Comparative Genomics and Description of Putative Virulence Factors of Melissococcus plutonius, the Causative Agent of European Foulbrood Disease in Honey Bees


 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Origin of Melissococcus plutonius Strains
2.2. Growth Conditions and Isolation of DNA from Melissococcus plutonius
2.3. Genome Sequencing, Assembly and Annotation
2.4. Genome Analyses
2.5. cDNA Synthesis and Reverse Transcription PCR (RT-PCR)
3. Results
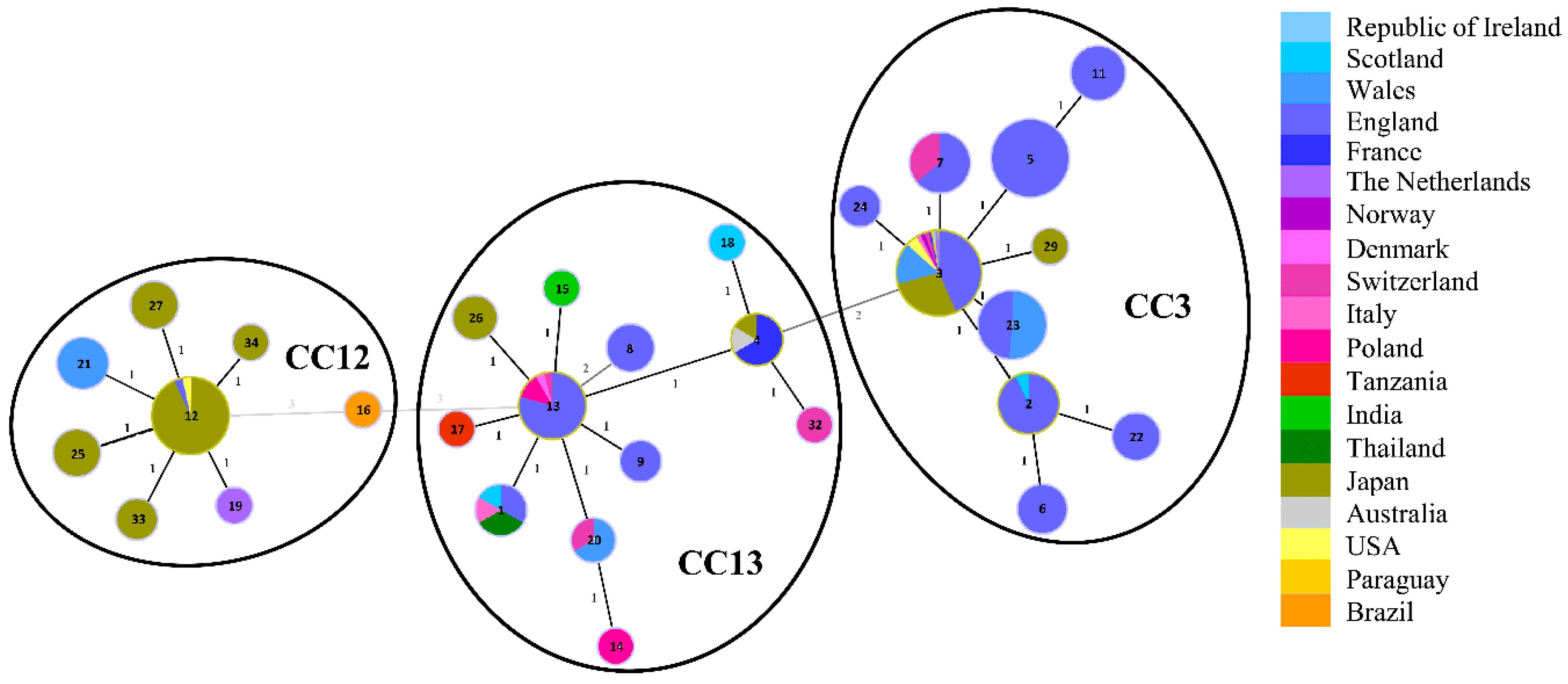
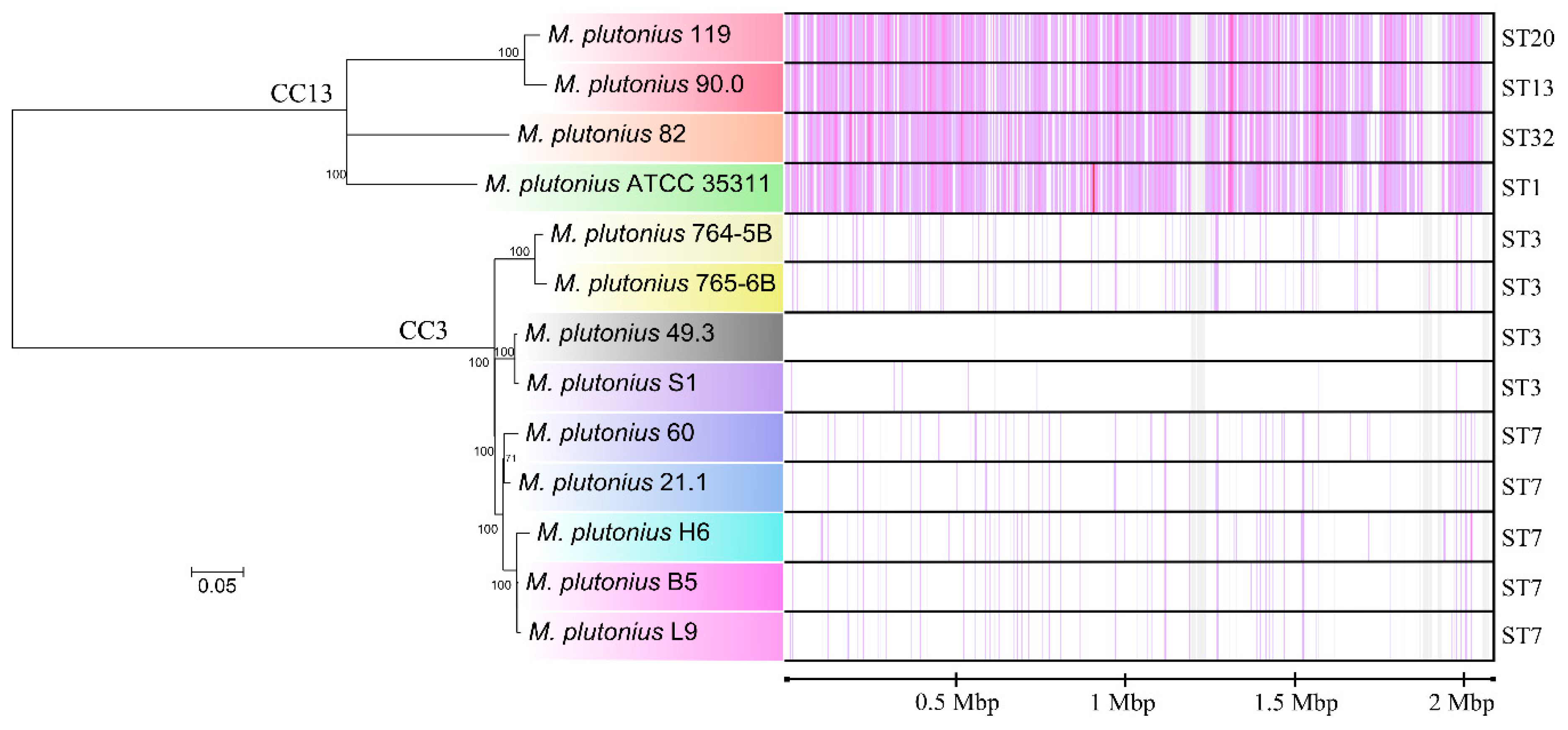
3.1. Sequence Types of Melissococcus plutonius Isolates and Clonal Complex Association
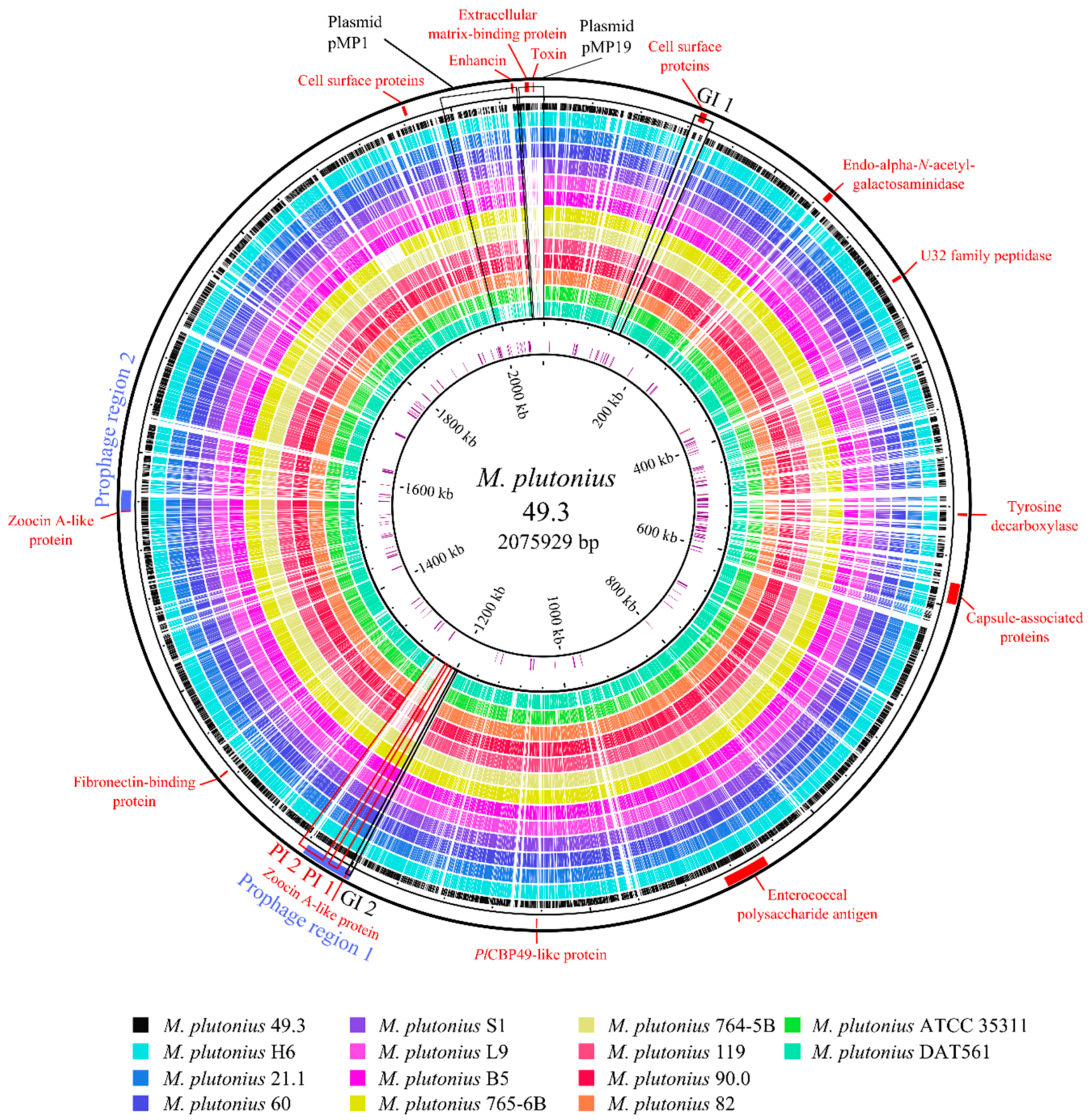
3.2. Genome Analysis—General Properties
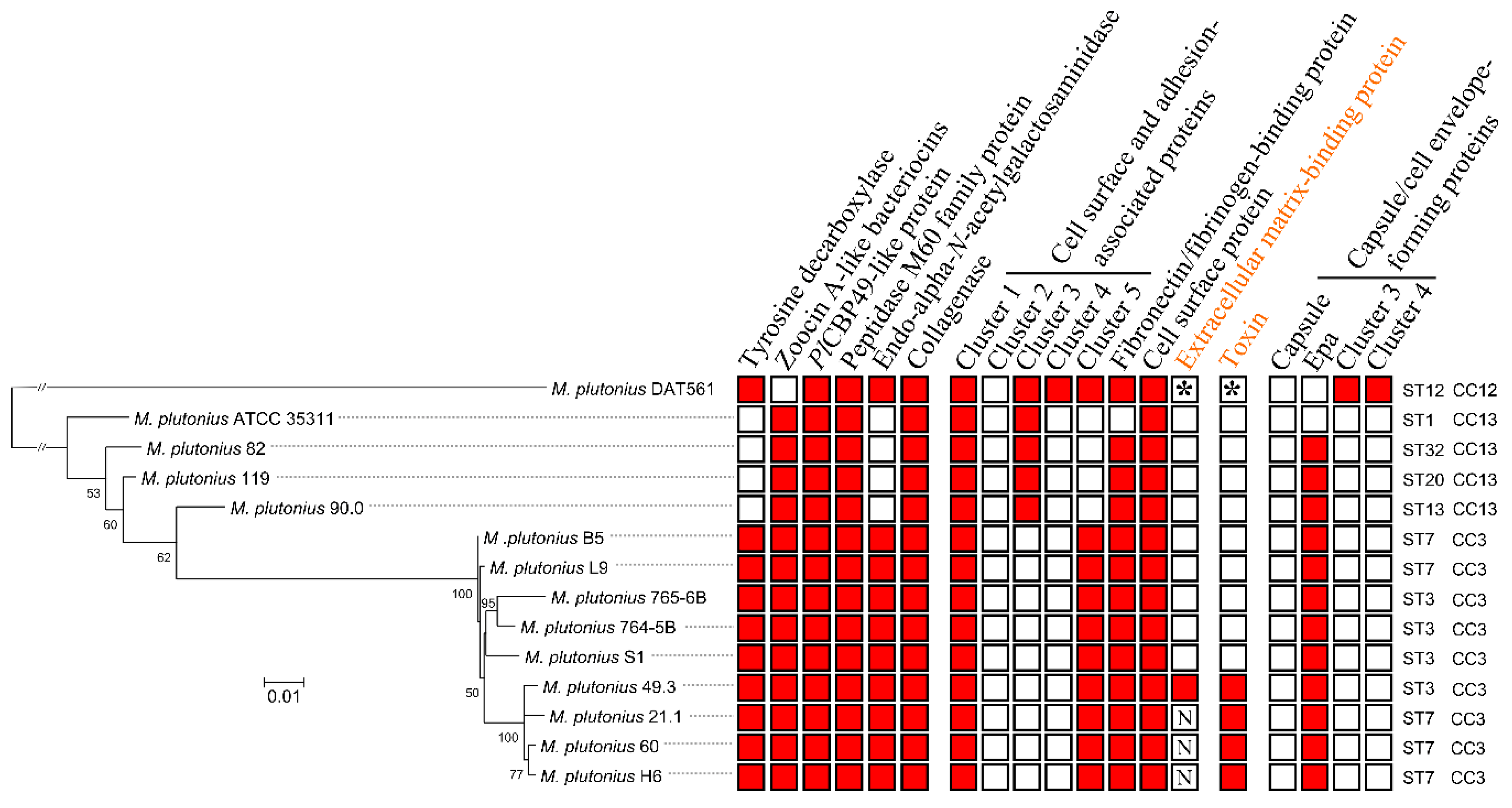
3.3. Genome Analysis—Detection of Putative Virulence Factors
3.4. Characterization of Genes Putatively Important for Melissococcus plutonius Survival and Pathogenicity
3.4.1. Bacteriocins
3.4.2. Tyramine
3.4.3. Larval Glycoprotein and Peritrophic Matrix-Degrading Enzymes
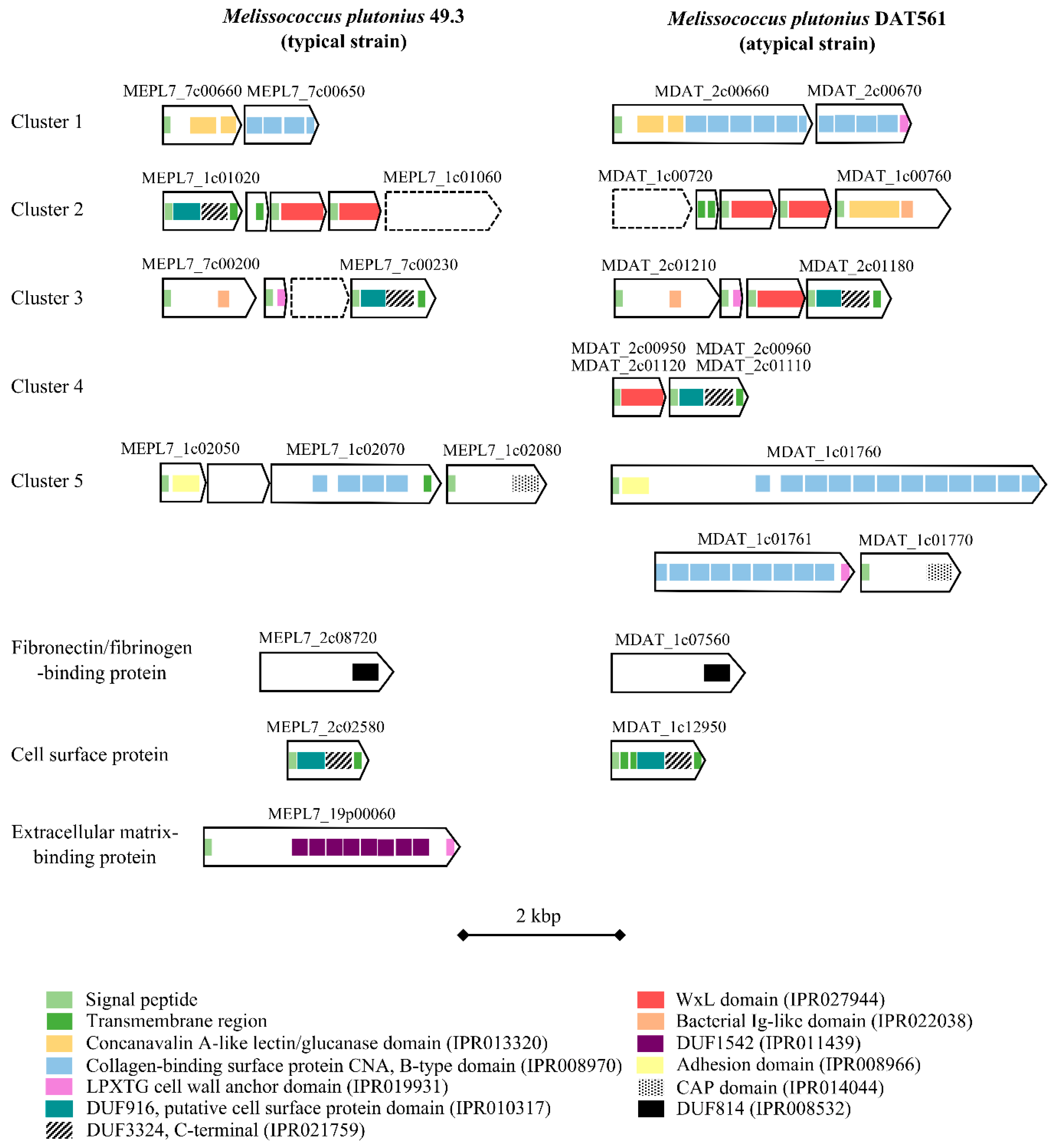
3.4.4. Bacterial Cell Curface- and Host Cell adhesion-associated Proteins
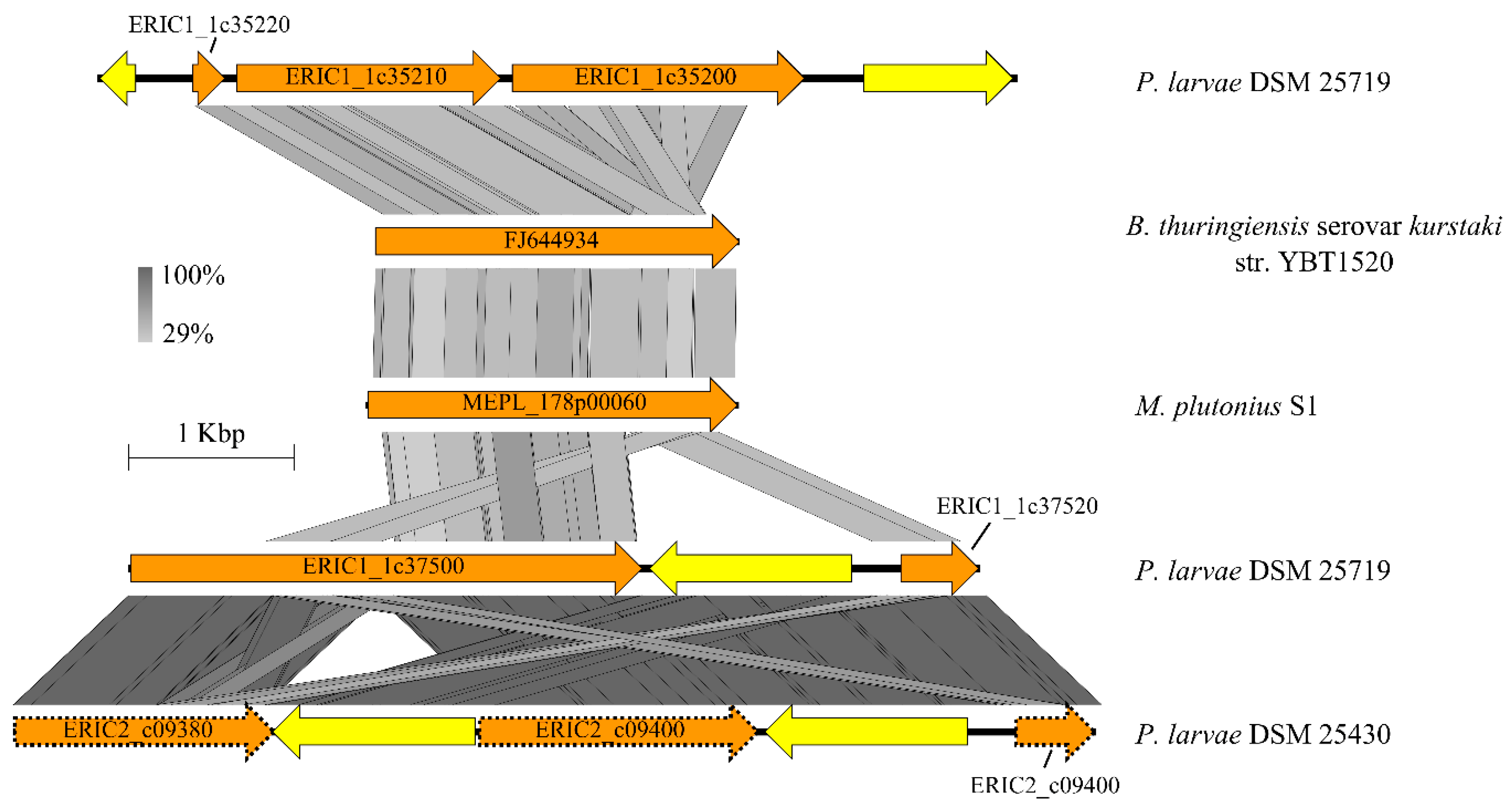
3.4.5. Toxin
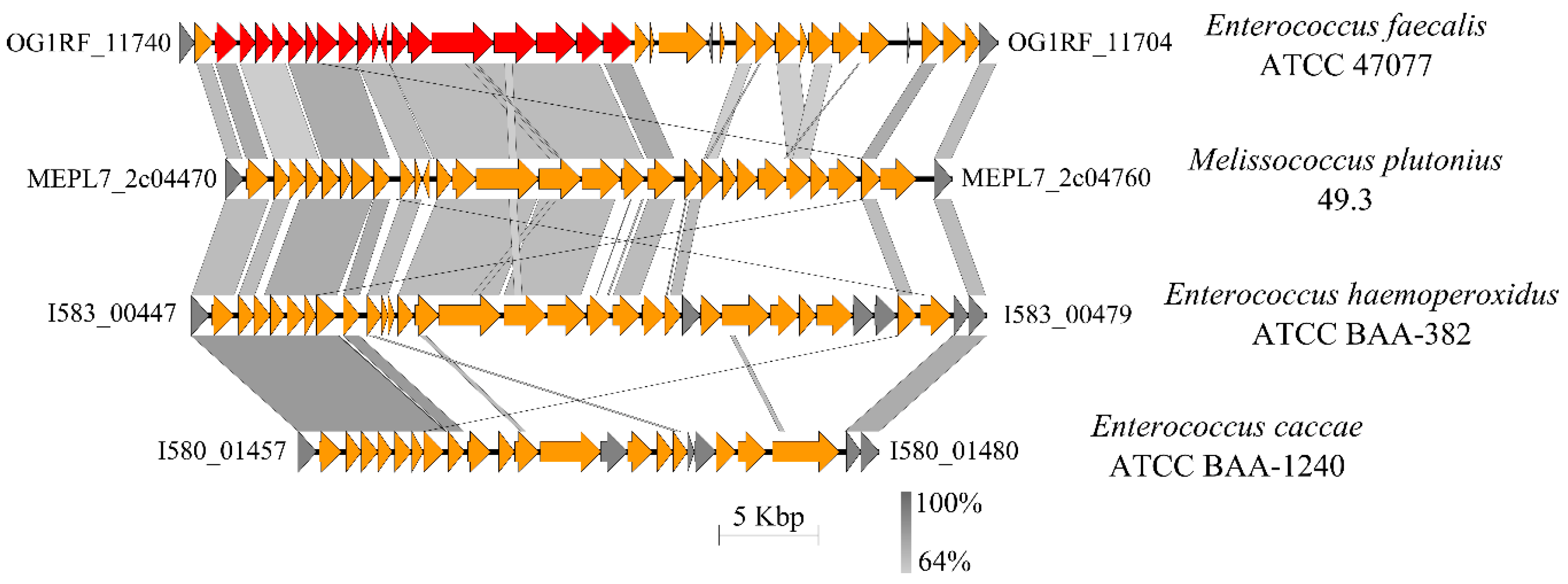
3.4.6. Capsule and Cell Envelope-Forming Proteins
3.4.7. Energy and Sugar Metabolism
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Morse, R.A.; Calderone, N.W. The value of honey bees as pollinators of U.S. crops in 2000. Bee Cult 2000, 128, 1–15. [Google Scholar]
