Heterogeneous Uptake of N2O5 in Sand Dust and Urban Aerosols Observed during the Dry Season in Beijing

 ,
,  ,
, 
Abstract
:1. Introduction
2. Methods
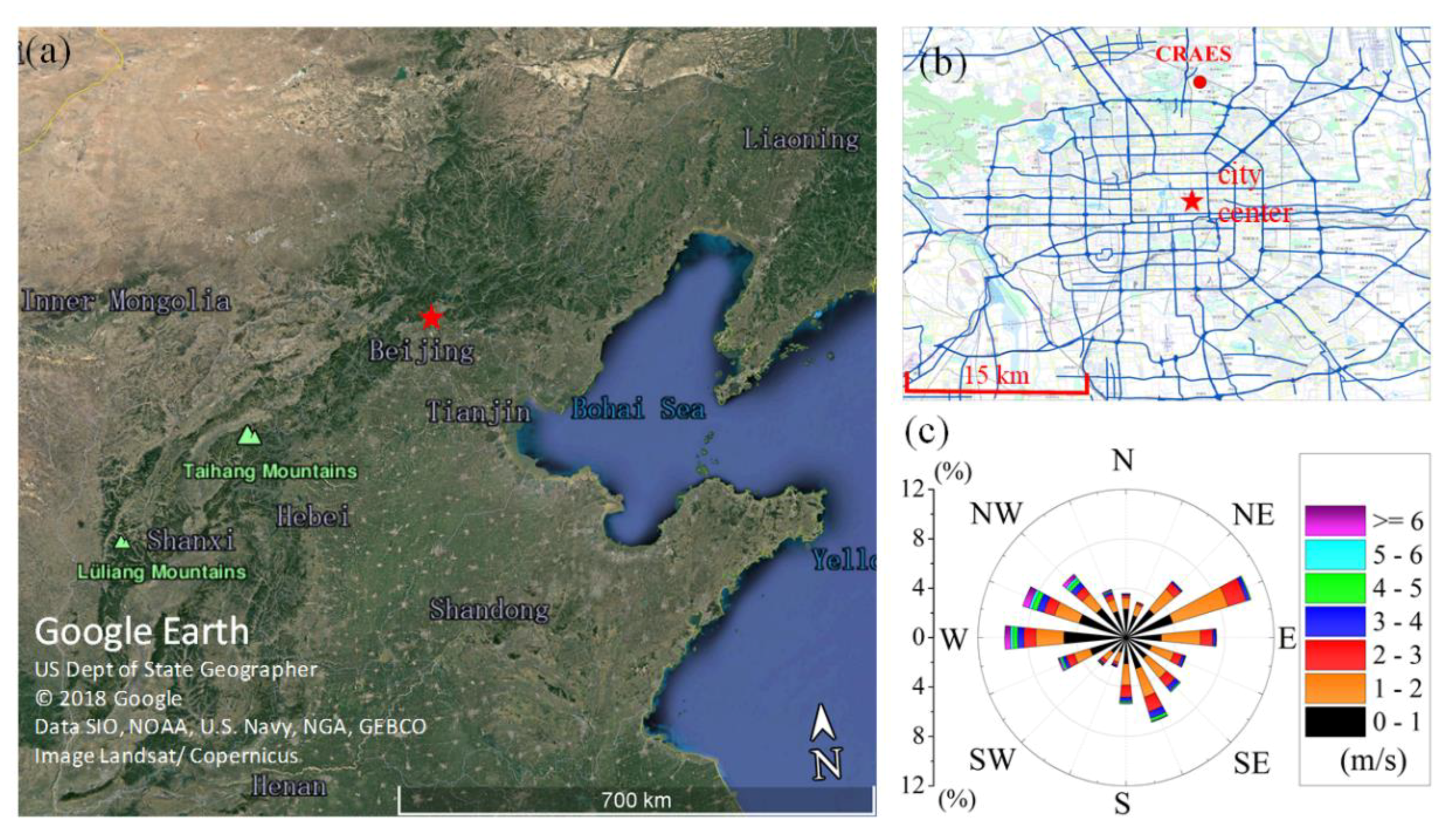
2.1. Measurement Site and Period
2.2. Instruments
2.2.1. Chemical Ionization Mass Spectrometry (CIMS) Setup
2.2.2. Other Measurements
2.3. Estimation of γ(N2O5) and φ(ClNO2)
= k1[NO2][O3] − k(NO3)[NO3] − k(N2O5)[N2O5]
d[NO3]/dt)/[N2O5].
