancers. Nature 1998, 396, 643–649. [Google Scholar] [CrossRef] [PubMed]
- Cahill, D.P.; Kinzler, K.W.; Vogelstein, B.; Lengauer, C. Genetic instability and darwinian selection in tumours. Trends Cell Biol. 1999, 9, M57–M60. [Google Scholar] [CrossRef]
- Negrini, S.; Gorgoulis, V.G.; Halazonetis, T.D. Genomic instability—An evolving hallmark of cancer. Nat. Rev. Mol. Cell Biol. 2010, 11, 220–228. [Google Scholar] [CrossRef] [PubMed]
- Thompson, S.L.; Compton, D.A. Chromosomes and cancer cells. Chromosome Res. 2011, 19, 433–444. [Google Scholar] [CrossRef] [PubMed]
- Swanton, C. Intratumor heterogeneity: Evolution through space and time. Cancer Res. 2012, 72, 4875–4882. [Google Scholar] [CrossRef] [PubMed]
- McGranahan, N.; Burrell, R.A.; Endesfelder, D.; Novelli, M.R.; Swanton, C. Cancer chromosomal instability: Therapeutic and diagnostic challenges. EMBO Rep. 2012, 13, 528–538. [Google Scholar] [CrossRef] [PubMed]
- Burrell, R.A.; McClelland, S.E.; Endesfelder, D.; Groth, P.; Weller, M.C.; Shaikh, N.; Domingo, E.; Kanu, N.; Dewhurst, S.M.; Gronroos, E.; et al. Replication stress links structural and numerical cancer chromosomal instability. Nature 2013, 494, 492–496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orr, B.; Compton, D.A. A double-edged sword: How oncogenes and tumor suppressor genes can contribute to chromosomal instability. Front. Oncol. 2013, 3, 164. [Google Scholar] [CrossRef] [PubMed]
- Sansregret, L.; Vanhaesebroeck, B.; Swanton, C. Determinants and clinical implications of chromosomal instability in cancer. Nat. Rev. Clin. Oncol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Sonik, A.; Stindl, R.; Rasnick, D.; Duesberg, P. Aneuploidy vs. gene mutation hypothesis of cancer: Recent study claims mutation but is found to support aneuploidy. Proc. Natl. Acad. Sci. USA 2000, 97, 3236–3241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zimonjic, D.; Brooks, M.W.; Popescu, N.; Weinberg, R.A.; Hahn, W.C. Derivation of human tumor cells in vitro without widespread genomic instability. Cancer Res. 2001, 61, 8838–8844. [Google Scholar] [PubMed]
- Webb, T. When theories collide: Experts develop different models for carcinogenesis. J. Natl. Cancer Inst. 2001, 93, 92–94. [Google Scholar] [CrossRef] [PubMed]
- Marx, J. Debate surges over the origins of genomic defects in cancer. Science 2002, 297, 544–546. [Google Scholar] [CrossRef] [PubMed]
- Elenbaas, B.; Spirio, L.; Koerner, F.; Fleming, M.D.; Zimonjic, D.B.; Donaher, J.L.; Popescu, N.C.; Hahn, W.C.; Weinberg, R.A. Human breast cancer cells generated by oncogenic transformation of primary mammary epithelial cells. Genes Dev. 2001, 15, 50–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schneider, B.L.; Kulesz-Martin, M. Destructive cycles: The role of genomic instability and adaptation in carcinogenesis. Carcinogenesis 2004, 25, 2033–2044. [Google Scholar] [CrossRef] [PubMed]
- Klein, A.; Li, N.; Nicholson, J.M.; McCormack, A.A.; Graessmann, A.; Duesberg, P. Transgenic oncogenes induce oncogene-independent cancers with individual karyotypes and phenotypes. Cancer Genet. Cytogenet. 2010, 200, 79–99. [Google Scholar] [CrossRef] [PubMed]
- Vincent, M.D. Cancer: Beyond speciation. Adv. Cancer Res. 2011, 112, 283–350. [Google Scholar] [PubMed]
- Garraway, L.A.; Lander, E.S. Lessons from the cancer genome. Cell 2013, 153, 17–37. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, M.S.; Stojanov, P.; Polak, P.; Kryukov, G.V.; Cibulskis, K.; Sivachenko, A.; Carter, S.L.; Stewart, C.; Mermel, C.H.; Roberts, S.A.; et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 2013, 499, 214–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A., Jr.; Kinzler, K.W. Cancer genome landscapes. Science 2013, 339, 1546–1558. [Google Scholar] [CrossRef] [PubMed]
- Duesberg, P.; Rasnick, D. Aneuploidy, the somatic mutation that makes cancer a species of its own. Cell Motil. Cytoskeleton 2000, 47, 81–107. [Google Scholar] [CrossRef]
- Vincent, M.D. The animal within: Carcinogenesis and the clonal evolution of cancer cells are speciation events sensu stricto. Evolution 2010, 64, 1173–1183. [Google Scholar] [CrossRef] [PubMed]
- Heng, H.H.; Stevens, J.B.; Bremer, S.W.; Liu, G.; Abdallah, B.Y.; Ye, C.J. Evolutionary mechanisms and diversity in cancer. Adv. Cancer Res. 2011, 112, 217–253. [Google Scholar] [PubMed]
- Stepanenko, A.A.; Kavsan, V.M. Evolutionary karyotypic theory of cancer versus conventional cancer gene mutation theory. Biopolym. Cell 2012, 28, 267–280. [Google Scholar] [CrossRef] [Green Version]
- Huxley, J. Cancer biology: Comparative and genetic. Biol. Rev. 1956, 31, 474–514. [Google Scholar] [CrossRef]
- Valen, L.M.V.; Maiorana, V.C. HeLa, a new microbial species. Evolut. Theory 1991, 10, 71–74. [Google Scholar]
- Murgia, C.; Pritchard, J.K.; Kim, S.Y.; Fassati, A.; Weiss, R.A. Clonal origin and evolution of a transmissible cancer. Cell 2006, 126, 477–487. [Google Scholar] [CrossRef] [PubMed]
- Pearse, A.M.; Swift, K.; Hodson, P.; Hua, B.; McCallum, H.; Pyecroft, S.; Taylor, R.; Eldridge, M.D.; Belov, K. Evolution in a transmissible cancer: A study of the chromosomal changes in devil facial tumor (DFT) as it spreads through the wild Tasmanian devil population. Cancer Genet. Cytogenet. 2012, 205, 101–112. [Google Scholar] [CrossRef] [PubMed]
- Goldschmidt, R.; Fischer, A. Chromosomenstudien an Carcinomzellen in vitro. Z. Krebsforsch. 1929, 30, 281–285. [Google Scholar] [CrossRef]
- Makino, S. Further evidence favoring the concept of the stem cell in ascites tumors of rats. Ann. N. Y. Acad. Sci. 1956, 63, 818–830. [Google Scholar] [CrossRef] [PubMed]
- Duesberg, P.; McCormack, A. Immortality of cancers: A consequence of inherent karyotypic variations and selections for autonomy. Cell Cycle 2013, 12, 783–802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pitot, H.C. Fundamentals of Oncology, 4th ed.; Marcel Dekker, Inc.: New York, NY, USA, 2002. [Google Scholar]
- Goldschmidt, R.B. The Material Basis of Evolution; Yale University Press.: New Haven, CT, USA, 1940; pp. 205–206. [Google Scholar]
- Hauschka, T.S. The chromosomes in ontogeny and oncogeny. Cancer Res. 1961, 21, 957–974. [Google Scholar] [PubMed]
- O’Brien, S.; Menotti-Raymond, M.; Murphy, W.; Nash, W.; Wirnberg, J.; Stanyon, R.; Copeland, N.; Jenkins, N.; Womack, J.; Marshall Graves, J. The promise of comparative genomics in mammals. Science 1999, 286, 458–481. [Google Scholar] [CrossRef] [PubMed]
- Theissen, G. Saltational evolution: Hopeful monsters are here to stay. Theory Biosci. 2009, 128, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Forsdyke, D.R. Speciation: Goldschmidt’s Heresy, once supported by Gould and Dawkins, is again reinstated. Biol. Theory 2017, 12, 4–12. [Google Scholar] [CrossRef]
- Mellors, R.C.; Keane, J.F., Jr.; Papanicolaou, G.N. Nucleic acid content of the squamous cancer cell. Science 1952, 116, 265–269. [Google Scholar] [CrossRef] [PubMed]
- Levan, A.; Biesele, J.J. Role of chromosomes in cancerogenesis, as studied in serial tissue culture of mammalian cells. Ann. N. Y. Acad. Sci. 1958, 71, 1022–1053. [Google Scholar] [CrossRef] [PubMed]
- Yerganian, G.; Shein, H.M.; Enders, J.F. Chromosomal disturbances observed in human fetal renal cells transformed in vitro by simian virus 40 and carried in culture. Cytogenetics 1962, 1, 314–324. [Google Scholar] [CrossRef] [PubMed]
- Wolman, S.; Hirschhorn, K.; Todaro, G. Early chromosomal changes in SV40-infected human fibroblast cultures. Cytogenet. Genome Res. 1964, 3, 45–61. [Google Scholar] [CrossRef]
- Oshimura, M.; Hesterberg, T.W.; Barrett, J.C. An early, nonrandom karyotypic change in immortal Syrian hamster cell lines transformed by asbestos: Trisomy of chromosome 11. Cancer Genet. Cytogenet. 1986, 22, 225–237. [Google Scholar] [CrossRef]
- Oshimura, M.; Barrett, J. Chemically induced aneuploidy in mammalian cells: mechanisms and biological significance in cancer. Environ. Mutagen. 1986, 8, 129–159. [Google Scholar] [CrossRef] [PubMed]
- Awa, A.A. Persistent chromosome aberrations in the somatic cells of A-bomb survivors, Hiroshima and Nagasaki. J. Radiat. Res. 1991, 32, 265–274. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Yerganian, G.; Duesberg, P.; Kraemer, A.; Willer, A.; Rausch, C.; Hehlmann, R. Aneuploidy correlated 100% with chemical transformation of Chinese hamster cells. Proc. Natl. Acad. Sci. USA 1997, 94, 14506–14511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duesberg, P.; Li, R. Multistep carcinogenesis: A chain reaction of aneuploidizations. Cell Cycle 2003, 2, 202–210. [Google Scholar] [CrossRef] [PubMed]