- Aizen, M.A.; Garibaldi, L.A.; Cunningham, S.A.; Klein, A.M. Long-term global trends in crop yield and production reveal no current pollination shortage but increasing pollinator dependency. Curr. Biol. 2008, 18, 1572–1575. [Google Scholar] [CrossRef] [PubMed]
- Aizen, M.A.; Harder, L.D. The global stock of domesticated honey bees is growing slower than agricultural demand for pollination. Curr. Biol 2009, 19, 915–918. [Google Scholar] [CrossRef] [PubMed]
- Gallai, N.; Salles, J.-M.; Settele, J.; Vaissière, B.E. Economic valuation of the vulnerability of world agriculture confronted with pollinator decline. Ecol. Econ. 2009, 68, 810–821. [Google Scholar] [CrossRef] [Green Version]
- Forsgren, E. European foulbrood in honey bees. J. Invertebr. Pathol. 2010, 103, S5–S9. [Google Scholar] [CrossRef] [PubMed]
- Bailey, L. European foul brood: A disease of the larval honeybee (Apis mellifera L.) caused by a combination of Streptococcus pluton (Bacillus pluton White) and Bacterium eurydice White. Nature 1957, 180, 1214–1215. [Google Scholar] [CrossRef]
- Williams, D.L. A veterinary approach to the European honey bee (Apis mellifera). Vet. J. 2000, 160, 61–73. [Google Scholar] [CrossRef] [PubMed]
- Budge, G.E.; Shirley, M.D.F.; Jones, B.; Quill, E.; Tomkies, V.; Feil, E.J.; Brown, M.A.; Haynes, E.G. Molecular epidemiology and population structure of the honey bee brood pathogen Melissococcus plutonius. ISME J. 2014, 8, 1588–1597. [Google Scholar] [CrossRef] [PubMed]
