1. Introduction
The equation of state (EOS) concept relating force to generic density is central to all of material science, from microscopic to galactic scales [
1]. This statement includes—perhaps most crucially—fluid media of the unique Earth system [
2]. In the present work, we build a case for exploring certain new EOS that may be relevant to global climate—those constituted by detrital biopolymers occupying the sea surface microlayer. Interfacially active macromolecules reside ubiquitously along the atmospheric interface [
3], establishing measurable planar densities and exerting tangential forces regulatory of energy-mass transfer across the full planetary surface [
4,
5,
6]. Such effects occur primarily via a single monolayer of organic carbon, distributed unevenly yet systematically by the food web. This ultra-thin filter alters small eddy energies, the spectrum of friction elements, film behaviors during gas diffusion or bubble-burst salt injection, and more. Local influence of the organics has been a source of pure wonder for millennia, and the subject was often studied quite directly during the last half century of oceanographic research [
6,
7,
8,
9]. Here, we show that a combination of tools from surfactant physical chemistry plus contemporary systems modeling [
4,
10] allows us to probe unexpected couplings due to this tenuous but pervasive global medium. In fact, we are able to suggest a novel set of geophysical connections regarding exchange across fluid dynamic boundary layers. Estimates of altered climate-flux strengths and distributions are constructed in a coarse but world-wide fashion. Indications are that through food web processing of the dissolved organics, selected biosurfactants control planetary flows of momentum, heat (whether sensible or latent), and several types of biogeochemical tracer mass. The patterns have previously gone unreported and unrepresented, within models or otherwise.
Our development proceeds in the following order. Fundamentals of physical surfactant chemistry are reviewed first, because they will be somewhat unfamiliar to those trained in the Earth system sciences. A slightly earlier and heavily chemically-oriented publication is referred to several times, providing more of the molecular-level detail [
6]. Notorious micrometeorological flux complexities are next distilled down for the reader as a practical matter, to a simple neutral stability case allowing for demonstration. We thus restrict ourselves somewhat for pedagogical purposes, but the geographic applicability is broad in any case and results point immediately to several types of energy-mass flux [
2]. Roughness length is the key quantity, as always. Here however, we take the step of linking it to local intermolecular forces operating in a conceptual organic tangent plane [
4,
5,
6,
7,
8]. This allows for a global, systems-level biogeographic distribution to be proposed for surfactant properties and influence [
10]. Extensions to laminar layer transfer barriers and bubble film disruption are made empirically working from laboratory data [
5,
11], though the micro-fluid dynamic mechanisms involved are closely related.
At this stage, we introduce formally and rigorously the concept of a two dimensional (2D) or interfacial EOS. Force-to-surface concentration (density) relationships can thereby be parameterized conveniently and globally for the first time. The transition is accomplished based on a handful of carefully selected surrogate compounds drawn from the fresh protein and lipid families (among the wide diversity of total dissolved organics [
3,
6,
12]). Individual macromolecular carbon distributions are then estimated in the dissolved state from a blend of primitive to dynamic systems modeling methods [
3,
10,
13]. Local values are fed into the proposed global EOS to generate 2D pressure fields (tension reductions as defined by the surface chemist [
4,
5,
6]), and these values are themselves handed back into the flux parameterizations with roughness acting as the intermediary. The cycle is thus completed both computationally and conceptually. Biological pressure exerted by planetary ecodynamics, and leading to chemical definition within the organic microlayer, has the net effect of forcing of sea-air flows and interfacial mixing.
Final results are placed on a crude but complete and illustrative biogeographic grid, consistent with ocean ecogeographic principles published late in the last century [
6,
10]. Ultimately, we analyze outcomes from the exercise in multi-tabular and then schematic forms relative to large agglomerate biosystems, in order to enhance accessibility for the early results. We are able to demonstrate a likelihood that biological surfactant controls are significant over many areas of the sea—from the province to the basin scale and perhaps beyond. We end with an obvious but speculative point that deserves closer attention during the era of global change. In the early interpretation built up here, any and all marine bio-modulation occurs through global scale maintenance of just one microscopically thin material layer. Technically, the arguments imply a total vertical extent of about one carbon-carbon bond. Flux leveraging by marine life must therefore be characterized as supremely efficient, if it is real. Sensitivities and uncertainties are listed and evaluated across the board in the latter half of the text, and they are many [
3,
6]. Hence although it may seem promising that this first cut at the problem yields strong alterations to climate transfer, well-coordinated laboratory and field experiments will definitely be required to elucidate.