- Bloomfield, M.; Duesberg, P. Is cancer progression caused by gradual or simultaneous acquisitions of new chromosomes? Mol. Cytogenet. 2018, 11, 4. [Google Scholar] [CrossRef] [PubMed]
- Fabarius, A.; Li, R.; Yerganian, G.; Hehlmann, R.; Duesberg, P. Specific clones of spontaneously evolving karyotypes generate individuality of cancers. Cancer Genet. Cytogenet. 2008, 180, 89–99. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, J.M.; Duesberg, P. On the karyotypic origin and evolution of cancer cells. Cancer Genet. Cytogenet. 2009, 194, 96–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bloomfield, M.; McCormack, A.; Mandrioli, D.; Fiala, C.; Aldaz, C.M.; Duesberg, P. Karyotypic evolutions of cancer species in rats during the long latent periods after injection of nitrosourea. Mol. Cytogenet. 2014, 7, 71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bloomfield, M.; Duesberg, P. Inherent variability of cancer-specific aneuploidy generates metastases. Mol. Cytogenet. 2016, 9, 90. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Hehlmann, R.; Sachs, R.; Duesberg, P. Chromosomal alterations cause the high rates and wide ranges of drug resistance in cancer cells. Cancer Genet. Cytogenet. 2005, 163, 44–56. [Google Scholar] [CrossRef] [PubMed]
- Duesberg, P.; Iacobuzio-Donahue, C.; Brosnan, J.A.; McCormack, A.; Mandrioli, D.; Chen, L. Origin of metastases: Subspecies of cancers generated by intrinsic karyotypic variations. Cell Cycle 2012, 11, 1151–1166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shapiro, J.; Yung, W.-K.; Shapiro, W.R. Isolation, karyotype, and clonal growth of heterogeneous subpopulations of human malignant gliomas. Cancer Res. 1981, 41, 2349–2359. [Google Scholar] [PubMed]
- Heppner, G.H. Tumor heterogeneity. Cancer Res. 1984, 44, 2259–2265. [Google Scholar] [PubMed]
- Wolman, S.R. Cytogenetic heterogeneity: Its role in tumor evolution. Cancer Genet. Cytogenet. 1986, 19, 129–140. [Google Scholar] [CrossRef]
- Li, L.; McCormack, A.A.; Nicholson, J.M.; Fabarius, A.; Hehlmann, R.; Sachs, R.K.; Duesberg, P.H. Cancer-causing karyotypes: Chromosomal equilibria between destabilizing aneuploidy and stabilizing selection for oncogenic function. Cancer Genet. Cytogenet. 2009, 188, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Marusyk, A.; Polyak, K. Tumor heterogeneity: Causes and consequences. Biochim. Biophys. Acta 2010, 1805, 105–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marusyk, A.; Polyak, K. Cancer. Cancer cell phenotypes, in fifty shades of grey. Science 2013, 339, 528–529. [Google Scholar] [CrossRef] [PubMed]
- Burrell, R.A.; McGranahan, N.; Bartek, J.; Swanton, C. The causes and consequences of genetic heterogeneity in cancer evolution. Nature 2013, 501, 338–345. [Google Scholar] [CrossRef] [PubMed]
- Wikipedia. Tumor Heterogeneity. Available online: https://en.wikipedia.org/wiki/Tumour_heterogeneity (accessed on 8 August 2018).
- Gusev, Y.; Kagansky, V.; Dooley, W.C. Long-term dynamics of chromosomal instability in cancer: A transition probability model. Math. Comput. Model. 2001, 33, 1253–1273. [Google Scholar] [CrossRef]
- Makino, S. Cytogenetics of canine venereal tumors: Worldwide distribution and a common karyotype. In Chromosomes and Cancer; German, J., Ed.; John Wiley & Sons: New York, NY, USA, 1974; pp. 336–372. [Google Scholar]
- Murchison, E.P.; Wedge, D.C.; Alexandrov, L.B.; Fu, B.; Martincorena, I.; Ning, Z.; Tubio, J.M.; Werner, E.I.; Allen, J.; De Nardi, A.B.; et al. Transmissible dog cancer genome reveals the origin and history of an ancient cell lineage. Science 2014, 343, 437–440. [Google Scholar] [CrossRef] [PubMed]
- Wolman, S.R. Karyotypic progression in human tumors. Cancer Metastasis Rev. 1983, 2, 257–293. [Google Scholar] [CrossRef] [PubMed]
- Albertson, D.G.; Collins, C.; McCormick, F.; Gray, J.W. Chromosome aberrations in solid tumors. Nat. Genet. 2003, 34, 369–376. [Google Scholar] [CrossRef] [PubMed]
- Yoon, D.S.; Wersto, R.P.; Zhou, W.; Chrest, F.J.; Garrett, E.S.; Kwon, T.K.; Gabrielson, E. Variable levels of chromosomal instability and mitotic spindle checkpoint defects in breast cancer. Am. J. Pathol. 2002, 161, 391–397. [Google Scholar] [CrossRef]
- Wangsa, D.; Braun, R.; Schiefer, M.; Gertz, E.M.; Bronder, D.; Quintanilla, I.; Padilla-Nash, H.M.; Torres, I.; Hunn, C.; Warner, L.; et al. The evolution of single cell-derived colorectal cancer cell lines is dominated by the continued selection of tumor specific genomic imbalances, despite random chromosomal instability. Carcinogenesis 2018, 1, 13. [Google Scholar] [CrossRef] [PubMed]
- Foulds, L. Tumor progression: A review. Cancer Res. 1954, 14, 327–339. [Google Scholar] [PubMed]
- Duesberg, P.; Li, R.; Sachs, R.; Fabarius, A.; Upender, M.B.; Hehlmann, R. Cancer drug resistance: The central role of the karyotype. Drug Resist. Updat 2007, 10, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Heim, S.; Mitelman, F. Cancer Cytogenetics, 2nd ed.; Wiley-Liss: New York, NY, USA, 1995. [Google Scholar]
- Loeb, L.A.; Loeb, K.R.; Anderson, J.P. Multiple mutations and cancer. Proc. Natl. Acad. Sci. USA 2003, 100, 776–781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knouse, K.A.; Davioli, T.; Elledge, S.J.; Amon, A. Aneuploidy in Cancer: Seq-ing Answers to Old Questions. Annu. Rev. Cancer Biol. 2017, 1, 335–354. [Google Scholar] [CrossRef]
- Bocchetta, M.; Di Resta, I.; Powers, A.; Fresco, R.; Tosolini, A.; Testa, J.R.; Pass, H.I.; Rizzo, P.; Carbone, M. Human mesothelial cells are unusually susceptible to simian virus 40-mediated transformation and asbestos cocarcinogenicity. Proc. Natl. Acad. Sci. USA 2000, 97, 10214–10219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, M.J.D. Modes of Speciation; W H Freeman and Co.: San Francisco, CA, USA, 1978. [Google Scholar]
- Aldaz, C.M.; Chen, A.; Gollahon, L.S.; Russo, J.; Zappler, K. Nonrandom abnormalities involving chromosome 1 and Harvey-ras-1 alleles in rat mammary tumor progression. Cancer Res. 1992, 52, 4791–4798. [Google Scholar] [PubMed]
- Goepfert, T.M.; Moreno-Smith, M.; Edwards, D.G.; Pathak, S.; Medina, D.; Brinkley, W.R. Loss of chromosomal integrity drives rat mammary tumorigenesis. Int. J. Cancer 2007, 120, 985–994. [Google Scholar] [CrossRef] [PubMed]
- Stepanenko, A.; Andreieva, S.; Korets, K.; Mykytenko, D.; Huleyuk, N.; Vassetzky, Y.; Kavsan, V. Step-wise and punctuated genome evolution drive phenotype changes of tumor cells. Mutat. Res. 2015, 771, 56–69. [Google Scholar] [CrossRef] [PubMed]
- Duesberg, P.; Li, R.; Rasnick, D.; Rausch, C.; Willer, A.; Kraemer, A.; Yerganian, G.; Hehlmann, R. Aneuploidy precedes and segregates with chemical carcinogenesis. Cancer Genet. Cytogenet. 2000, 119, 83–93. [Google Scholar] [CrossRef]
- Fabarius, A.; Willer, A.; Yerganian, G.; Hehlmann, R.; Duesberg, P. Specific aneusomies in Chinese hamster cells at different stages of neoplastic transformation, initiated by nitrosomethylurea. Proc. Natl. Acad. Sci. USA 2002, 99, 6778–6783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Botchan, M.; Stringer, J.; Mitchison, T.; Sambrook, J. Integration and excision of SV40 DNA from the chromosome of a transformed cell. Cell 1980, 20, 143–152. [Google Scholar] [CrossRef]
- Fabarius, A.; Hehlmann, R.; Duesberg, P.H. Instability of chromosome structure in cancer cells increases exponentially with degrees of aneuploidy. Cancer Genet. Cytogenet. 2003, 143, 59–72. [Google Scholar] [CrossRef]
- Paulsson, K.; Morse, H.; Fioretos, T.; Behrendtz, M.; Strombeck, B.; Johansson, B. Evidence for a single-step mechanism in the origin of hyperdiploid childhood acute lymphoblastic leukemia. Genes Chromosomes Cancer 2005, 44, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Gao, R.; Davis, A.; McDonald, T.O.; Sei, E.; Shi, X.; Wang, Y.; Tsai, P.C.; Casasent, A.; Waters, J.; Zhang, H.; et al. Punctuated copy number evolution and clonal stasis in triple-negative breast cancer. Nat. Genet. 2016, 48, 1119–1130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Felsenstein, J. The evolutionary advantage of recombination. Genetics 1974, 78, 737–756. [Google Scholar] [PubMed]
- Evans, H.J. Effects on chromosomes of carcinogenic rays and chemicals. In Chromosome Mutation and Neoplasia; German, J., Ed.; Alan R. Liss, Inc.: New York, NY, USA, 1983; pp. 253–279. [Google Scholar]
- Muller, H.J. Artificial transmutation of the gene. Science 1927, 66, 84–87. [Google Scholar] [CrossRef] [PubMed]