2.4. Parameterizations of γ(N2O5) and φ(ClNO2)
10−8 × RH3) × 10(0.04 × (T − 294)
3. Results and Discussion
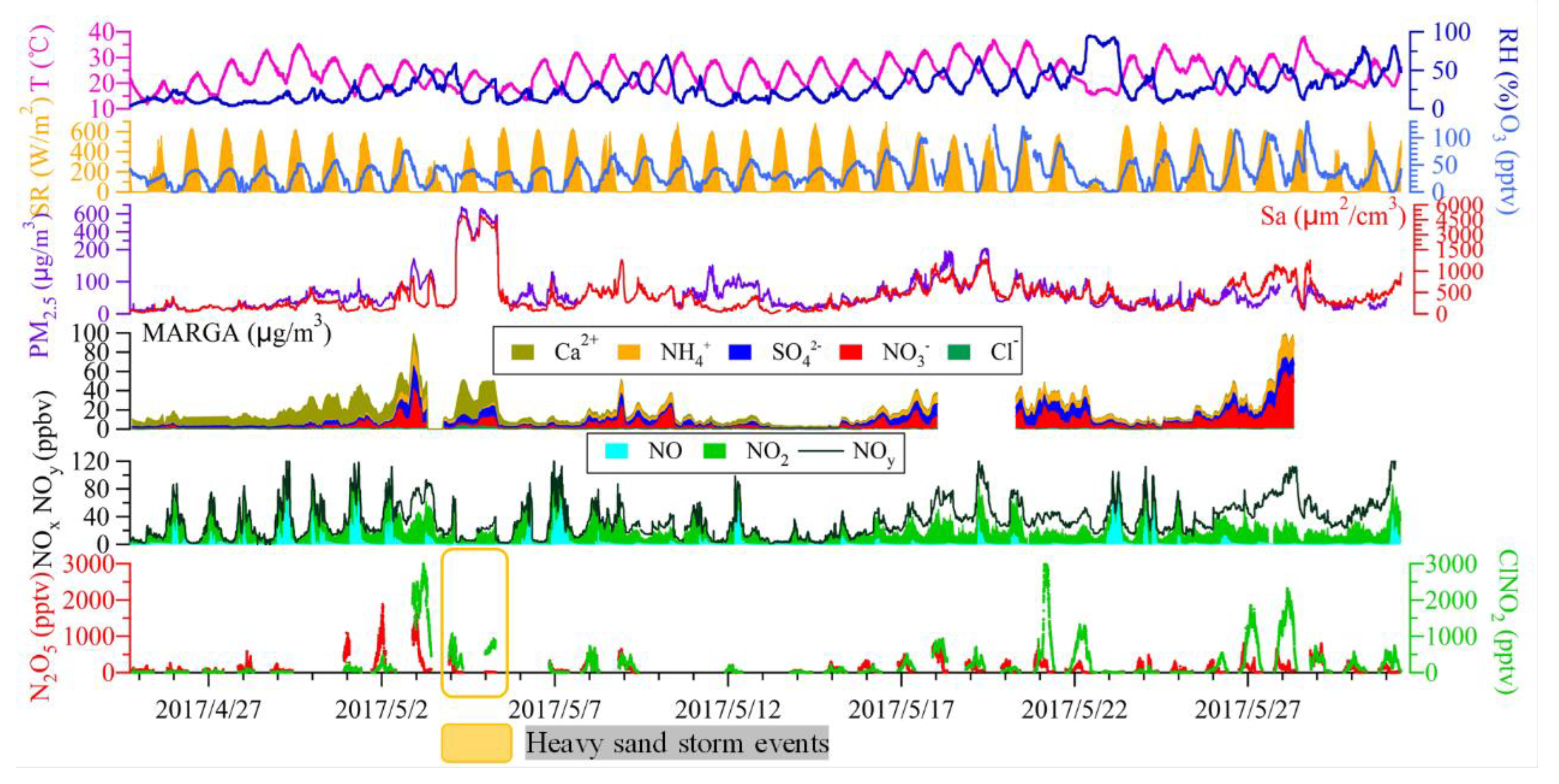
3.1. Overall Observations
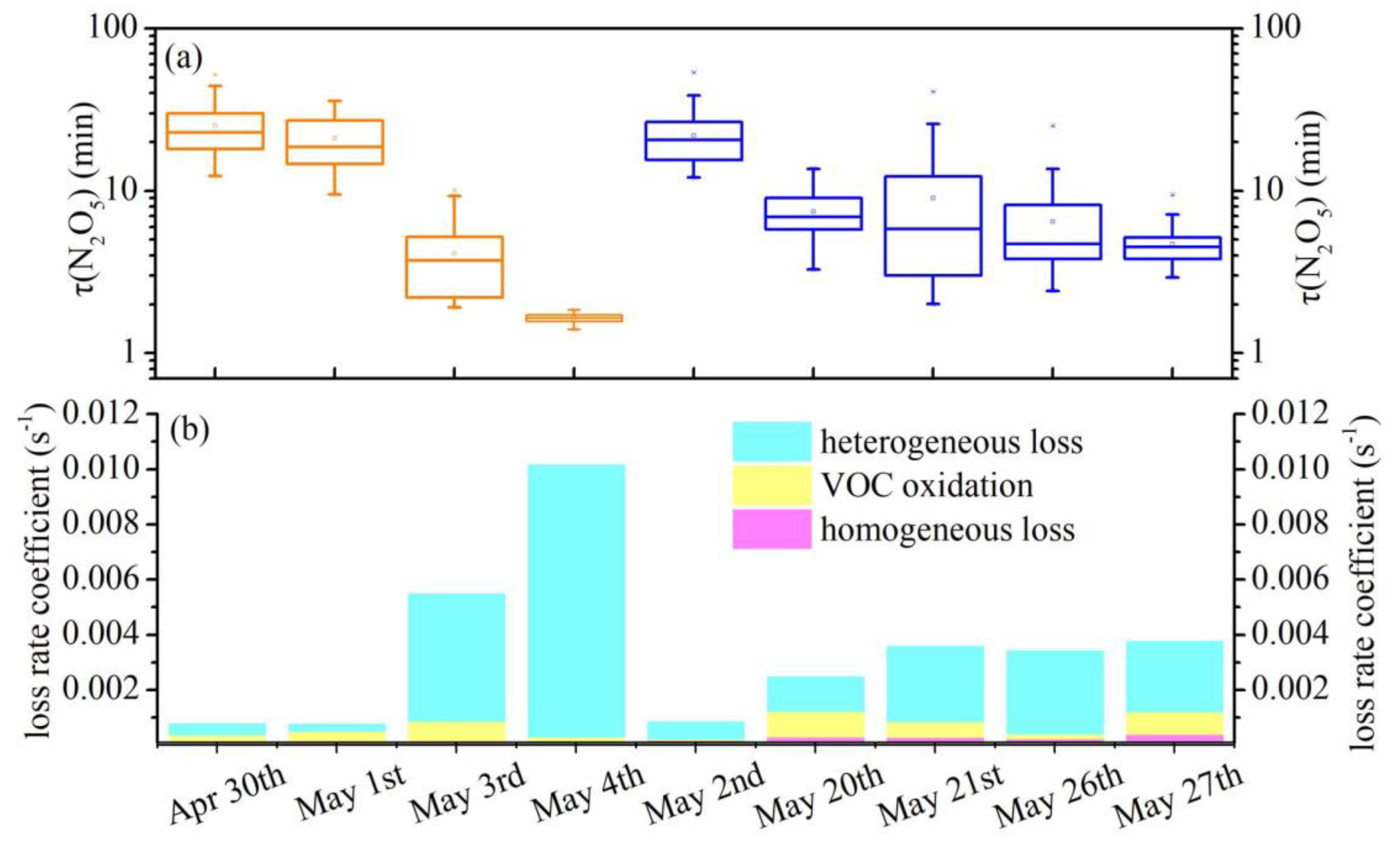
3.2. N2O5 Reactivity and Loss Pathways
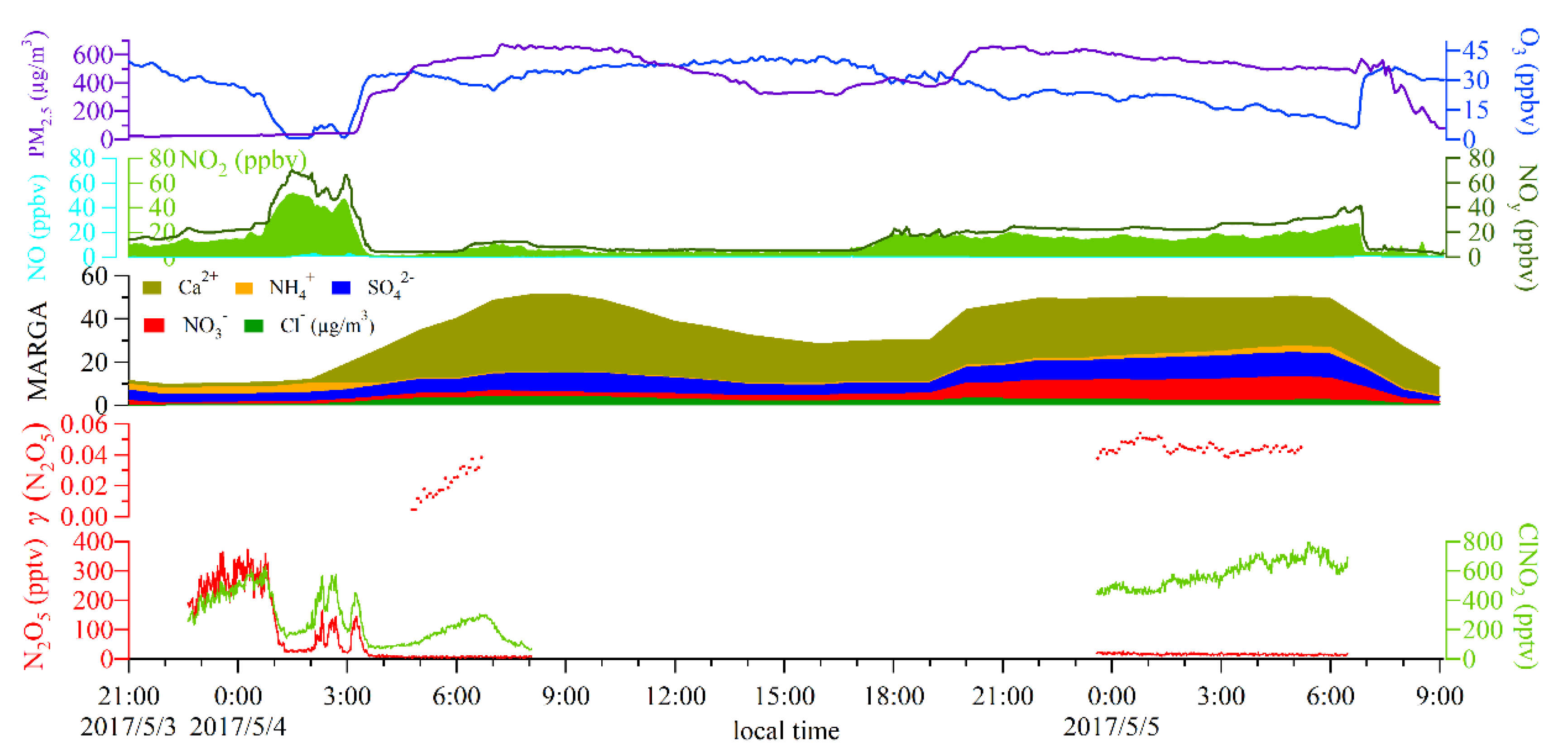
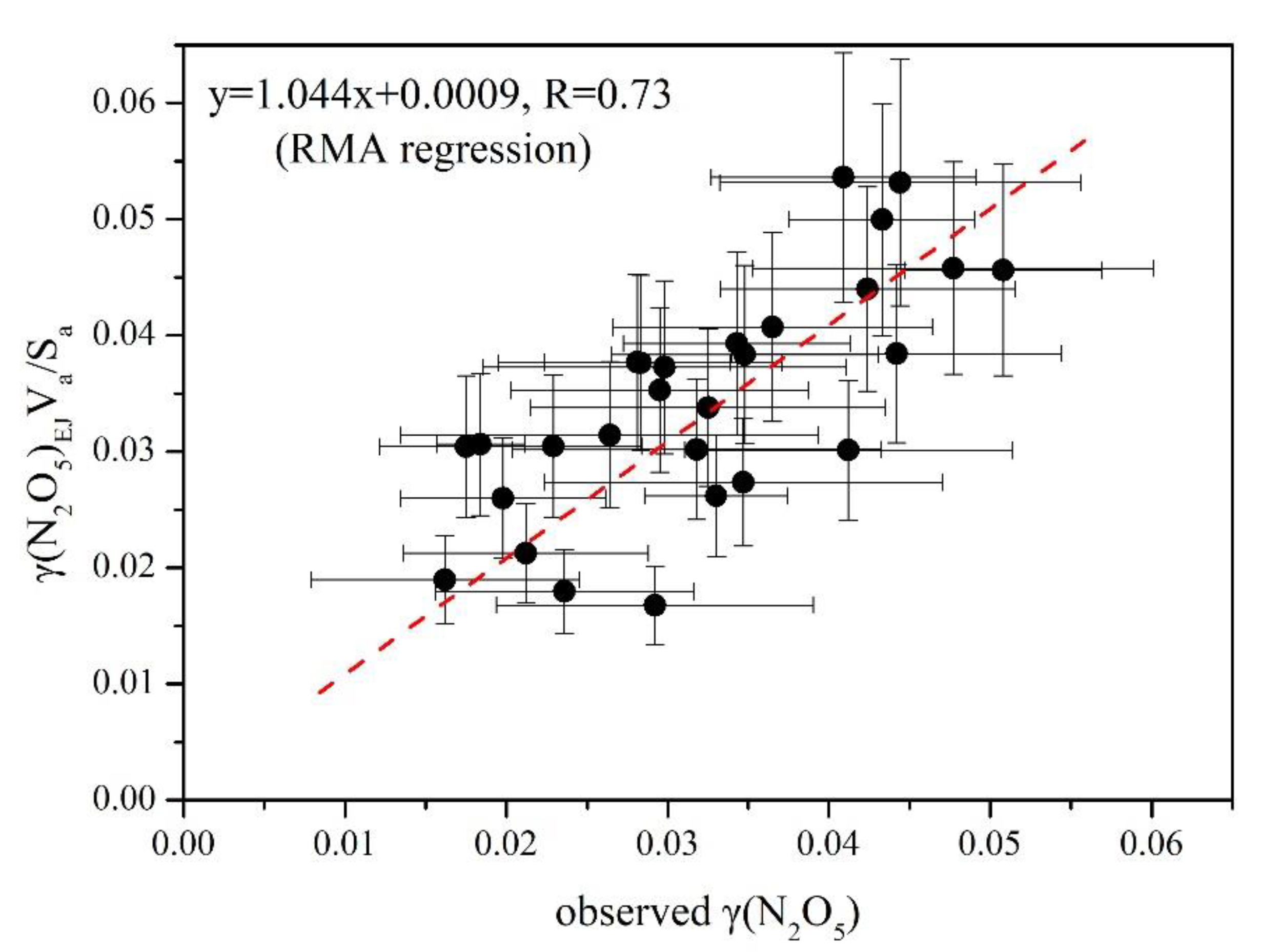
3.3. Derivation of γ(N2O5) and Evaluation of Parameterizations
- The air mass should be stable without dramatic changes in temperature, RH, and wind direction. Wind speed should be less than 3 m/s to minimize the effect of air mass transport.
- Ambient NO should be below 0.1 ppbv. Otherwise, little production of N2O5 would occur.
- ClNO2 should exhibit an increasing trend, indicating considerable uptake of N2O5. In cases with decreasing or fluctuating ClNO2, physical processes or changes of air mass may invalidate the method for the estimation of γ(N2O5).
- Sa should be above 200 μm2/cm3 to facilitate significant N2O5 uptake. In cases with Sa below 200 μm2/cm3, we found that the derived γ(N2O5) was either abnormally high (for example, above 0.1), or even negative. This phenomenon indicates that the method we adopted may be invalid in low aerosol loadings.
3.4. Influencing Factors of γ(N2O5) and Implications
4. Concluding Remark
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Data Availability
References
- Seinfeld, J.H.; Pandis, S.N. Atmospheric Chemistry and Physics: From Air Pollution to Climate Change; John Wiley & Sons: Hoboken, NJ, USA, 2016. [Google Scholar]
- Chang, W.L.; Bhave, P.V.; Brown, S.S.; Riemer, N.; Stutz, J.; Dabdub, D. Heterogeneous atmospheric chemistry, ambient measurements, and model calculations of N2O5: A review. Aerosol Sci. Technol. 2011, 45, 665–695. [Google Scholar] [CrossRef]
- Brown, S.S.; Stark, H.; Ryerson, T.B.; Williams, E.J.; Nicks, D.K.; Trainer, M.; Fehsenfeld, F.C.; Ravishankara, A. Nitrogen oxides in the nocturnal boundary layer: Simultaneous in situ measurements of NO3, N2O5, NO2, NO, and O3. J. Geophys. Res. Atmos. 2003, 108. [Google Scholar] [CrossRef]
- Brown, S.S.; Stutz, J. Nighttime radical observations and chemistry. Chem. Soc. Rev. 2012, 41, 6405–6447. [Google Scholar] [CrossRef]
- Atkinson, R.; Arey, J. Atmospheric degradation of volatile organic compounds. Chem. Rev. 2003, 103, 4605–4638. [Google Scholar] [CrossRef]
- Mentel, T.F.; Bleilebens, D.; Wahner, A. A study of nighttime nitrogen oxide oxidation in a large reaction chamber—The fate of NO2, N2O5, HNO3, and O3 at different humidities. Atmos. Environ. 1996, 30, 4007–4020. [Google Scholar] [CrossRef]
- Finlayson-Pitts, B.; Ezell, M.; Pitts, J. Formation of chemically active chlorine compounds by reactions of atmospheric NaCl particles with gaseous N2O5 and ClONO2. Nature 1989, 337, 241–244. [Google Scholar] [CrossRef]