The material we now develop is highly interdisciplinary in nature, running the gamut from surface physicochemistry through upper ocean biology to micrometeorology and beyond -as reflected, for instance, in the keyword list just below our abstract. We endeavor to equip a diverse audience with sufficient preliminary information entering into all individual sections. But further, an
Appendix A is included within which most symbols and specialized terms are defined explicitly. Some readers may find it helpful to scan these end notes before proceeding.
2. Background
Our preferred source regarding surfactant physical chemistry is the 1960s era textbook, Davies and Rideal [
4], which places the interaction of organics with interfacial mixing in an engineering context. However, the information translates readily to natural water systems. We suggest the reader supplement with Reichl [
1] on statistical physics to appreciate the generality of equation of state concepts, both within and between bulk media. The mechanisms by which natural organic monolayers operate through physical chemistry to suppress transfer constitute an extremely complex example of microfluidics. Elliott et al. [
6] review the more accessible treatments and interpret heuristically. A well-established geophysical alternative is offered in Frew [
5], a very thorough monograph chapter discussing surfactant effects not only on ripples, but also relative to laminar layer barriers with their variable thicknesses. Lewis and Schwartz [
14] characterize the stabilization of bubble films by coatings relative to drainage during foam residence time. The process of marginal regeneration is involved and reduces atmospheric salt input.
Most readers will be aware that surface tension is defined as the energy required to form new interface between bulk media, for example, in units of J/m2 (joules per area), but more conveniently, mJ/m2. A reference value to keep in mind is 72 for pure liquid water. However, the tension may be thought of in terms of net forces required per unit distance along the tangent, and can thus be expressed alternatively in mN/m (1 joule = 1 newton meter). Multiple mechanisms are in play as water molecules buffet one another and their hydrogen bonding network morphs and stretches. For a given organic surfactant, a balance is attained between the loss of solvent attractions and the somewhat weaker Van der Waals pull among carbon chains penetrating above the dividing plane, plus a boost to area formation due to the Coulombic repulsion of charged, embedded head groups.
Electrostatic assistance to surface formation is a partial analog of three-dimensional pressure, as it tends to increase area as the analog to volume. But net force differences are often reflected in a lowering of tension. Absolute value for the change is denoted the “surface pressure” and given the symbol π. It figures prominently in much of the oceanographic surfactant literature, and we adopt the convention here. Alterations to π per unit fractional area constitute a local (logarithmic) derivative termed the elasticity or modulus ε. Eddies approaching the ocean interface are affected where π differs on their upward and downward arms per ε. This may occur because tangent macromolecules are shifted together into troughs, increasing their effective 2D concentration (coverage) Γ—another important variable that we carry in units of mg/m
2. The Γ is just surfactant density, and its reciprocal is an average area—the 2D thermochemical counterpart of volume—here denoted as A. The concept of tangent pressure is a tricky one, and it can sometimes seem counterintuitive. We recommend referring to diagrams in Davies and Rideal [
4] as updated by Frew [
5] then summarized in Elliott et al. [
6] if more detail is needed.
The height and number of oceanic capillary waves may be reduced by surfactant forces reflected in π and ε, as represented in our quick review. Such transformations occur regularly across the planet during turbulence–boundary interaction. One can think of energy withdrawal into the ubiquitous organic macromolecular system leading to suppressed penetration of the border by millimeter scale eddy-objects. The thicknesses of laminar layer barriers to diffusion are susceptible as well. Bubble microfilms stabilized by the surfactants tend to lose their saline interiors if they are preserved through marginal regeneration, supported by the presence of the biomacromolecules. Because the point is so crucial we reiterate that the interactions described are enormously intricate at a fluid dynamic level, but heuristic reviews are at hand in [
4,
5,
6] and elsewhere. At the same time, the effects are very real; see [
6,
7,
8,
9] in the bibliography and citations therein.