- Heim, S.; Mitelman, F. Cancer Cytogenetics, 3rd ed.; Wiley-Blackwell: Hoboken, NJ, USA, 2009. [Google Scholar]
- McCormack, A.; Fan, J.L.; Duesberg, M.; Bloomfield, M.; Fiala, C.; Duesberg, P. Individual karyotypes at the origins of cervical carcinomas. Mol. Cytogenet. 2013, 6, 44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McMichael, H.; Wagner, J.E.; Nowell, P.C.; Hungerford, D.A. Chromosome studies of virus-induced rabbit papillomas and derived primary carcinomas. J. Natl. Cancer Inst. 1963, 31, 1197–1215. [Google Scholar] [PubMed]
- Sandberg, A.A. The chromosomes and causation of human cancer and leukemia. Cancer Res. 1966, 26, 2064–2081. [Google Scholar] [PubMed]
- Sandberg, A.A. The Chromosomes in Human Cancer and Leukemia, 2nd ed.; Elsevier Science Publishing: New York, NY, USA, 1990. [Google Scholar]
- Fialkow, P.J. Clonal origin of human tumors. Annu. Rev. Med. 1979, 30, 135–143. [Google Scholar] [CrossRef] [PubMed]
- Sidransky, D.; Frost, P.; Von Eschenbach, A.; Oyasu, R.; Preisinger, A.C.; Vogelstein, B. Clonal origin bladder cancer. N. Engl. J. Med. 1992, 326, 737–740. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.L.; Rago, C.; Silliman, N.; Ptak, J.; Markowitz, S.; Willson, J.K.; Parmigiani, G.; Kinzler, K.W.; Vogelstein, B.; Velculescu, V.E. Prevalence of somatic alterations in the colorectal cancer cell genome. Proc. Natl. Acad. Sci. USA 2002, 99, 3076–3080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vincent, M.D. On the nature of cancer, and why it matters. In Proceedings of the 3rd Conference on Aneuploidy and Cancer: Clinical and Experimental Aspects, Berkeley, CA, USA, 26–29 January 2017. [Google Scholar]
- Reisman, L.E.; Zuelzer, W.W.; Thompson, R.I. Further Observation on the Role of Aneuploidy in Acute Leukemia. Cancer Res. 1964, 24, 1448–1455. [Google Scholar] [PubMed]
- Reisman, L.E.; Mitani, M.; Zuelzer, W.W. Chromosome studies in leukemia. I. Evidence for the origin of leukemic stem lines from aneuploid mutants. N. Engl. J. Med. 1964, 270, 591–597. [Google Scholar] [CrossRef] [PubMed]
- Lamb, D. Correlation of chromosome counts with histological appearances and prognosis in transitional-cell carcinoma of bladder. Br. Med. J. 1967, 1, 273–277. [Google Scholar] [CrossRef] [PubMed]
- Atkin, N.B. Modal deoxyribonucleic acid value and survival in carcinoma of the breast. Br. Med. J. 1972, 1, 271–272. [Google Scholar] [CrossRef] [PubMed]
- Spriggs, A.I. Cytogenetics of cancer and precancerous states of the cervix uteri. In Chromosomes and Cancer; John Wiley: Hoboken, NJ, USA, 1974; pp. 423–450. [Google Scholar]
- Jakobsen, A.; Bichel, P.; Sell, A. Correlation of DNA distribution and cytological differentiation of human cervical carcinomas. Virchows Arch. B Cell. Pathol. Incl. Mol. Pathol. 1979, 31, 75–79. [Google Scholar] [CrossRef] [PubMed]
- Auer, G.; Eriksson, E.; Azavedo, E.; Caspersson, T.; Wallgren, A. Prognostic significance of nuclear DNA content in mammary adenocarcinomas in humans. Cancer Res. 1984, 44, 394–396. [Google Scholar] [PubMed]
- Lee, S.E.; Currin, S.M.; Paulson, D.F.; Walther, P.J. Flow cytometric determination of ploidy in prostatic adenocarcinoma: A comparison with seminal vesicle involvement and histopathological grading as a predictor of clinical recurrence. J. Urol. 1988, 140, 769–774. [Google Scholar] [CrossRef]
- Forsslund, G.; Esposti, P.L.; Nilsson, B.; Zetterberg, A. The prognostic significance of nuclear DNA content in prostatic carcinoma. Cancer 1992, 69, 1432–1439. [Google Scholar] [CrossRef]
- Bardi, G.; Johansson, B.; Pandis, N.; Bak-Jensen, E.; Orndal, C.; Heim, S.; Mandahl, N.; Andren-Sandberg, A.; Mitelman, F. Cytogenetic aberrations in colorectal adenocarcinomas and their correlation with clinicopathologic features. Cancer 1993, 71, 306–314. [Google Scholar] [CrossRef] [Green Version]
- Shackney, S.E.; Berg, G.; Simon, S.R.; Cohen, J.; Amina, S.; Pommersheim, W.; Yakulis, R.; Wang, S.; Uhl, M.; Smith, C.A.; et al. Origins and clinical implications of aneuploidy in early bladder cancer. Cytometry 1995, 22, 307–316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hemmer, J.; Thein, T.; Van Heerden, W.F. The value of DNA flow cytometry in predicting the development of lymph node metastasis and survival in patients with locally recurrent oral squamous cell carcinoma. Cancer 1997, 79, 2309–2313. [Google Scholar] [CrossRef] [Green Version]
- Grimwade, D.; Walker, H.; Oliver, F.; Wheatley, K.; Harrison, C.; Harrison, G.; Rees, J.; Hann, I.; Stevens, R.; Burnett, A.; et al. The importance of diagnostic cytogenetics on outcome in AML: Analysis of 1612 patients entered into the MRC AML 10 trial. The Medical Research Council Adult and Children’s Leukaemia Working Parties. Blood 1998, 92, 2322–2333. [Google Scholar] [PubMed]
- Grimwade, D.; Walker, H.; Harrison, G.; Oliver, F.; Chatters, S.; Harrison, C.J.; Wheatley, K.; Burnett, A.K.; Goldstone, A.H. The predictive value of hierarchical cytogenetic classification in older adults with acute myeloid leukemia (AML): Analysis of 1065 patients entered into the United Kingdom Medical Research Council AML11 trial. Blood 2001, 98, 1312–1320. [Google Scholar] [CrossRef] [PubMed]
- Choma, D.; Daures, J.P.; Quantin, X.; Pujol, J.L. Aneuploidy and prognosis of non-small-cell lung cancer: A meta-analysis of published data. Br. J. Cancer 2001, 85, 14–22. [Google Scholar] [CrossRef] [PubMed]
- Schoch, C.; Haferlach, T.; Haase, D.; Fonatsch, C.; Loffler, H.; Schlegelberger, B.; Staib, P.; Sauerland, M.C.; Heinecke, A.; Buchner, T.; et al. Patients with de novo acute myeloid leukaemia and complex karyotype aberrations show a poor prognosis despite intensive treatment: a study of 90 patients. Br. J. Haematol. 2001, 112, 118–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Micci, F.; Bjerkehagen, B.; Heim, S. Pairwise comparison of genomic imbalances between primary and recurrent well differentiated liposarcomas. Cancer Genet. Cytogenet. 2007, 178, 163–167. [Google Scholar] [CrossRef] [PubMed]
- Fabarius, A.; Leitner, A.; Hochhaus, A.; Muller, M.C.; Hanfstein, B.; Haferlach, C.; Gohring, G.; Schlegelberger, B.; Jotterand, M.; Reiter, A.; et al. Impact of additional cytogenetic aberrations at diagnosis on prognosis of CML: Long-term observation of 1151 patients from the randomized CML Study IV. Blood 2011, 118, 6760–6768. [Google Scholar] [CrossRef] [PubMed]
- Böcking, A. Comparability of tumor-cytogenetics and -DNA-cytometry. Mol. Cytogenet. 2015, 8, 28. [Google Scholar] [CrossRef] [PubMed]
- Rubin, C.E.; Haggitt, R.C.; Burmer, G.C.; Brentnall, T.A.; Stevens, A.C.; Levine, D.S.; Dean, P.J.; Kimmey, M.; Perera, D.R.; Rabinovitch, P.S. DNA aneuploidy in colonic biopsies predicts future development of dysplasia in ulcerative colitis. Gastroenterology 1992, 103, 1611–1620. [Google Scholar] [CrossRef]
- Lee, A.J.; Endesfelder, D.; Rowan, A.J.; Walther, A.; Birkbak, N.J.; Futreal, P.A.; Downward, J.; Szallasi, Z.; Tomlinson, I.P.; Howell, M.; et al. Chromosomal instability confers intrinsic multidrug resistance. Cancer Res. 2011, 71, 1858–1870. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Karyotypes | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 | 15 | 16 | 17 | 18 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| No of Chrs. | 42 | 42 | 42 | 42 | 42 | 42 | 42 | 42 | 42 | 42 | 40 | 35 | 39 | 40 | 37 | 41 | 41 | 46 |
| Chromosomes | Chromosome Copy Number | |||||||||||||||||
| 1 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 1 | 2 | 2 | 2 | 2 | 2 | 2 |
| 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 3 |
| 3 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 1 | 2 | 2 | 2 | 2 | 2 | 2 |
| 4 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 1 | 2 | 2 | 2 | 2 | 2 | 2 |
| 5 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 3 | 2 | 1 | 2 | 2 | 2 | 2 | 2 | 2 |
| 6 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 1 | 1 | 1 | 2 | 2 | 2 | 2 | 2 |
| 7 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 1 | 2 | 2 | 2 |
| 8 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 3 | 2 | 2 | 2 | 2 | 1 | 2 | 2 | 3 |
| 9 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 1 | 2 | 2 | 3 |
| 10 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 3 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 |
| 11 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 1 | 2 |
| 12 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 1 | 1 | 1 | 2 | 2 | 2 | 2 |
| 13 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 1 | 1 | 1 | 2 | 2 | 3 |