- Brown, S.; Dibb, J.E.; Stark, H.; Aldener, M.; Vozella, M.; Whitlow, S.; Williams, E.; Lerner, B.M.; Jakoubek, R.; Middlebrook, A. Nighttime removal of NOx in the summer marine boundary layer. Geophys. Res. Lett. 2004, 31. [Google Scholar] [CrossRef]
- Behnke, W.; George, C.; Scheer, V.; Zetzsch, C. Production and decay of ClNO2 from the reaction of gaseous N2O5 with NaCl solution: Bulk and aerosol experiments. J. Geophys. Res. Atmos. 1997, 102, 3795–3804. [Google Scholar] [CrossRef]
- McDuffie, E.E.; Fibiger, D.L.; Dubé, W.P.; Lopez-Hilfiker, F.; Lee, B.H.; Thornton, J.A.; Shah, V.; Jaeglé, L.; Guo, H.; Weber, R.J. Heterogeneous N2O5 uptake during winter: Aircraft measurements during the 2015 WINTER campaign and critical evaluation of current parameterizations. J. Geophys. Res. Atmos. 2018, 123, 4345–4372. [Google Scholar] [CrossRef]
- Tham, Y.J.; Wang, Z.; Li, Q.; Wang, W.; Wang, X.; Lu, K.; Ma, N.; Yan, C.; Kecorius, S.; Wiedensohler, A. Heterogeneous N2O5 uptake coefficient and production yield of ClNO2 in polluted northern China: Roles of aerosol water content and chemical composition. Atmos. Chem. Phys. 2018, 18, 13155–13171. [Google Scholar] [CrossRef]
- Kane, S.M.; Caloz, F.; Leu, M.-T. Heterogeneous uptake of gaseous N2O5 by (NH4) 2SO4, NH4HSO4, and H2SO4 aerosols. J. Phys. Chem. A 2001, 105, 6465–6470. [Google Scholar] [CrossRef]
- Hallquist, M.; Stewart, D.J.; Stephenson, S.K.; Cox, R.A. Hydrolysis of N2O5 on sub-micron sulfate aerosols. Phys. Chem. Chem. Phys. 2003, 5, 3453–3463. [Google Scholar] [CrossRef]
- Bertram, T.; Thornton, J. Toward a general parameterization of N2O5 reactivity on aqueous particles: The competing effects of particle liquid water, nitrate and chloride. Atmos. Chem. Phys. 2009, 9, 8351–8363. [Google Scholar] [CrossRef]
- Roberts, J.M.; Osthoff, H.D.; Brown, S.S.; Ravishankara, A.; Coffman, D.; Quinn, P.; Bates, T. Laboratory studies of products of N2O5 uptake on Cl− containing substrates. Geophys. Res. Lett. 2009, 36. [Google Scholar] [CrossRef]
- Anttila, T.; Kiendler-Scharr, A.; Tillmann, R.; Mentel, T.F. On the reactive uptake of gaseous compounds by organic-coated aqueous aerosols: Theoretical analysis and application to the heterogeneous hydrolysis of N2O5. J. Phys. Chem. A 2006, 110, 10435–10443. [Google Scholar] [CrossRef]
- Escorcia, E.N.; Sjostedt, S.J.; Abbatt, J.P. Kinetics of N2O5 Hydrolysis on Secondary Organic Aerosol and Mixed Ammonium Bisulfate− Secondary Organic Aerosol Particles. J. Phys. Chem. A 2010, 114, 13113–13121. [Google Scholar] [CrossRef] [PubMed]
- Mogili, P.K.; Kleiber, P.D.; Young, M.A.; Grassian, V.H. N2O5 hydrolysis on the components of mineral dust and sea salt aerosol: Comparison study in an environmental aerosol reaction chamber. Atmos. Environ. 2006, 40, 7401–7408. [Google Scholar] [CrossRef]