- Arai, R.; Tominaga, K.; Wu, M.; Okura, M.; Ito, K.; Okamura, N.; Onishi, H.; Osaki, M.; Sugimura, Y.; Yoshiyama, M.; Takamatsu, D. Diversity of Melissococcus plutonius from honeybee larvae in Japan and experimental reproduction of European foulbrood with cultured atypical isolates. PLoS ONE 2012, 7, e33708. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Yamazaki, Y.; Shiraishi, A.; Kobayashi, S.; Harada, M.; Yoshiyama, M.; Osaki, M.; Okura, M.; Takamatsu, D. Virulence differences among Melissococcus plutonius strains with different genetic backgrounds in Apis mellifera larvae under an improved experimental condition. Sci. Rep. 2016, 6, 33329. [Google Scholar] [CrossRef] [PubMed]
- Okumura, K.; Arai, R.; Okura, M.; Kirikae, T.; Takamatsu, D.; Osaki, M.; Miyoshi-Akiyama, T. Complete genome sequence of Melissococcus plutonius ATCC 35311. J. Bacteriol. 2011, 193, 4029–4030. [Google Scholar] [CrossRef] [PubMed]
- Okumura, K.; Arai, R.; Okura, M.; Kirikae, T.; Takamatsu, D.; Osaki, M.; Miyoshi-Akiyama, T. Complete genome sequence of Melissococcus plutonius DAT561, a strain that shows an unusual growth profile and is representative of an endemic cluster in Japan. J. Bacteriol. 2012, 194, 3014. [Google Scholar] [CrossRef] [PubMed]
- Bailey, L.; Collins, M.D. Reclassification of ‘Streptococcus pluton’ (White) in a new genus Melissococcus, as Melissococcus pluton nom. rev.; comb. nov. J. Appl. Bacteriol. 1982, 53, 215–217. [Google Scholar] [CrossRef]
- Bailey, L. The isolation and cultural characteristics of Streptococcus pluton and further oberservations on Bacterium eurydice. J. Gen. Microbiol. 1957, 17, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Chevreux, B. MIRA: An Automated Genome and EST Assembler. Ph.D. Thesis, Ruprecht-Karls University Heidelberg, Heidelberg, Germany, 2005. [Google Scholar]