| 14 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 |
| 15 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 1 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 |
| 16 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 1 |
| 17 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 1 | 1 | 2 | 3 |
| 18 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 1 | 2 | 1 | 2 | 2 | 2 | 2 | 2 | 2 |
| 19 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 |
| 20 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 1 | 1 | 2 | 2 | 2 | 2 | 2 | 2 | 2 |
| X | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 |
| Y | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| (a) | ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Karyotypes 1 Month Post-SV40 | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 | 15 | 16 | 17 | 18 | 19 | 20 |
| No. of Chrs. | 46 | 46 | 46 | 46 | 46 | 46 | 46 | 46 | 45 | 45 | 47 | 49 | 45 | 45 | 47 | 47 | 44 | 48 | 46 | 42 |
| Chromosomes | Chromosome Copy Number | |||||||||||||||||||
| 1 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 1 | 2 | 2 | 2 | 2 | 2 | 2 | 2 |
| 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 3 | 2 | 2 | 2 |
| 3 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 1 |
| 4 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 |
| 5 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 3 | 3 | 2 | 2 | 3 | 2 |
| 6 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 1 |
| 7 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 1 | 2 | 2 | 2 | 2 | 2 | 2 |
| 8 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 1 |
| 9 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 3 | 2 | 2 |
| 10 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 |
| 11 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 1 | 2 |
| 12 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 |
| 13 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 3 | 2 |
| 14 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 |
| 15 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 3 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 |
| 16 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 1 | 2 | 2 | 2 |
| 17 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 3 | 2 | 2 | 2 | 2 | 1 | 2 | 2 | 2 |
| 18 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 1 | 2 |
| 19 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 3 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 |
| 20 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 1 | 3 | 2 | 2 |
| 21 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 1 | 2 | 2 | 3 | 2 | 1 | 2 | 2 | 1 | 2 | 2 | 2 |
| 22 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 |
| X | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 1 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 1 |
| Y | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| der(7;21)? | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 |
| der(7) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| i(21q) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 |
| (b) | ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Karyotypes 2 Months Post-SV40 | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 | 15 | 16 | 17 | 18 | 19 | 20 |
| No. of Chrs. | 46 | 45 | 46 | 46 | 43 | 46 | 46 | 47 | 47 | 46 | 47 | 47 | 47 | 47 | 48 | 49 | 60 | 75 | 75 | 83 |
| Chromosomes | Chromosome Copy Number | |||||||||||||||||||
| 1 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 1 | 1 | 2 | 2 | 2 | 2 | 2 | 2 | 3 | 4 | 4 |
| 2 | 2 | 1 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 3 | 1 | 4 | 3 |
| 3 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 3 | 2 | 2 | 2 | 2 | 2 | 3 | 4 | 3 |
| 4 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 3 | 2 | 4 | 2 | 3 | 4 |
| 5 | 2 | 2 | 2 | 2 | 2 | 3 | 2 | 3 | 2 | 2 | 1 | 2 | 2 | 3 | 2 | 3 | 1 | 5 | 3 | 4 |
| 6 | 2 | 1 | 2 | 2 | 2 | 2 | 2 | 2 | 3 | 2 | 2 | 2 | 2 | 2 | 3 | 2 | 2 | 3 | 4 | 2 |
| 7 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 4 | 2 | 3 |
| 8 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 3 | 3 | 2 | 3 |
| 9 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 1 | 2 | 2 | 2 | 2 | 2 | 2 | 3 | 3 | 2 | 3 |
| 10 | 2 | 2 | 1 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 3 | 2 | 2 | 2 | 2 | 2 | 2 | 4 | 4 | 4 |
| 11 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 3 | 3 | 3 |
| 12 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 3 | 4 | 3 | 4 |
| 13 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 4 | 3 | 3 |
| 14 | 2 | 2 | 2 | 1 | 1 | 1 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 4 | 4 | 4 |
| 15 | 2 | 2 | 2 | 2 | 1 | 2 | 1 | 2 | 2 | 2 | 1 | 2 | 2 | 2 | 2 | 2 | 4 | 4 | 3 | 4 |
| 16 | 2 | 2 | 2 | 2 | 1 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 4 | 2 | 3 | 2 |
| 17 | 2 | 2 | 2 | 2 | 2 | 1 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 1 | 4 | 4 | 3 |
| 18 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 1 | 3 | 2 | 2 | 3 | 2 | 2 |
| 19 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 1 | 1 | 2 | 2 | 2 | 1 | 3 | 2 | 3 | 3 | 4 |
| 20 | 2 | 2 | 1 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 3 | 4 | 4 | 4 | 4 |
| 21 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 1 | 2 | 3 | 3 |
| 22 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 4 | 2 | 3 | 2 |
| X | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 1 | 3 | 3 | 4 |
| Y | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| der(1q) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| der(18) long | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 1 | 0 |
| der(9;6;9;6) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 |
| der(17;18) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 |
| der(22;7) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 |
| der(16;8;3) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 |
| der(8;3) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 |
| der(22;16) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 |
| min(3?) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 |
| der(8) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 |
| der(1;17) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 |
| der(17;12) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 |
| der(22) small | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 |
| min(19) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 |
| der(19q) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| der(9;13?;9) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| der(16q) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 | 0 | 0 |
| der(19;14) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| der(1) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| dic(5;15) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| i(14q) | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| dic(2;6) | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| der(13) small | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| der(15;3) | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| der(12;9) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 |
| der(10q) | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| der(20) long | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| der(17) | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| (c) | ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Karyotypes 6 Months Post-SV40 | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 | 15 | 16 | 17 | 18 | 19 | 20 |
| No. of Chrs. | 44 | 40 | 47 | 43 | 44 | 43 | 45 | 41 | 44 | 45 | 44 | 45 | 47 | 43 | 42 | 51 | 65 | 83 | 73 | 84 |
| Chromosomes | Chromosome Copy Number | |||||||||||||||||||
| 1 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 1 | 2 | 2 | 2 | 3 | 4 | 4 | 3 |
| 2 | 2 | 2 | 2 | 2 | 1 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 1 | 2 | 3 | 4 | 4 | 2 | 3 |
| 3 | 2 | 1 | 2 | 2 | 1 | 2 | 2 | 1 | 2 | 2 | 1 | 2 | 1 | 1 | 2 | 2 | 2 | 2 | 4 | 4 |
| 4 | 2 | 2 | 2 | 2 | 1 | 2 | 2 | 2 | 2 | 1 | 2 | 2 | 3 | 2 | 2 | 2 | 4 | 4 | 2 | 3 |
| 5 | 3 | 3 | 3 | 3 | 3 | 3 | 2 | 1 | 2 | 2 | 1 | 2 | 2 | 1 | 3 | 2 | 2 | 2 | 3 | 4 |
| 6 | 2 | 0 | 2 | 1 | 2 | 2 | 2 | 2 | 2 | 2 | 1 | 2 | 1 | 2 | 2 | 2 | 2 | 2 | 3 | 5 |
| 7 | 2 | 2 | 2 | 1 | 2 | 1 | 1 | 1 | 2 | 2 | 2 | 2 | 1 | 2 | 2 | 1 | 1 | 4 | 0 | 2 |
| 8 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 1 | 1 | 2 | 2 | 1 | 2 | 1 | 3 | 2 | 4 | 2 |