- Tang, M.; Telford, P.; Pope, F.; Rkiouak, L.; Abraham, N.; Archibald, A.; Braesicke, P.; Pyle, J.; McGregor, J.; Watson, I. Heterogeneous reaction of N2O5 with airborne TiO2 particles and its implication for stratospheric particle injection. Atmos. Chem. Phys. 2014, 14, 6035–6048. [Google Scholar] [CrossRef]
- Karagulian, F.; Santschi, C.; Rossi, M. The heterogeneous chemical kinetics of N2O5 on CaCO3 and other atmospheric mineral dust surrogates. Atmos. Chem. Phys. 2006, 6, 1373–1388. [Google Scholar] [CrossRef]
- Wagner, C.; Hanisch, F.; Holmes, N.; Coninck, H.d.; Schuster, G.; Crowley, J. The interaction of N2O5 with mineral dust: Aerosol flow tube and Knudsen reactor studies. Atmos. Chem. Phys. 2008, 8, 91–109. [Google Scholar] [CrossRef]
- Wagner, C.; Schuster, G.; Crowley, J. An aerosol flow tube study of the interaction of N2O5 with calcite, Arizona dust and quartz. Atmos. Environ. 2009, 43, 5001–5008. [Google Scholar] [CrossRef]
- Seisel, S.; Börensen, C.; Vogt, R.; Zellner, R. Kinetics and mechanism of the uptake of N2O5 on mineral dust at 298 K. Atmos. Chem. Phys. 2005, 5, 3423–3432. [Google Scholar] [CrossRef]
- Tang, M.; Thieser, J.; Schuster, G.; Crowley, J. Kinetics and mechanism of the heterogeneous reaction of N2O5 with mineral dust particles. Phys. Chem. Chem. Phys. 2012, 14, 8551–8561. [Google Scholar] [CrossRef] [PubMed]
- Evans, M.; Jacob, D.J. Impact of new laboratory studies of N2O5 hydrolysis on global model budgets of tropospheric nitrogen oxides, ozone, and OH. Geophys. Res. Lett. 2005, 32. [Google Scholar] [CrossRef]
- Davis, J.M.; Bhave, P.V.; Foley, K.M. Parameterization of N2O5 reaction probabilities on the surface of particles containing ammonium, sulfate, and nitrate. Atmos. Chem. Phys. 2008, 8, 5295–5311. [Google Scholar] [CrossRef]
- Brown, S.; Ryerson, T.; Wollny, A.; Brock, C.; Peltier, R.; Sullivan, A.; Weber, R.; Dube, W.; Trainer, M.; Meagher, J. Variability in nocturnal nitrogen oxide processing and its role in regional air quality. Science 2006, 311, 67–70. [Google Scholar] [CrossRef] [PubMed]
- Bertram, T.H.; Thornton, J.A.; Riedel, T.P.; Middlebrook, A.M.; Bahreini, R.; Bates, T.S.; Quinn, P.K.; Coffman, D.J. Direct observations of N2O5 reactivity on ambient aerosol particles. Geophys. Res. Lett. 2009, 36. [Google Scholar] [CrossRef]
- Phillips, G.J.; Thieser, J.; Tang, M.; Sobanski, N.; Schuster, G.; Fachinger, J.; Drewnick, F.; Borrmann, S.; Bingemer, H.; Lelieveld, J. Estimating N2O5 uptake coefficients using ambient measurements of NO3, N2O5, ClNO2 and particle-phase nitrate. Atmos. Chem. Phys. 2016, 16, 13231–13249. [Google Scholar] [CrossRef]
- Wagner, N.; Riedel, T.; Young, C.; Bahreini, R.; Brock, C.; Dubé, W.; Kim, S.; Middlebrook, A.; Öztürk, F.; Roberts, J. N2O5 uptake coefficients and nocturnal NO2 removal rates determined from ambient wintertime measurements. J. Geophys. Res. Atmos. 2013, 118, 9331–9350. [Google Scholar] [CrossRef]