- Staden, R.; Beal, K.F.; Bonfield, J.K. The Staden package, 1998. Methods Mol. Biol. 2000, 132, 115–130. [Google Scholar] [CrossRef] [PubMed]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed]
- Seemann, T. Prokka: Rapid prokaryotic genome annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef] [PubMed]
- Gasteiger, E.; Jung, E.; Bairoch, A. SWISS-PROT: Connecting biomolecular knowledge via a protein database. Curr. Issues Mol. Biol. 2001, 3, 47–55. [Google Scholar] [CrossRef] [PubMed]
- O’Donovan, C.; Martin, M.J.; Gattiker, A.; Gasteiger, E.; Bairoch, A.; Apweiler, R. High-quality protein knowledge resource: SWISS-PROT and TrEMBL. Brief Bioinform. 2002, 3, 275–284. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.; Binns, D.; Chang, H.Y.; Fraser, M.; Li, W.; McAnulla, C.; McWilliam, H.; Maslen, J.; Mitchell, A.; Nuka, G.; et al. InterProScan 5: Genome-scale protein function classification. Bioinformatics 2014, 30, 1236–1240. [Google Scholar] [CrossRef] [PubMed]
- Markowitz, V.M.; Mavromatis, K.; Ivanova, N.N.; Chen, I.M.; Chu, K.; Kyrpides, N.C. IMG ER: A system for microbial genome annotation expert review and curation. Bioinformatics 2009, 25, 2271–2278. [Google Scholar] [CrossRef] [PubMed]
- Lagesen, K.; Hallin, P.; Rødland, E.A.; Staerfeldt, H.H.; Rognes, T.; Ussery, D.W. RNAmmer: Consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 2007, 35, 3100–3108. [Google Scholar] [CrossRef] [PubMed]