| 9 | 2 | 1 | 2 | 2 | 2 | 2 | 2 | 1 | 2 | 1 | 2 | 1 | 1 | 2 | 2 | 2 | 2 | 4 | 3 | 5 |
| 10 | 2 | 1 | 2 | 2 | 1 | 2 | 1 | 2 | 2 | 2 | 2 | 1 | 1 | 2 | 2 | 2 | 3 | 4 | 4 | 4 |
| 11 | 2 | 2 | 2 | 1 | 1 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 1 | 3 | 2 | 3 | 4 |
| 12 | 2 | 2 | 1 | 1 | 1 | 1 | 2 | 0 | 2 | 1 | 2 | 1 | 2 | 1 | 1 | 2 | 3 | 4 | 4 | 2 |
| 13 | 2 | 2 | 2 | 2 | 2 | 1 | 1 | 1 | 1 | 2 | 2 | 2 | 2 | 1 | 1 | 1 | 3 | 4 | 1 | 3 |
| 14 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 3 | 3 | 2 | 2 | 2 | 2 | 4 | 2 | 2 |
| 15 | 1 | 1 | 2 | 1 | 2 | 1 | 1 | 1 | 1 | 1 | 2 | 1 | 2 | 1 | 2 | 2 | 1 | 2 | 2 | 2 |
| 16 | 2 | 2 | 2 | 2 | 2 | 1 | 1 | 2 | 2 | 2 | 2 | 2 | 2 | 1 | 2 | 2 | 3 | 4 | 2 | 4 |
| 17 | 2 | 1 | 2 | 1 | 2 | 2 | 2 | 0 | 1 | 1 | 2 | 2 | 2 | 2 | 2 | 2 | 1 | 4 | 2 | 4 |
| 18 | 2 | 2 | 2 | 2 | 2 | 2 | 1 | 2 | 2 | 2 | 1 | 1 | 1 | 2 | 1 | 2 | 4 | 4 | 2 | 3 |
| 19 | 1 | 2 | 2 | 1 | 2 | 1 | 2 | 2 | 2 | 2 | 2 | 2 | 1 | 2 | 2 | 1 | 4 | 2 | 2 | 3 |
| 20 | 2 | 2 | 2 | 1 | 2 | 2 | 2 | 1 | 2 | 2 | 2 | 2 | 1 | 2 | 2 | 1 | 2 | 2 | 2 | 3 |
| 21 | 1 | 1 | 2 | 2 | 1 | 2 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 0 | 0 | 2 | 2 | 2 | 3 |
| 22 | 0 | 0 | 2 | 1 | 1 | 1 | 1 | 0 | 2 | 1 | 1 | 1 | 2 | 1 | 1 | 2 | 0 | 2 | 2 | 3 |
| X | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 3 | 4 | 4 | 3 |
| Y | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| der(15;22) | 1 | 1 | 0 | 1 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 2 | 0 | 0 |
| der(19;21) | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 |
| der(6;21) | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| der(22;6) | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| der(12;9) | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| der(6;8) | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| der(11;20) | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| dic(12;19) | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| dic(7;17) | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| dic(4;10) | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| der(3;21) | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| der(2;22) | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| der(11;12) | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| del(2q) | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| der(7;12) | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| dic(16;19) | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| der(7;15) | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| der(7;22) | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| der(12;20;21) | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| dic(12;16) | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| der(5;9) | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| der(18;20) | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| der(16) | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| dic(5;9) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| dic(5;22) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| der(15;7) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| dic(16;20) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| dic(12;22;17) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| der(12;7) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| der(10;22) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| der(10;21) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| der(3) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| der(5;15) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| der(17;21) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| der(8;21) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| der(4;17) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| del(4p) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 |
| der(5;12) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| der(12) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| del(9q) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| dic(5;6) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| dic(5;22)long | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| i(18) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| der(3;21) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| der(9;11) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| der(5;21) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| der(12;10) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| der(10;12) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| der(7;18) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| der(4;22) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| der(19;11) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| der(5)short | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| der(9;15) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| dic(20;6;21) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| del(6q) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| der(1) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| der(21;17) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| der(2;5) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 |
| der(3;15) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 |
| der(16;21) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 1 | 0 |
| (der(8;11) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 |
| der(8;12) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 |
| der(X;22) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 |
| del(5q) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 |
| der(21;22) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 |
| der(5;6) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 |
| der(8;13) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 |
| der(6;8;14) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 |
| der(17;6) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 |
| min(16) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 |
| der(11;14) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 |
| der(6;8) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 |
| der(7;5;21) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 |
| der(2;5)short | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 |
| der(20;6) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 |
| mar(12) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 |
| der(9;10) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 |
| der(17;22) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 |
| der(21;7) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 |
| der(3;22) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 |
| dic(X;6) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 |
| der(5;20;21) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 |
| del(5q)short | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 |
| der(17)long | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 |
| der(3;5) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 | 0 | 0 |
| dic(5;6)short | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 | 0 | 0 |
| dic(8;11) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 | 0 | 0 |
| der(20;21) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 | 0 | 0 |
| der(19;5;19) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 |
| der(18;2) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 |
| der(2;3) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 |
| der(12;2;19) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 |
| der(4;11;4) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 |
| der(5;17) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 |
| der(7;21) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 |
| der(10;19) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 |
| der(14;22) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 |
| der(17;14) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 |
| del(7q) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 |
| der(14;15) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 |
| der(16) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 |
| der(1;18) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 |
| der(1;16) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 |
| der(6;14) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 |
| der(6;7) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 |
| der(5;7) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 |
| der(8;13)short | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 |