Observations of environmental slicking are extremely well documented within and outside of the oceanographic community, dating back many centuries—in fact, into classical history [
6,
9]. Some familiar names that will be encountered are Aristotle, Plutarch, Franklin, and Rayleigh. Early true pioneers of the concepts treated so briefly here include Gibbs and the impressive amateur (female) scientist, Agnes Pockels. Her work was built upon in the early twentieth century by Langmuir, resulting in a Nobel Prize. A key outcome well known to these historical figures and the international shipping industry (but most often neglected by climate scientists) is that detailed chemical structure of the spread organics matters tremendously. It follows that the same may be true of 2D organic mixing state if one thinks in terms of tangent solutions. Surfactants may be classified by their π-A behavior as planar “solids, liquids, and gases” in the obvious analogy to P-V in three dimensions [
1,
4]. Archetypal examples include (1) the fatty acids which are mainly lipidic and anchored by negatively charged head groups (s) but note that double bonding within the hydrocarbon tail dramatically softens the π versus A profile, and (2) proteins with their amino acid monomeric skeletons and nonpolar R group arms protruding into the atmosphere above (l). Extreme combinations of the liquid properties give gas-like π(A) relationships that are quite hyperbolic. For more information on the useful interface-to-bulk parallels, see the series [
1,
4,
5,
6,
7,
8].
Contributions we hope to make in the text below flow naturally from the development in this section. We are about to parameterize the global surfactant influence of detrital macromolecules via carefully chosen 2D s,l,g (solid, liquid, gas) representatives that mimic behaviors reported for the open sea. This leads in turn to the prospect for estimation of potential climate flux shifts. Both potential strengths and biogeographic patterns can be sketched in broad outline.
3. Micrometeorology with Chemistry
Since our focus must necessarily fall on surfactant issues, we take the liberty of performing an extreme distillation (truncation) of micrometeorology over the open ocean. This could be considered dangerous, of course, because the field is so vast. However, it is necessary for purposes of dealing with an interdisciplinary audience. We closely follow early forms of Prandtl mixing length theory [
15,
16], stopping short of introducing atmospheric buoyancy through Monin Obukhov similarity. All arguments are therefore strictly limited to neutral conditions, but these are nonetheless useful because they are familiar, easily comprehended, and ubiquitous. Following ample precedent, it is further assumed that influences upon the coefficients for transfer of momentum, heat, and water vapor will be closely related to one another [
2,
17]. Thus it is only necessary to comment on the biogeochemistry of drag at this stage. In fact, the simplifications we adopt are often followed in modern wind stress studies [
17]. While published accounts attribute flux variability to a network of causes including anemometer difficulties, gustiness, directionality, gravity waves, and other purely physical factors, the chemistry option is usually not considered. Effectively, we claim here that momentum stress, friction velocity, and roughness length over the sea may be related not only to one another but also—in a systematic manner—to the underlying dissolved organic field within the microlayer. Dissolved carbon will be broken down into its macromolecular content and further, ultimately providing surfactant distributions.
Under these theoretical restrictions/additions, the momentum drag coefficient takes the compact, traditional form of:
with extensions to the other turbulent flux proportionalities
CH and
CE being direct [
2,
17]. Please note that we elect to duplicate the use of the symbol upper case
C for Prandtl coefficients in the current section and later on to signify marine chemical carbon atom concentrations, as opposed to insisting on some new, internally consistent nomenclature to deal simultaneously with ocean chemistry. The aim for the moment is to maximize readability for either the meteorology or geochemistry communities. But there should be little chance for confusion. The former usage will always be identifiable by the subscript capital
D standing for drag. Heat and evaporation influences are assumed to be similar [
2].
The roughness length z
o is the only operative variable—normally, one finds it related in some exclusively micrometeorological fashion to the friction velocity or stress [
16,
17]—however, during the peak of marine surfactant studies several decades ago, (analytical) chemical oceanography groups were able to show clearly that ripples and capillary wave distributions must also be determined, in part, by microlayer composition. By this they meant not only the mere presence of organic surfactants but also their bonding, branching, heteroatomic content, as well as general trace elemental and functional constituencies [
6,
7,
8,
9]. Barger et al. [
8] soon performed multi-kilometer spreading (manipulation) experiments along the eastern seaboard, both without and with concentrated slicks. Their work confirmed that roughness suppression could be pronounced. Observable reductions to friction element size could reach orders of magnitude, requiring a log scale to portray them adequately.