- Morgan, W.; Ouyang, B.; Allan, J.; Aruffo, E.; Di Carlo, P.; Kennedy, O.; Lowe, D.; Flynn, M.; Rosenberg, P.; Williams, P. Influence of aerosol chemical composition on N2O5 uptake: Airborne regional measurements in northwestern Europe. Atmos. Chem. Phys. 2015, 15, 973–990. [Google Scholar] [CrossRef]
- Wang, Z.; Wang, W.; Tham, Y.J.; Li, Q.; Wang, H.; Wen, L.; Wang, X.; Wang, T. Fast heterogeneous N2O5 uptake and ClNO2 production in power plant plumes observed in the nocturnal residual layer over the North China Plain. Atmos. Chem. Phys. 2017, 17, 12361–12378. [Google Scholar] [CrossRef]
- Riedel, T.; Bertram, T.; Ryder, O.; Liu, S.; Day, D.; Russell, L.; Gaston, C.; Prather, K.; Thornton, J. Direct N2O5 reactivity measurements at a polluted coastal site. Atmos. Chem. Phys. 2012, 12, 2959–2968. [Google Scholar] [CrossRef]
- Wang, T.; Nie, W.; Gao, J.; Xue, L.K.; Gao, X.M.; Wang, X.F.; Qiu, J.; Poon, C.N.; Meinardi, S.; Blake, D.; et al. Air quality during the 2008 Beijing Olympics: Secondary pollutants and regional impact. Atmos. Chem. Phys. 2010, 10, 7603–7615. [Google Scholar] [CrossRef]
- Gao, J.; Zhang, Y.; Zhang, M.; Zhang, J.; Wang, S.; Tao, J.; Wang, H.; Luo, D.; Chai, F.; Ren, C. Photochemical properties and source of pollutants during continuous pollution episodes in Beijing, October, 2011. J. Environ. Sci. 2014, 26, 44–53. [Google Scholar] [CrossRef]
- Sun, Y.; Zhuang, G.; Wang, Y.; Zhao, X.; Li, J.; Wang, Z.; An, Z. Chemical composition of dust storms in Beijing and implications for the mixing of mineral aerosol with pollution aerosol on the pathway. J. Geophys. Res. Atmos. 2005, 110. [Google Scholar] [CrossRef] [Green Version]
- Wang, T.; Tham, Y.J.; Xue, L.; Li, Q.; Zha, Q.; Wang, Z.; Poon, S.C.; Dubé, W.P.; Blake, D.R.; Louie, P.K. Observations of nitryl chloride and modeling its source and effect on ozone in the planetary boundary layer of southern China. J. Geophys. Res. Atmos. 2016, 121, 2476–2489. [Google Scholar] [CrossRef]
- Tham, Y.J.; Wang, Z.; Li, Q.; Yun, H.; Wang, W.; Wang, X.; Xue, L.; Lu, K.; Ma, N.; Bohn, B.; et al. Significant concentrations of nitryl chloride sustained in the morning: Investigations of the causes and impacts on ozone production in a polluted region of northern China. Atmos. Chem. Phys. 2016, 16, 14959–14977. [Google Scholar] [CrossRef]
- Yun, H.; Wang, W.; Wang, T.; Xia, M.; Yu, C.; Wang, Z.; Poon, S.C.N.; Yue, D.; Zhou, Y. Nitrate formation from heterogeneous uptake of dinitrogen pentoxide during a severe winter haze in southern China. Atmos. Chem. Phys. 2018, 2018, 23. [Google Scholar] [CrossRef]
- Jongejan, P.; Bai, Y.; Veltkamp, A.; Wye, G.; Slaninaa, J. An automated field instrument for the determination of acidic gases in air. Int. J. Environ. Anal. Chem. 1997, 66, 241–251. [Google Scholar] [CrossRef]