| der(5;9)short | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 |
| der(16;18) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 |
| dic(5;2;17;9) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 |
| der(X;21) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 |
| (a) | ||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| F10 Karyotypes | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 | 15 | 16 | 17 | 18 | 19 | 20 | 21 | 22 | 23 | 24 | 25 | 26 | 27 | 28 | 29 | 30 |
| No. of Chrs. | 41 | 41 | 41 | 41 | 39 | 42 | 42 | 41 | 41 | 41 | 40 | 42 | 41 | 42 | 40 | 42 | 41 | 41 | 41 | 42 | 43 | 41 | 41 | 41 | 39 | 41 | 42 | 81 | 76 | 84 |
| Chromosomes | Chromosome Copy Number | |||||||||||||||||||||||||||||
| 1 | 2 | 1 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 1 | 2 | 2 | 4 | 3 | 4 |
| 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 3 | 4 | 4 |
| 3 | 1 | 0 | 1 | 1 | 0 | 1 | 1 | 1 | 1 | 2 | 1 | 0 | 1 | 1 | 1 | 1 | 2 | 1 | 1 | 2 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 3 | 2 | 3 |
| 4 | 2 | 2 | 2 | 2 | 1 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 4 | 3 | 3 |
| 5 | 1 | 1 | 2 | 1 | 1 | 2 | 2 | 2 | 2 | 2 | 1 | 2 | 1 | 2 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 2 | 1 | 2 | 1 | 2 | 1 | 2 | 2 | 4 |
| 6 | 2 | 2 | 2 | 2 | 1 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 1 | 2 | 1 | 2 | 1 | 2 | 2 | 2 | 2 | 2 | 2 | 4 | 4 | 4 |
| 7 | 2 | 2 | 2 | 2 | 1 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 1 | 2 | 4 | 4 | 3 |
| 8 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 4 | 4 | 4 |
| 9 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 4 | 4 | 4 |
| 10 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 1 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 4 | 3 | 4 |
| 11 | 2 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 2 | 1 | 2 | 1 | 2 | 2 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 2 | 2 |
| 12 | 1 | 1 | 1 | 1 | 1 | 2 | 1 | 1 | 1 | 1 | 1 | 2 | 1 | 2 | 0 | 0 | 1 | 1 | 1 | 2 | 2 | 1 | 1 | 1 | 0 | 0 | 1 | 4 | 2 | 4 |
| 13 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 1 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 4 | 3 | 4 |
| 14 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 1 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 3 | 4 | 4 |
| 15 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 4 | 3 | 4 |
| 16 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 4 | 4 | 4 |
| 17 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 1 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 4 | 3 | 4 |
| 18 | 2 | 1 | 2 | 2 | 2 | 2 | 3 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 4 | 4 | 4 |
| 19 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 1 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 4 | 4 | 3 |
| 20 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 4 | 4 | 4 |
| X | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 4 | 4 | 3 |
| Y | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| der(3;12) | 1 | 1 | 1 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 1 | 1 | 0 | 1 | 0 | 0 | 0 | 0 | 1 | 0 | 1 | 1 | 0 | 0 | 2 | 0 |
| i(5q) | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 1 | 1 | 1 | 0 | 0 | 1 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 | 0 | 0 |
| der(5;11) | 0 | 0 | 1 | 0 | 1 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 1 | 1 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 1 | 0 | 1 | 0 | 0 | 0 | 2 | 0 |
| der(3;11) | 0 | 0 | 0 | 1 | 0 | 1 | 1 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 | 0 | 1 |
| der(5;12) | 0 | 0 | 0 | 1 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| der(3;5) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 1 | 1 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 |
| der(5;5) | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 1 | 0 | 0 | 1 | 0 | 1 | 1 | 0 | 0 | 0 | 0 |
| der(5;9) | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| der(11;12) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 |
| der(5;7) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 |
| der(5) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| der(3;1;19) | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| dic(1;5;5) | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| der(1;5) | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| der(1;11) | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| dic(4;6) | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| dic(3;5;5) | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| der(7) | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| der(i(5q);11) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| der(1;3) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| der(12;19) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| i(3q) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| der(11;1) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| dic(5;12) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 1 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 |
| dic(6;17) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| dic(2;5) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 |
| dic(5;13) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 |
| der(3;10) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 |
| tricentric(5;5;5) | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| der(X;4) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 |
| der(5;19) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 |
| der(19;5) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 |
| i(4q) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 |
| der(4) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 |
| der(5,6) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| der(3,11) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 1 | 1 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 |
| der(1,5,6) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| der(6,9,6) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| der(5,6,5,20?) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| der(6,5,20) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| der(4,11) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 |
| der(7,12) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 |
| dic(2;6;5;6;5;6;5) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 |
| der(2;11?,5) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 |
| (b) | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| C1 Karyotype | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 | 15 | 16 |
| No. of Chrms. | 41 | 42 | 42 | 46 | 42 | 42 | 45 | 42 | 42 | 42 | 42 | 44 | 40 | 44 | 46 | 41 |
| Chromosomes | Chromosome Copy Number | |||||||||||||||
| 1 | 2 | 2 | 2 | 2 | 1 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 1 | 2 |
| 2 | 1 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 1 | 2 | 2 | 2 | 2 | 2 |
| 3 | 1 | 1 | 1 | 1 | 1 | 1 | 2 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 |
| 4 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 1 | 1 | 2 | 2 | 2 |
| 5 | 1 | 1 | 1 | 2 | 2 | 2 | 1 | 1 | 1 | 1 | 1 | 2 | 1 | 1 | 1 | 1 |
| 6 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 |
| 7 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 1 |
| 8 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 |
| 9 | 1 | 1 | 2 | 2 | 2 | 2 | 2 | 2 | 1 | 1 | 1 | 1 | 2 | 1 | 1 | 1 |
| 10 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 1 | 2 | 2 | 2 | 2 |
| 11 | 2 | 1 | 2 | 2 | 0 | 0 | 2 | 1 | 1 | 0 | 1 | 1 | 2 | 1 | 1 | 1 |
| 12 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 |
| 13 | 2 | 2 | 2 | 1 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 |
| 14 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 |
| 15 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 |
| 16 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 |
| 17 | 2 | 2 | 2 | 2 | 2 | 1 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 |
| 18 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 |
| 19 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 |
| 20 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 1 | 2 | 2 | 2 | 2 |
| X | 1 | 1 | 2 | 2 | 2 | 2 | 2 | 1 | 1 | 1 | 2 | 1 | 2 | 1 | 1 | 1 |
| Y | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| i(5q) | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 2 | 0 | 1 | 1 | 1 | 0 |
| der(5;12) | 2 | 2 | 2 | 1 | 0 | 1 | 2 | 2 | 2 | 2 | 2 | 2 | 1 | 2 | 2 | 2 |
| der(12;11) | 0 | 1 | 0 | 0 | 1 | 0 | 1 | 1 | 1 | 2 | 1 | 0 | 0 | 1 | 0 | 1 |