Results are summarized in
Table 1, with pre-World War II approximations for global average conditions offered as background ([
15] citing Rossby). Clean interfacial values (slick-less) are offered as the reference level. One of our major steps is to connect the lower two rows from this table in log linear form at a given wind speed, as follows:
Marine interfacial roughness will be lower-bounded here at 10 nm to reflect realities of molecular structure. The water molecule alone occupies 3 angstroms, and a single hydrogen bonded couple thus spans close to a nanometer. Variable surface pressure π is capped (forced to saturate) at the threshold 3 mN/m (mJ/m
2), except in sensitivity cases. This serves as a reasonable initial guess because laboratory surfactant responses typically asymptote at this level [
4,
5,
6,
7,
11]. Barger et al. relied in [
8] upon very low altitude spray dispersal (from a motorboat) of the common commercial agent oleyl alcohol in order to achieve their spread. Beading eventually confirmed total coverage. In the parlance of surface chemistry and the above tangent forces, the long chain (but unsaturated) alcohol is best classified as a two-dimensional liquid.
Jarvis et al. [
7] show that realistic global monolayer behaviors can be reproduced in the laboratory and then mapped to open water through the surrogate combination of a single 2D liquid plus one solid counterpart. Easily available analogs albumin and stearic acid are sufficient to accomplish the task [
6,
7]. We now proceed to construct a globally viable equation of planar state working from this mixture, and the resulting regional interfacial pressure values are fed back into the momentum transfer. The shifts in gas and salt flux are estimated more directly (but graphically) from empirical data in Frew and Modini et al. [
5,
11].
4. Organic Equation of State for the Global Marine Boundary
The format of any low dimensionality EOS must necessarily be dictated by intimate connections between interfacial chemistry and neighboring solute concentrations. Hence the resulting expression set cannot be as streamlined or elegant as, for example, an ideal bulk gas law or the Van der Waals curve. Externalities must be accounted for. But the mechanical analogies are nonetheless precise, as shown in the appendices of our previous publication Elliott et al. [
6]. Adsorption equilibria relate the amount of some less soluble, organic compound residing along the water–air division to the corresponding aqueous concentrations. Tangent mixing states are likely a factor over real seawater, but we begin by merely linearizing and the results conform to measurements (e.g., [
7]).
Our particular arithmetic choices include the two simplest isothermal forms, Langmuir (L) and Freundlich (F). The L form can be derived from a crude dynamic model, while F is extraordinarily flexible and therefore useful, despite its empiricism [
4]. An amount of some organic compound spread along the interface is often termed the “excess” by physical chemists because it resides, by definition and from the standpoint of the laboratory analyst, outside of standard solution. The L and F equations are readily combined to parameterize surface mass concentration and the tension quantity by defining effective fractional “coverages” and then adjusting to experimental outcomes [
6]. We adopt the decades old but seminal and comprehensive studies of Graham and Philips [
18] and Christadoulou and Rosano [
19] here for albumin and stearic data, respectively. These are supplemented with data from Brzozowska et al. [
6,
20] to provide kinetic estimates of low, micromolar level dissolved concentrations just below the boundary. We refer to the resulting equations for obvious reasons as a “Power Langmuir”, and further exposition is provided in Elliott et al. [
6].
The θ as used here retain a known and convenient form from ancestral isothermal approaches, but they act strictly as weighting functions; our system must therefore be considered highly operational and should eventually be supplemented from first principles with surfactant theory, perhaps of a statistical mechanical nature.
The
C in the present case are just the pair of concentrations for proxies albumin and stearate [
7] expressed in terms of total dissolved carbon atoms (micromolar). Vector notation is used to encourage expansion in next generation versions. Behavior or process types
t are limited to the adsorption equilibrium itself supporting surface accumulation, and depression of the tension yielding π. Reference
C converted into Langmuir-style equilibrium constants are identified with phenomenological half maxima as a start-up expedient. The sole restriction on our adjustable exponents
n is that they should be positive—integers are not required, or rather
n = 1/m is acceptable. The reader will see that
n < 1 flattens its profile while
n > 1 sharpens, and we exploit this tradeoff to deal with the extremes of 2D phase behaviors (divergent elasticity for 2D gases and liquids or total film collapse for solids). The symbol Γ refers explicitly to the excess, usually quoted in mg/m
2. Note that its reciprocal is just a normalized area. Surface pressure π is treated similarly and the reader is reminded that it represents the difference between surfactant and pure water tension [
4,
5,
6,
7].