- Rumsey, I.; Cowen, K.; Walker, J.; Kelly, T.; Hanft, E.; Mishoe, K.; Rogers, C.; Proost, R.; Beachley, G.; Lear, G. An assessment of the performance of the Monitor for AeRosols and GAses in ambient air (MARGA): A semi-continuous method for soluble compounds. Atmos. Chem. Phys. 2014, 14, 5639–5658. [Google Scholar] [CrossRef]
- Zhang, H.; Li, H.; Zhang, Q.; Zhang, Y.; Zhang, W.; Wang, X.; Bi, F.; Chai, F.; Gao, J.; Meng, L. Atmospheric Volatile Organic Compounds in a Typical Urban Area of Beijing: Pollution Characterization, Health Risk Assessment and Source Apportionment. Atmosphere 2017, 8, 61. [Google Scholar] [CrossRef]
- Gao, J.; Wang, T.; Zhou, X.; Wu, W.; Wang, W. Measurement of aerosol number size distributions in the Yangtze River delta in China: Formation and growth of particles under polluted conditions. Atmos. Environ. 2009, 43, 829–836. [Google Scholar] [CrossRef]
- Wang, X.; Wang, H.; Xue, L.; Wang, T.; Wang, L.; Gu, R.; Wang, W.; Tham, Y.J.; Wang, Z.; Yang, L.; et al. Observations of N2O5 and ClNO2 at a polluted urban surface site in North China: High N2O5 uptake coefficients and low ClNO2 product yields. Atmos. Environ. 2017, 156, 125–134. [Google Scholar] [CrossRef]
- Lewis, E.R. An examination of Köhler theory resulting in an accurate expression for the equilibrium radius ratio of a hygroscopic aerosol particle valid up to and including relative humidity 100%. J. Geophys. Res. Atmos. 2008, 113. [Google Scholar] [CrossRef] [Green Version]
- Kanaya, Y.; Cao, R.; Akimoto, H.; Fukuda, M.; Komazaki, Y.; Yokouchi, Y.; Koike, M.; Tanimoto, H.; Takegawa, N.; Kondo, Y. Urban photochemistry in central Tokyo: 1. Observed and modeled OH and HO2 radical concentrations during the winter and summer of 2004. J. Geophys. Res. Atmos. 2007, 112. [Google Scholar] [CrossRef]
- McLaren, R.; Wojtal, P.; Majonis, D.; McCourt, J.; Halla, J.; Brook, J. NO3 radical measurements in a polluted marine environment: Links to ozone formation. Atmos. Chem. Phys. 2010, 10, 4187–4206. [Google Scholar] [CrossRef]
- Wahner, A.; Mentel, T.F.; Sohn, M. Gas-phase reaction of N2O5 with water vapor: Importance of heterogeneous hydrolysis of N2O5 and surface desorption of HNO3 in a large Teflon chamber. Geophys. Res. Lett. 1998, 25, 2169–2172. [Google Scholar] [CrossRef]
- Wexler, A.S. Atmospheric aerosol models for systems including the ions H+, NH4+, Na+, SO42−, NO3−, Cl−, Br−, and H2O. J. Geophys. Res. 2002, 107. [Google Scholar] [CrossRef]
- Riemer, N.; Vogel, H.; Vogel, B.; Anttila, T.; Kiendler-Scharr, A.; Mentel, T. Relative importance of organic coatings for the heterogeneous hydrolysis of N2O5 during summer in Europe. J. Geophys. Res. Atmos. 2009, 114. [Google Scholar] [CrossRef]
- Draxler, R.R.; Hess, G. An overview of the HYSPLIT_4 modelling system for trajectories. Aust. Meteorol. Mag. 1998, 47, 295–308. [Google Scholar]