- Lowe, T.M.; Eddy, S.R. tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997, 25, 955–964. [Google Scholar] [CrossRef] [PubMed]
- Parks, D.H.; Imelfort, M.; Skennerton, C.T.; Hugenholtz, P.; Tyson, G.W. CheckM: Assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 2014, 25, 1043–1055. [Google Scholar] [CrossRef] [PubMed]
- Larsen, M.V.; Cosentino, S.; Rasmussen, S.; Friis, C.; Hasman, H.; Marvig, R.L.; Jelsbak, L.; Sicheritz-Pontén, T.; Ussery, D.W.; Aarestrup, F.M.; Lund, O. Multilocus sequence typing of total-genome-sequenced bacteria. J. Clin. Microbiol. 2012, 50, 1355–1361. [Google Scholar] [CrossRef] [PubMed]
- Haynes, E.; Helgason, T.; Young, J.P.; Thwaites, R.; Budge, G.E. A typing scheme for the honeybee pathogen Melissococcus plutonius allows detection of disease transmission events and a study of the distribution of variants. Environ. Microbiol. Rep. 2013, 5, 525–529. [Google Scholar] [CrossRef] [PubMed]
- Francisco, A.P.; Vaz, C.; Monteiro, P.T.; Melo-Cristino, J.; Ramirez, M.; Carrio, J.A. PHYLOViZ: Phylogenetic inference and data visualization for sequence based typing methods. BMC Bioinform. 2012, 13, 87. [Google Scholar] [CrossRef] [PubMed]
- Lechner, M.; Findeiss, S.; Steiner, L.; Marz, M.; Stadler, P.F.; Prohaska, S.J. Proteinortho: Detection of (co-)orthologs in large-scale analysis. BMC Bioinform. 2011, 12, 124. [Google Scholar] [CrossRef] [PubMed]
- Leimbach, A. BAC-Genomics-Scripts. 2014. Available online: https://github.com/aleimba/bac-genomics-scripts (accessed on 31 July 2018).