| der(3;12) | 0 | 0 | 1 | 1 | 1 | 1 | 0 | 0 | 0 | 0 | 1 | 0 | 1 | 0 | 1 | 0 |
| der(12;3;X) | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 1 | 1 | 1 | 0 | 1 | 0 | 1 | 0 | 1 |
| der(X;3) | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 1 | 1 | 1 | 0 | 1 | 0 | 1 | 1 | 0 |
| der(3;9) | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| der(12;9;X) | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| mar(2) | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| mar(9) | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 |
| mar(13) | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| mar(11) | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| mar(12) | 0 | 0 | 0 | 1 | 1 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 |
| der(11,1,5) | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| der(4,12) | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| der(4,16) | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| der(12,9) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| der(2,5) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 |
| der(9,11) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 |
| mar(7) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 |
| der(5, 7) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 |
| (c) | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| C2 Karyotype | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 |
| No. of Chrs. | 43 | 41 | 42 | 38 | 41 | 40 | 40 | 40 | 43 |
| Chromosomes | Chromosomes Copy Number | ||||||||
| 1 | 2 | 2 | 1 | 2 | 2 | 2 | 1 | 2 | 2 |
| 2 | 3 | 3 | 2 | 2 | 3 | 2 | 2 | 2 | 3 |
| 3 | 1 | 1 | 0 | 1 | 1 | 1 | 1 | 1 | 1 |
| 4 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 1 | 2 |
| 5 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 2 |
| 6 | 3 | 2 | 2 | 1 | 2 | 2 | 2 | 2 | 2 |
| 7 | 2 | 1 | 2 | 1 | 1 | 1 | 1 | 2 | 1 |
| 8 | 2 | 2 | 1 | 1 | 2 | 2 | 2 | 2 | 2 |
| 9 | 1 | 2 | 1 | 1 | 1 | 1 | 2 | 1 | 2 |
| 10 | 2 | 2 | 1 | 2 | 2 | 2 | 2 | 2 | 2 |
| 11 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 |
| 12 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| 13 | 2 | 2 | 3 | 2 | 2 | 2 | 2 | 2 | 2 |
| 14 | 2 | 2 | 3 | 2 | 2 | 2 | 2 | 2 | 2 |
| 15 | 2 | 2 | 2 | 1 | 2 | 2 | 1 | 2 | 2 |
| 16 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 |
| 17 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 |
| 18 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 |
| 19 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 |
| 20 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 |
| X | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 |
| Y | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| der(5;9)long | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 |
| der(5;7) | 1 | 0 | 1 | 1 | 1 | 1 | 1 | 1 | 1 |
| der(11;12) | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 |
| der(3;12) | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 |
| der(5;7) | 0 | 1 | 0 | 1 | 1 | 1 | 1 | 0 | 0 |
| der(5) | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| der(5;5) | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 |
| dic(1;11) | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 |
| t(3;8) | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 |
| t(8;3) | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 |
| der(6;8) | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 |
| der(1) | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 |
| der(4;5) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 |
| der(10;7) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 |
| (d) | |||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| C6 Karyotype | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 | 15 | 16 | 17 | 18 | 19 | 20 | 21 | 22 | 23 | 24 | 25 | 26 | 27 | 28 | 29 |
| No. of Chrs. | 42 | 41 | 42 | 41 | 41 | 41 | 41 | 41 | 42 | 37 | 41 | 43 | 42 | 43 | 39 | 42 | 42 | 43 | 42 | 42 | 43 | 42 | 43 | 39 | 42 | 42 | 43 | 42 | 42 |
| Chromosomes | Chromosome Copy Number | ||||||||||||||||||||||||||||
| 1 | 2 | 2 | 2 | 1 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 |
| 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 1 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 |
| 3 | 1 | 0 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 2 | 1 | 1 | 1 | 2 | 1 | 1 | 1 | 2 | 2 | 1 | 1 | 1 | 2 | 1 | 1 | 1 | 2 |
| 4 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 1 | 2 | 2 | 2 | 2 | 1 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 1 | 2 | 2 | 2 | 2 | 2 |
| 5 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 |
| 6 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 |
| 7 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 1 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 1 | 2 | 2 | 2 | 2 |
| 8 | 2 | 1 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 1 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 1 | 2 | 2 | 2 | 2 | 2 |
| 9 | 1 | 0 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 |
| 10 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 1 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 |
| 11 | 1 | 0 | 1 | 1 | 1 | 1 | 2 | 0 | 2 | 1 | 1 | 1 | 1 | 0 | 2 | 1 | 1 | 1 | 2 | 0 | 1 | 1 | 0 | 2 | 1 | 1 | 1 | 2 | 0 |
| 12 | 0 | 0 | 1 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 1 | 1 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 1 |
| 13 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 |
| 14 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 1 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 1 | 2 | 2 | 2 |
| 15 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 |
| 16 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 |
| 17 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 |
| 18 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 3 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 3 | 2 | 2 | 2 | 2 |
| 19 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 |
| 20 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 1 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 |
| X | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 2 | 2 | 1 | 1 | 1 | 2 | 2 | 1 | 2 | 1 | 2 | 1 | 1 | 1 | 2 | 2 | 1 | 2 | 1 |
| Y | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| i(5q) | 1 | 1 | 1 | 1 | 0 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 |
| der(5;12) | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 0 | 1 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 0 | 1 | 2 | 2 | 2 | 2 |
| der(11;12) | 1 | 1 | 1 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 1 | 1 | 1 | 2 | 1 | 1 | 1 | 1 | 0 | 1 | 1 | 1 | 2 | 1 | 1 | 1 | 1 | 0 | 1 |
| der(12;3;X) | 1 | 1 | 1 | 1 | 1 | 1 | 0 | 1 | 0 | 1 | 0 | 0 | 1 | 1 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 1 | 1 | 0 | 1 | 0 | 0 | 0 | 0 |
| der(X;3) | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 0 | 1 | 0 | 0 | 1 | 1 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 1 | 1 | 0 | 0 | 0 | 1 | 0 | 0 |
| der(9;12?) | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| der(9) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 |
| der(2;5) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 |
| der(3;12) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 1 | 1 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 1 | 1 |
| dup(9) | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| der(6;12) | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| der(6) | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| der(3;11) | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| dup(8) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 |
| der(9;12;11) | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| der(i5(q);7) | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| der(7) | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| dic(9;11) | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| der(9;3?) | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| der(14) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 |
| der(X;3;9) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 |
| der(3;9) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| der(X;9;12) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| der(2) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| (e) | |||||||
|---|---|---|---|---|---|---|---|
| C7 Karyotype | 1 | 2 | 3 | 4 | 5 | 6 | 7 |
| No. of Chrs. | 39 | 41 | 41 | 40 | 42 | 41 | 42 |
| Chromosomes | Chromosome Copy Number | ||||||
| 1 | 2 | 2 | 2 | 2 | 2 | 2 | 2 |