- Osthoff, H.D.; Roberts, J.M.; Ravishankara, A.R.; Williams, E.J.; Lerner, B.M.; Sommariva, R.; Bates, T.S.; Coffman, D.; Quinn, P.K.; Dibb, J.E.; et al. High levels of nitryl chloride in the polluted subtropical marine boundary layer. Nat. Geosci. 2008, 1, 324–328. [Google Scholar] [CrossRef]
- Thornton, J.A.; Kercher, J.P.; Riedel, T.P.; Wagner, N.L.; Cozic, J.; Holloway, J.S.; Dube, W.P.; Wolfe, G.M.; Quinn, P.K.; Middlebrook, A.M.; et al. A large atomic chlorine source inferred from mid-continental reactive nitrogen chemistry. Nature 2010, 464, 271–274. [Google Scholar] [CrossRef]
- Mielke, L.H.; Furgeson, A.; Osthoff, H.D. Observation of ClNO2 in a mid-continental urban environment. Environ. Sci. Technol. 2011, 45, 8889–8896. [Google Scholar] [CrossRef]
- Wang, H.; Lu, K.; Chen, X.; Zhu, Q.; Chen, Q.; Guo, S.; Jiang, M.; Li, X.; Shang, D.; Tan, Z. High N2O5 Concentrations Observed in Urban Beijing: Implications of a Large Nitrate Formation Pathway. Environ. Sci. Technol. Lett. 2017, 4, 416–420. [Google Scholar] [CrossRef]
- Breton, M.L.; Hallquist, Å.M.; Pathak, R.K.; Simpson, D.; Wang, Y.; Johansson, J.; Zheng, J.; Yang, Y.; Shang, D.; Wang, H. Chlorine oxidation of VOCs at a semi-rural site in Beijing: Significant chlorine liberation from ClNO2 and subsequent gas-and particle-phase Cl–VOC production. Atmos. Chem. Phys. 2018, 18, 13013–13030. [Google Scholar] [CrossRef]
- Wang, H.; Lu, K.; Guo, S.; Wu, Z.; Shang, D.; Tan, Z.; Wang, Y.; Breton, M.L.; Lou, S.; Tang, M. Efficient N2O5 uptake and NO3 oxidation in the outflow of urban Beijing. Atmos. Chem. Phys. 2018, 18, 9705–9721. [Google Scholar] [CrossRef]
- Zhou, W.; Zhao, J.; Ouyang, B.; Mehra, A.; Xu, W.; Wang, Y.; Bannan, T.J.; Worrall, S.D.; Priestley, M.; Bacak, A. Production of N2O5 and ClNO2 in summer in urban Beijing, China. Atmos. Chem. Phys. 2018, 18, 11581–11597. [Google Scholar] [CrossRef]
- Brown, S.S.; Dubé, W.P.; Fuchs, H.; Ryerson, T.B.; Wollny, A.G.; Brock, C.A.; Bahreini, R.; Middlebrook, A.M.; Neuman, J.A.; Atlas, E. Reactive uptake coefficients for N2O5 determined from aircraft measurements during the Second Texas Air Quality Study: Comparison to current model parameterizations. J. Geophys. Res. Atmos. 2009, 114. [Google Scholar] [CrossRef]
- Stone, D.; Evans, M.; Walker, H.; Ingham, T.; Vaughan, S.; Ouyang, B.; Kennedy, O.; McLeod, M.; Jones, R.; Hopkins, J. Radical chemistry at night: Comparisons between observed and modelled HOx, NO3 and N2O5 during the RONOCO project. Atmos. Chem. Phys. 2014, 14, 1299–1321. [Google Scholar] [CrossRef]
- Tang, M.; Thieser, J.; Schuster, G.; Crowley, J. Uptake of NO3 and N2O5 to Saharan dust, ambient urban aerosol and soot: A relative rate study. Atmos. Chem. Phys. 2010, 10, 2965–2974. [Google Scholar] [CrossRef]