- Stamatakis, A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huson, D.H.; Scornavacca, C. Dendroscope 3: An interactive tool for rooted phylogenetic trees and networks. Syst. Biol. 2012, 61, 1061–1067. [Google Scholar] [CrossRef] [PubMed]
- Treangen, T.J.; Ondov, B.D.; Koren, S.; Phillippy, A.M. The Harvest suite for rapid core-genome alignment and visualization of thousands of intraspecific microbial genomes. Genome Biol. 2014, 15, 524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Croucher, N.J.; Page, A.J.; Connor, T.R.; Delaney, A.J.; Keane, J.A.; Bentley, S.D.; Parkhill, J.; Harris, S.R. Rapid phylogenetic analysis of large samples of recombinant bacterial whole genome sequences using Gubbins. Nucleic Acids Res. 2015, 43, e15. [Google Scholar] [CrossRef] [PubMed]
- Angiuoli, S.V.; Salzberg, S.L. Mugsy: fast multiple alignment of closely related whole genomes. Bioinformatics 2011, 27, 334–342. [Google Scholar] [CrossRef] [PubMed]
- Hadfield, J.; Croucher, N.J.; Goater, R.J.; Abudahab, K.; Aanensen, D.M.; Harris, S.R. Phandango: An interactive viewer for bacterial population genomics. Bioinformatics 2018, 34, 292–293. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Liang, Y.; Lynch, K.H.; Dennis, J.J. Wishart, D.S. PHAST: A fast phage search tool. Nucleic Acids Res. 2011, 39, W347–W352. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Xiong, Z.; Sun, L.; Yang, J.; Jin, Q. VFDB 2012 update: Toward the genetic diversity and molecular evolution of bacterial virulence factors. Nucleic Acids Res. 2012, 40, D641–D645. [Google Scholar] [CrossRef] [PubMed]
- Van Heel, A.J.; de Jong, A.; Montalbán-López, M.; Kok, J.; Kuipers, O.P. BAGEL3: Automated identification of genes encoding bacteriocins and (non-)bactericidal posttranslationally modified peptides. Nucleic Acids Res. 2013, 41, W448–W453. [Google Scholar] [CrossRef] [PubMed]
- Hammami, R.; Zouhir, A.; Le Lay, C.; Ben Hamida, J.; Fliss, I. BACTIBASE second release: A database and tool platform for bacteriocin characterization. BMC Microbiol. 2010, 10, 22. [Google Scholar] [CrossRef] [PubMed]
- Rawlings, N.D.; Waller, M.; Barrett, A.J.; Bateman, A. MEROPS: The database of proteolytic enzymes, their substrates and inhibitors. Nucleic Acids Res. 2014, 42, D503–D509. [Google Scholar] [CrossRef] [PubMed]
- Alikhan, N.-F.; Petty, N.K.; Ben Zakour, N.L.; Beatson, S.A. BLAST Ring Image Generator (BRIG): Simple prokaryote genome comparisons. BMC Genom. 2011, 12, 402. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, M.J.; Petty, N.K.; Beatson, S.A. Easyfig: A genome comparison visualizer. Bioinformatics 2011, 27, 1009–1010. [Google Scholar] [CrossRef] [PubMed]
- Govan, V.A.; Brözel, V.; Allsopp, M.H.; Davison, S.A. PCR detection method for rapid identification of Melissococcus pluton in honeybee larvae. Appl. Environ. Microbiol. 1998, 64, 1983–1985. [Google Scholar] [PubMed]
- Takamatsu, D.; Morinishi, K.; Arai, R.; Sakamoto, A.; Okura, M.; Osaki, M. Typing of Melissococcus plutonius isolated from European and Japanese honeybees suggests spread of sequence types across borders and between different Apis species. Vet. Microbiol 2014, 171, 221–226. [Google Scholar] [CrossRef] [PubMed]
- Tutar, Y. Pseudogenes. Comp. Funct. Genom. 2012, 2012, 424526. [Google Scholar] [CrossRef] [PubMed]
- Okumura, K.; Takamatsu, D.; Okura, M. Complete genome sequence of Melissococcus plutonius DAT561, a strain that shows an unusual growth profile, obtained by PacBio sequencing. Genome Announc. 2018, 6, e00431-18. [Google Scholar] [CrossRef] [PubMed]
- Lewkowski, O.; Erler, S. Virulence of Melissococcus plutonius and secondary invaders associated with European foulbrood disease of the honey bee. MicrobiologyOpen 2018, e649. [Google Scholar] [CrossRef] [PubMed]
- Zacharof, M.P.; Lovitt, R.W. Bacteriocins produced by lactic acid bacteria a review article. APCBEE Procedia 2012, 2, 50–56. [Google Scholar] [CrossRef]
- Joris, B.; Englebert, S.; Chu, C.P.; Kariyama, R.; Daneo-Moore, L.; Shockman, G.D.; Ghuysen, J.-M. Modular design of the Enterococcus hirae muramidase-2 and Streptococcus faecalis autolysin. FEMS Microbiol. Lett. 1992, 70, 257–264. [Google Scholar] [CrossRef] [PubMed]
- Connil, N.; Breton, Y.L.; Dousset, X.; Auffray, Y.; Rince, A.; Prévost, H. Identification of the Enterococcus faecalis tyrosine decarboxylase operon involved in tyramine production. Appl. Environ. Microbiol. 2002, 68, 3537–3544. [Google Scholar] [CrossRef] [PubMed]
- Kanbar, G.; Engels, W.; Nicholson, G.J.; Hertle, R.; Winkelmann, G. Corrigendum to: Tyramine functions as a toxin in honey bee larvae during Varroa-transmitted infection by Melissococcus pluton. FEMS Microbiol. Lett. 2005, 245, 193. [Google Scholar] [CrossRef]