| 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 |
| 3 | 0 | 1 | 1 | 1 | 1 | 1 | 1 |
| 4 | 2 | 2 | 2 | 2 | 2 | 2 | 2 |
| 5 | 1 | 1 | 1 | 1 | 1 | 1 | 1 |
| 6 | 1 | 1 | 1 | 1 | 2 | 2 | 2 |
| 7 | 2 | 2 | 2 | 2 | 2 | 2 | 2 |
| 8 | 2 | 2 | 2 | 2 | 2 | 2 | 2 |
| 9 | 2 | 2 | 2 | 2 | 2 | 2 | 2 |
| 10 | 2 | 2 | 2 | 2 | 2 | 2 | 2 |
| 11 | 1 | 1 | 1 | 1 | 1 | 1 | 1 |
| 12 | 1 | 2 | 2 | 2 | 2 | 2 | 2 |
| 13 | 2 | 2 | 2 | 2 | 2 | 2 | 2 |
| 14 | 2 | 2 | 2 | 2 | 2 | 2 | 2 |
| 15 | 2 | 2 | 2 | 2 | 2 | 2 | 2 |
| 16 | 1 | 2 | 2 | 2 | 2 | 2 | 2 |
| 17 | 2 | 2 | 2 | 2 | 2 | 2 | 2 |
| 18 | 2 | 2 | 2 | 2 | 2 | 2 | 2 |
| 19 | 2 | 2 | 2 | 2 | 2 | 2 | 2 |
| 20 | 2 | 2 | 2 | 2 | 2 | 2 | 2 |
| X | 2 | 2 | 2 | 2 | 2 | 2 | 2 |
| Y | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| dic(5;5;6) | 1 | 0 | 0 | 0 | 1 | 1 | 0 |
| der(3;11) | 1 | 1 | 1 | 1 | 1 | 1 | 1 |
| dic(5;6) | 0 | 1 | 1 | 1 | 0 | 0 | 0 |
| der(6;5) | 0 | 1 | 1 | 0 | 0 | 0 | 1 |
| der(3;12) | 1 | 0 | 0 | 0 | 0 | 0 | 0 |
| dic(6;16) | 1 | 0 | 0 | 0 | 0 | 0 | 0 |
| der(5;5) | 0 | 0 | 0 | 0 | 1 | 0 | 0 |
| der(5) | 0 | 0 | 0 | 0 | 0 | 0 | 1 |
| (f) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| C8 Karyotype | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 |
| No. of Chrs. | 38 | 38 | 40 | 42 | 40 | 41 | 42 | 41 | 40 | 41 |
| Chromosomes | Chromosome Copy Number | |||||||||
| 1 | 2 | 2 | 2 | 3 | 2 | 2 | 2 | 2 | 3 | 2 |
| 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 |
| 3 | 1 | 0 | 1 | 1 | 1 | 0 | 1 | 1 | 1 | 1 |
| 4 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 |
| 5 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 |
| 6 | 2 | 2 | 2 | 2 | 2 | 2 | 1 | 2 | 2 | 2 |
| 7 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 |
| 8 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 |
| 9 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 |
| 10 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 |
| 11 | 1 | 1 | 1 | 0 | 1 | 1 | 1 | 2 | 1 | 1 |
| 12 | 1 | 0 | 0 | 1 | 0 | 2 | 0 | 0 | 0 | 0 |
| 13 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 1 | 2 |
| 14 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 |
| 15 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 |
| 16 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 |
| 17 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 |
| 18 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 |
| 19 | 2 | 2 | 1 | 2 | 2 | 2 | 2 | 2 | 2 | 2 |
| 20 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 |
| X | 2 | 2 | 2 | 2 | 2 | 1 | 2 | 2 | 2 | 2 |
| Y | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| der(5,5) | 0 | 1 | 1 | 1 | 1 | 0 | 1 | 1 | 1 | 1 |
| der(3,12) | 0 | 1 | 1 | 0 | 1 | 0 | 1 | 1 | 1 | 1 |
| der(5,12) | 0 | 0 | 1 | 1 | 0 | 2 | 1 | 1 | 0 | 1 |
| der(11,12) | 0 | 0 | 1 | 2 | 1 | 0 | 1 | 0 | 0 | 1 |
| der(3,11) | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 |
| der(6) | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 |
| der(X) | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 |
| (g) | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| C9 Karyotype | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 |
| No. of Chrs. | 40 | 40 | 39 | 41 | 40 | 41 | 41 | 40 | 41 |
| Chromosomes | Chromosome Copy Number | ||||||||
| 1 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 |
| 2 | 2 | 2 | 1 | 2 | 2 | 2 | 2 | 2 | 2 |
| 3 | 1 | 1 | 1 | 1 | 1 | 1 | 0 | 1 | 1 |
| 4 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 |
| 5 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 |
| 6 | 2 | 1 | 1 | 2 | 1 | 1 | 2 | 1 | 1 |
| 7 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 |
| 8 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 |
| 9 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 |
| 10 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 |
| 11 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 |
| 12 | 2 | 2 | 1 | 2 | 2 | 2 | 2 | 2 | 2 |
| 13 | 1 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 |
| 14 | 2 | 2 | 2 | 2 | 1 | 2 | 2 | 2 | 2 |
| 15 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 |
| 16 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 |
| 17 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 |
| 18 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 |
| 19 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 |
| 20 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 |
| X | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 |
| Y | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| der(3;11) | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 |
| der(5;5) | 0 | 0 | 0 | 0 | 1 | 1 | 1 | 0 | 1 |
| dic(5;5;6) | 0 | 1 | 1 | 1 | 0 | 0 | 0 | 1 | 0 |
| dic(5;5;13) | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| dic(2;6) | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 |
| dic(6;13) | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 |
| dic(6;6) | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 |
| der(3;6) | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 |
| der(6) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 |
| Cytogenetic Analyses of a Neoplastic Primary Rat Clone F10 and Six Sub-Clones | |||||||
|---|---|---|---|---|---|---|---|
| F10 Stem Line | C1 | C2 | C6 | C7 | C8 | C9 | |
| Avg Chr. No. ± SD | 45.1 ± 11.8 | 42.7 ± 1.72 | 40.9 ± 1.52 | 41.6 ± 1.33 | 40.9 ± 0.99 | 40.3 ± 1.35 | 40.3 ± 0.67 |
| Chromosomes | copy no (% clonal) | ||||||
| 1 | 2 (83) | 2 (88) | 2 (78) | 2 (97) | 2 (100) | 2 (80) | 2 (100) |
| 2 | 2 (90) | 2 (88) | 2 (56) | 2 (97) | 2 (100) | 2 (100) | 2 (89) |
| 3 | 1 (70) | 1 (94) | 1 (89) | 1 (76) | 1 (86) | 1 (80) | 1 (89) |
| 4 | 2 (87) | 2 (88) | 2 (89) | 2 (90) | 2 (100) | 2 (100) | 2 (100) |
| 5 | 1 (53) | 1 (75) | 1 (89) | 1 (100) | 1 (100) | 1 (100) | 1 (100) |
| 6 | 2 (77) | 2 (100) | 2 (78) | 2 (100) | 1 (57) | 2 (90) | 1 (67) |
| 7 | 2 (83) | 2 (94) | 1 (67) | 2 (93) | 2 (100) | 2 (100) | 2 (100) |
| 8 | 2 (90) | 2 (100) | 2 (78) | 2 (90) | 2 (100) | 2 (100) | 2 (100) |
| 9 | 2 (90) | 1 (56) | 1 (67) | 2 (62) | 2 (100) | 2 (100) | 2 (100) |
| 10 | 2 (87) | 2 (94) | 2 (89) | 2 (97) | 2 (100) | 2 (100) | 2 (100) |
| 11 | 1 (77) | 1 (50) | 1 (100) | 1 (59) | 1 (100) | 1 (80) | 1 (100) |
| 12 | 1 (60) | 0 (94) | 0 (100) | 0 (72) | 2 (86) | 0 (70) | 2 (89) |
| 13 | 2 (87) | 2 (94) | 2 (89) | 2 (100) | 2 (100) | 2 (90) | 2 (89) |
| 14 | 2 (87) | 2 (100) | 2 (89) | 2 (93) | 2 (100) | 2 (100) | 2 (89) |
| 15 | 2 (90) | 2 (100) | 2 (78) | 2 (100) | 2 (100) | 2 (100) | 2 (100) |
| 16 | 2 (90) | 2 (100) | 2 (100) | 2 (100) | 2 (86) | 2 (100) | 2 (100) |
| 17 | 2 (87) | 2 (94) | 2 (100) | 2 (100) | 2 (100) | 2 (100) | 2 (100) |
| 18 | 2 (83) | 2 (100) | 2 (100) | 2 (93) | 2 (100) | 2 (100) | 2 (100) |
| 19 | 2 (87) | 2 (100) | 2 (100) | 2 (100) | 2 (100) | 2 (90) | 2 (100) |
| 20 | 2 (90) | 2 (94) | 2 (100) | 2 (97) | 2 (100) | 2 (100) | 2 (100) |
| X | 2 (90) | 1 (56) | 2 (100) | 1 (69) | 2 (100) | 2 (90) | 2 (100) |
| Y | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| der(3;12) | 1 (40) | 1 (44) | 1 (100) | 1 (23) | 1 (14) | 1 (70) | - |
| i(5q) | 1 (33) | 1 (81) | 1 (100) | 1 (97) | - | - | - |
| der(5;11) | 1 (30) | - | - | - | - | - | - |
| der(3;11) | 1 (20) | - | - | 1 (3) | 1 (100) | 1 (10) | 1 (100) |
| der(5;12) | 1 (13) | 2 (75) | - | 2 (86) | - | 1 (50) | - |
| der(5;5) | 1 (20) | - | 1 (11) | - | 1 (14) | 1 (80) | 1 (44) |
| der(5;9) | 1 (3) | - | 1 (89) | - | - | - | - |
| der(11;12) | 1 (7) | 1 (50) | 1 (100) | 1 (66) | - | 0 (50) | - |
| der(5;7) | 1 (3) | 1 (6) | 1 (56) | - | - | - | - |
| der(5) | 1 (7) | - | 1 (11) | - | 1 (14) | - | - |
| der(X;3) | - | 1 (44) | - | 1 (52) | - | - | - |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hirpara, A.; Bloomfield, M.; Duesberg, P. Speciation Theory of Carcinogenesis Explains Karyotypic Individuality and Long Latencies of Cancers. Genes 2018, 9, 402. https://doi.org/10.3390/genes9080402
Hirpara A, Bloomfield M, Duesberg P. Speciation Theory of Carcinogenesis Explains Karyotypic Individuality and Long Latencies of Cancers. Genes. 2018; 9(8):402. https://doi.org/10.3390/genes9080402
Chicago/Turabian StyleHirpara, Ankit, Mathew Bloomfield, and Peter Duesberg. 2018. "Speciation Theory of Carcinogenesis Explains Karyotypic Individuality and Long Latencies of Cancers" Genes 9, no. 8: 402. https://doi.org/10.3390/genes9080402
APA StyleHirpara, A., Bloomfield, M., & Duesberg, P. (2018). Speciation Theory of Carcinogenesis Explains Karyotypic Individuality and Long Latencies of Cancers. Genes, 9(8), 402. https://doi.org/10.3390/genes9080402