- McDuffie, E.E.; Fibiger, D.L.; Dubé, W.P.; Lopez Hilfiker, F.; Lee, B.H.; Jaeglé, L.; Guo, H.; Weber, R.J.; Reeves, J.M.; Weinheimer, A.J. ClNO2 yields from aircraft measurements during the 2015 WINTER campaign and critical evaluation of the current parameterization. J. Geophys. Res. Atmos. 2018, 123, 12994–13015. [Google Scholar]
- Tang, M.; Cziczo, D.J.; Grassian, V.H. Interactions of water with mineral dust aerosol: Water adsorption, hygroscopicity, cloud condensation, and ice nucleation. Chem. Rev. 2016, 116, 4205–4259. [Google Scholar] [CrossRef]
- Tang, I.N. On the equilibrium partial pressures of nitric acid and ammonia in the atmosphere. Atmos. Environ. (1967) 1980, 14, 819–828. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Category | Periods | γ(N2O5) | φ(ClNO2) | Notes | |
|---|---|---|---|---|---|
| From | To | ||||
| Urban air masses | 16th May 23:00 | 17th May 04:00 | 0.022 ± 0.005 | 0.065 | |
| 17th May 21:00 | 18th May 01:00 | 0.013 ± 0.006 | 0.048 | ||
| 18th May 21:00 | 19th May 04:00 | 0.030 ± 0.009 | 0.055 (21:26~22:41) | ClNO2 yield changed | |
| 0.117 (01:06~03:31) | |||||
| 20th May 21:00 | 21st May 04:00 | 0.032 ± 0.007 | 0.082 | ||
| 22nd May 0:00 | 22nd May 04:00 | 0.035 ± 0.010 | 0.312 | ||
| 27th May 20:00 | 28th May 04:00 | 0.042 ± 0.008 | 0.084 (20:01~21:36) | ClNO2 yield changed | |
| 0.319 (01:36~03:01) | |||||
| 28th May 20:00 | 28th May 23:00 | 0.023 ± 0.007 | 0.142 | ||
| Heavy sand storm events | 4th May 04:00 | 4th May 06:00 | 0.019 ± 0.012 | 0.677 | Sand storm arrived |
| 4th May 23:00 | 5th May 05:00 | 0.044 ± 0.002 | 0.129 | Sand storm continued | |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xia, M.; Wang, W.; Wang, Z.; Gao, J.; Li, H.; Liang, Y.; Yu, C.; Zhang, Y.; Wang, P.; Zhang, Y.; et al. Heterogeneous Uptake of N2O5 in Sand Dust and Urban Aerosols Observed during the Dry Season in Beijing. Atmosphere 2019, 10, 204. https://doi.org/10.3390/atmos10040204
Xia M, Wang W, Wang Z, Gao J, Li H, Liang Y, Yu C, Zhang Y, Wang P, Zhang Y, et al. Heterogeneous Uptake of N2O5 in Sand Dust and Urban Aerosols Observed during the Dry Season in Beijing. Atmosphere. 2019; 10(4):204. https://doi.org/10.3390/atmos10040204
Chicago/Turabian StyleXia, Men, Weihao Wang, Zhe Wang, Jian Gao, Hong Li, Yutong Liang, Chuan Yu, Yuechong Zhang, Peng Wang, Yujie Zhang, and et al. 2019. "Heterogeneous Uptake of N2O5 in Sand Dust and Urban Aerosols Observed during the Dry Season in Beijing" Atmosphere 10, no. 4: 204. https://doi.org/10.3390/atmos10040204
APA StyleXia, M., Wang, W., Wang, Z., Gao, J., Li, H., Liang, Y., Yu, C., Zhang, Y., Wang, P., Zhang, Y., Bi, F., Cheng, X., & Wang, T. (2019). Heterogeneous Uptake of N2O5 in Sand Dust and Urban Aerosols Observed during the Dry Season in Beijing. Atmosphere, 10(4), 204. https://doi.org/10.3390/atmos10040204