- Kanbar, G.; Engels, W.; Nicholson, G.J.; Hertle, R.; Winkelmann, G. Tyramine functions as a toxin in honey bee larvae during Varroa-transmitted infection by Melissococcus pluton. FEMS Microbiol. Lett. 2004, 234, 149–154. [Google Scholar] [CrossRef] [PubMed]
- Terra, W.R. The origin and functions of the insect peritrophic membrane and peritrophic gel. Arch Insect Biochem. Physiol. 2001, 47, 47–61. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Gonzalez, E.; Poppinga, L.; Fünfhaus, A.; Hertlein, G.; Hedtke, K.; Jakubowska, A.; Genersch, E. Paenibacillus larvae chitin-degrading protein PlCBP49 is a key virulence factor in American Foulbrood of honey bees. PLoS Pathog. 2014, 10, e1004284. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.; Zhong, J.; Granados, R.R. A baculovirus enhancin alters the permeability of a mucosal midgut peritrophic matrix from lepidopteran larvae. J. Insect Physiol. 1999, 45, 159–166. [Google Scholar] [CrossRef]
- Tellam, R.L.; Wijffels, G.; Willadsen, P. Peritrophic matrix proteins. Insect Biochem. Mol. Biol. 1999, 29, 87–101. [Google Scholar] [CrossRef]
- Fang, S.; Wang, L.; Guo, W.; Zhang, X.; Peng, D.; Luo, C.; Yu, Z.; Sun, M. Bacillus thuringiensis bel protein enhances the toxicity of Cry1Ac protein to Helicoverpa armigera larvae by degrading insect intestinal mucin. Appl. Environ. Microbiol. 2009, 75, 5237–5243. [Google Scholar] [CrossRef] [PubMed]
- Toprak, U.; Harris, S.; Baldwin, D.; Theilmann, D.; Gillott, C.; Hegedus, D.D.; Erlandson, M.A. Role of enhancin in Mamestra configurata nucleopolyhedrovirus virulence: selective degradation of host peritrophic matrix proteins. J. Gen. Virol. 2012, 93, 744–753. [Google Scholar] [CrossRef] [PubMed]
- Djukic, M.; Brzuszkiewicz, E.; Fünfhaus, A.; Voss, J.; Gollnow, K.; Poppinga, L.; Liesegang, H.; Garcia-Gonzalez, E.; Genersch, E.; Daniel, R. How to kill the honey bee larva: genomic potential and virulence mechanisms of Paenibacillus larvae. PLoS ONE 2014, 9, e90914. [Google Scholar] [CrossRef] [PubMed]
- Clarke, S.R.; Harris, L.G.; Richards, R.G.; Foster, S.J. Analysis of Ebh, a 1.1-megadalton cell wall-associated fibronectin-binding protein of Staphylococcus aureus. Infect. Immun. 2002, 70, 6680–6687. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, K.; Jularic, M.; Horsburgh, S.M.; Hirschhausen, N.; Neumann, C.; Bertling, A.; Schulte, A.; Foster, S.; Kehrel, B.E.; Peters, G.; Heilmann, C. Molecular characterization of a novel Staphylococcus aureus surface protein (SasC) involved in cell aggregation and biofilm accumulation. PLoS ONE 2009, 4, e7567. [Google Scholar] [CrossRef] [PubMed]
- Djukic, M.; Poehlein, A.; Thürmer, A.; Daniel, R. Genome sequence of Brevibacillus laterosporus LMG 15441, a pathogen of invertebrates. J. Bacteriol. 2011, 193, 5535–5536. [Google Scholar] [CrossRef] [PubMed]
- Campos, M.A.; Vargas, M.A.; Regueiro, V.; Llompart, C.M.; Albertí, S.; Bengoechea, J.A. Capsule polysaccharide mediates bacterial resistance to antimicrobial peptides. Infect. Immun. 2004, 72, 7107–7114. [Google Scholar] [CrossRef] [PubMed]
- Schembri, M.A.; Dalsgaard, D.; Klemm, P. Capsule shields the function of short bacterial adhesins. J. Bacteriol. 2004, 186, 1249–1257. [Google Scholar] [CrossRef] [PubMed]
- Palmer, K.L.; Godfrey, P.; Griggs, A.; Kos, V.N.; Zucker, J.; Desjardins, C.; Cerqueira, G.; Gevers, D.; Walker, S.; Wortman, J.; Feldgarden, M.; Haas, B.; Birren, B.; Gilmore, M.S. Comparative genomics of enterococci: variation in Enterococcus faecalis, clade structure in E. faecium, and defining characteristics of E. gallinarum and E. casseliflavus. MBio 2012, 3, e00318-11. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Jiang, L.; Murray, B.E.; Weinstock, G.M. Enterococcus faecalis antigens in human infections. Infect. Immun. 1997, 65, 4207–4215. [Google Scholar] [PubMed]
- Xu, Y.; Murray, B.E.; Weinstock, G.M. A cluster of genes involved in polysaccharide biosynthesis from Enterococcus faecalis OG1RF. Infect. Immun. 1998, 66, 4313–4323. [Google Scholar] [PubMed]
- Hancock, L.E.; Murray, B.E.; Sillanpää, J. Enterococcal Cell Wall Components and Structures. In Enterococci: From Commensals to Leading Causes of Drug Resistant Infection; Gilmore, M.S., Clewell, D.B., Ike, Y., Shankar, N., Eds.; Massachusetts Eye and Ear Infirmary: Boston, MA, USA, 2012. [Google Scholar]
- Rigottier-Gois, L.; Madec, C.; Navickas, A.; Matos, R.C.; Akary-Lepage, E.; Mistou, M.Y.; Serror, P. The surface rhamnopolysaccharide epa of Enterococcus faecalis is a key determinant of intestinal colonization. J. Infect. Dis. 2015, 211, 62–71. [Google Scholar] [CrossRef] [PubMed]
- Engel, P.; Martinson, V.G.; Moran, N.A. Functional diversity within the simple gut microbiota of the honey bee. Proc. Natl. Acad. Sci. USA 2012, 109, 11002–11007. [Google Scholar] [CrossRef] [PubMed]
- Erler, S.; Lewkowski, O.; Poehlein, A.; Forsgren, E. The curious case of Achromobacter eurydice, a Gram-variable pleomorphic bacterium associated with European foulbrood disease in honeybees. Microb. Ecol. 2018, 75, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Luo, C.; Tsementzi, D.; Kyrpides, N.; Read, T.; Konstantinidis, K.T. Direct comparisons of Illumina vs. Roche 454 sequencing technologies on the same microbial community DNA sample. PLoS ONE 2012, 7, e30087. [Google Scholar] [CrossRef]
- Teng, F.; Singh, K.V.; Bourgogne, A.; Zeng, J.; Murray, B.E. Further characterization of the epa gene cluster and Epa polysaccharides of Enterococcus faecalis. Infect. Immun. 2009, 77, 3759–3767. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Gonzalez, E.; Genersch, E. Honey bee larval peritrophic matrix degradation during infection with Paenibacillus larvae, the aetiological agent of American foulbrood of honey bees, is a key step in pathogenesis. Environ. Microbiol. 2013, 15, 2894–2901. [Google Scholar] [CrossRef] [PubMed]
- Takamatsu, D.; Sato, M.; Yoshiyama, M. Infection of Melissococcus plutonius clonal complex 12 strain in European honeybee larvae is essentially confined to the digestive tract. J. Vet. Med. Sci. 2016, 78, 29–34. [Google Scholar] [CrossRef] [PubMed]
- Takamatsu, D.; Osawa, A.; Nakamura, K.; Yoshiyama, M.; Okura, M. High-level resistance of Melissococcus plutonius clonal complex 3 strains to antimicrobial activity of royal jelly. Environ. Microbiol. Rep. 2017, 9, 562–570. [Google Scholar] [CrossRef] [PubMed]
- Gubler, H.U. Bakteriologische Untersuchungen über die gutartige Faulbrut der Honigbiene (Apis mellifica L.). Pathol. Bakteriol. 1954, 17, 507–513. [Google Scholar] [CrossRef]
- Ritter, W. Bienen Gesund Erhalten; Eugen Ulmer: Stuttgart, Germany, 2012. [Google Scholar]
- Jolley, K.A.; Maiden, M.C. BIGSdb: Scalable analysis of bacterial genome variation at the population level. BMC Bioinform. 2010, 11, 595. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strain | Origin 1 | ST (CC) 2 | Classification | Genome Size [Mbp] | CDS 3 | Pseudogenes | Plasmids |
|---|---|---|---|---|---|---|---|
| 49.3 | CH | ST3 (CC3) | Typical strain | 2.076 | 1638 | 140 | pMP1, pMP19 |
| S1 | CH | ST3 (CC3) | Typical strain | 2.074 | 1609 | 151 | pMP1 |
| 21.1 | CH | ST7 (CC3) | Typical strain | 2.077 | 1629 | 145 | pMP1, pMP19 |
| 60 | CH | ST7 (CC3) | Typical strain | 2.072 | 1633 | 143 | pMP1, pMP19 |
| B5 | CH | ST7 (CC3) | Typical strain | 2.101 | 1686 | 146 | pMP1, pMP43 |
| H6 | CH | ST7 (CC3) | Typical strain | 2.075 | 1633 | 145 | pMP1, pMP19 |
| L9 | CH | ST7 (CC3) | Typical strain | 2.059 | 1616 | 145 | pMP1 |
| 82 | CH | ST32 (CC13) | Typical strain | 2.048 | 1614 | 129 | pMP1 |
| 90.0 | CH | ST13 (CC13) | Typical strain | 2.067 | 1642 | 131 | pMP1 |
| 119 | CH | ST20 (CC13) | Typical strain | 2.040 | 1614 | 127 | pMP1 |
| 764-5B | NO | ST3 (CC3) | Typical strain | 2.046 | 1605 | 145 | pMP1 |
| 765-6B | NO | ST3 (CC3) | Typical strain | 2.021 | 1589 | 142 | pMP1 |
| ATCC 35311 | GB-ENG | ST1 (CC13) | Typical strain | 2.069 | 1594 | 156 | pMP1 |
| DAT561 | JP | ST12 (CC12) | Atypical strain | 2.045 | 1595 | 75 | pMP1, pMP19 4 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Djukic, M.; Erler, S.; Leimbach, A.; Grossar, D.; Charrière, J.-D.; Gauthier, L.; Hartken, D.; Dietrich, S.; Nacke, H.; Daniel, R.; et al. Comparative Genomics and Description of Putative Virulence Factors of Melissococcus plutonius, the Causative Agent of European Foulbrood Disease in Honey Bees. Genes 2018, 9, 419. https://doi.org/10.3390/genes9080419
Djukic M, Erler S, Leimbach A, Grossar D, Charrière J-D, Gauthier L, Hartken D, Dietrich S, Nacke H, Daniel R, et al. Comparative Genomics and Description of Putative Virulence Factors of Melissococcus plutonius, the Causative Agent of European Foulbrood Disease in Honey Bees. Genes. 2018; 9(8):419. https://doi.org/10.3390/genes9080419
Chicago/Turabian StyleDjukic, Marvin, Silvio Erler, Andreas Leimbach, Daniela Grossar, Jean-Daniel Charrière, Laurent Gauthier, Denise Hartken, Sascha Dietrich, Heiko Nacke, Rolf Daniel, and et al. 2018. "Comparative Genomics and Description of Putative Virulence Factors of Melissococcus plutonius, the Causative Agent of European Foulbrood Disease in Honey Bees" Genes 9, no. 8: 419. https://doi.org/10.3390/genes9080419
APA StyleDjukic, M., Erler, S., Leimbach, A., Grossar, D., Charrière, J. -D., Gauthier, L., Hartken, D., Dietrich, S., Nacke, H., Daniel, R., & Poehlein, A. (2018). Comparative Genomics and Description of Putative Virulence Factors of Melissococcus plutonius, the Causative Agent of European Foulbrood Disease in Honey Bees. Genes, 9(8), 419. https://doi.org/10.3390/genes9080419