Taken together, our isotherms for the fundamental tangent properties Γ and π, both expressed as f(
C), comprise a relationship for normalized interfacial force versus area (π(A(
C)) since A = Γ
−1). Thus the equations in this section constitute an analog to PV relations in three dimensions, which are of course the best known equations of state [
1]. There is in fact an ideal 2D gas law with a certain pedagogical value; the curious reader is referred to [
4,
6] for more information. However, it does not have immediate practical value for us here. Overall our formulae are designed to parametrically account for collective aqueous surface formation energetics due to intermolecular organic spacing—the aforementioned hydrogen bond breaking, impedance due to Van der Waals attractions, Coulombic repulsion of acid head group, etc., are all included by default. Details of the tangent physical chemistry are offered at monograph length in Davies and Rideal [
4] along with numerous similar textbooks published more recently. Additionally, we have just reviewed such material extensively in our initial extrapolation of π across the global ocean [
6]. It is perhaps worth reiterating the ever-presence of dissolved organic drivers, because they embody and carry information from mixed layer detrital cycling. This connection is the essence of biological control, dictating the planar composition of a ubiquitous planetary monolayer.
All ideas in this section flow naturally from formal Gibbsian thermochemistry as it may be extended to the water-air interface, but with tension-area partials added to those of the more familiar state variables entropy and volume. Gibbs was, in fact, the first to derive an independent (single species) surfactant isotherm based on early expressions for solute chemical potential [
4,
6]. At this point, the physicochemical tools on hand are fundamental and all that remains is to characterize
C just beneath the interface of the planet. We will then loop back on our logic in the order of
C, π,
zo, flux (relative).
5. System Distributions: Real Surfactants and Their Effects
Within the mixed layer of the upper ocean, macromolecular carbon partitions into a number of organo-functional and reactive-transport steady states. Grazing production, advection-diffusion, and chemical losses (usually unidentified) are generally all involved for any family of fresh biopolymers. A good guess is that initial removal will be dominated by bacterial consumption, though in some cases photochemistry may play a role [
12]. Hydrolysis and random re-condensation eventually degrade the original configurations, and such processes complicate matters considerably. Here we follow the methods of Ogunro et al. [
3] in order to avoid such issues. For example, we rely on a traditional average protein-polysaccharide-lipid content for typical phytoplanktonic cells (60-20-20 carbon atom %), and beyond disruption-spillage we globalize all residence times.
The actual seawater biopolymer suite also includes lipopolysaccharides, amino sugars, humics, fulvics, massive hetero-polycondensates, nucleic acids, fragments of all the above and more. Collectively this solute assemblage is most often referred to as the dissolved organic carbon or DOC. In the interest of efficiency, we adopt a simple triage algorithm to zoom in on strong sources of surface activity [
6]. Total DOC content of most mixed layer water is readily established by drying with conversion to CO
2, and it tends to lie between 10–100 µM. Therefore carbon concentrations for any individual functional type likely fall in the range micromolar, perhaps reaching tens thereof at maximum. Except for proteins and lipids, all the classes mentioned are characterized by adsorption half saturation levels orders of magnitude higher. Hence, the protein-lipid fraction far outcompetes other forms by occupying most available sea-air boundary niches. We presume that other moieties make only a negligible contribution to monolayer mass. At this point, it is perhaps worthwhile for the reader to glance briefly at reference half saturation values that will be adopted in the EOS (
Table 2, also containing other parameters). The upshot of our triage strategy is just this: a few µM of appropriately selected macromolecules are expected to wield disproportionate influence on surface energetics. The task now is merely to quantify.
The Ogunro kinetics are supplemented for present purposes with much more recently computed background fields taken from the marine Network Common Data Form (NetCDF) output of the Community Earth System Model (CESM). CESM is a state-of-the-art international-biogeochemical climate code. Cross-checking was performed against early (oceanographic) station-oriented runs conducted in a single (numerical vertical) dimension, since plotted scenarios happened to correspond closely to our aggregate eco-zones [
10,
13]. Class specific protein and lipid data constitute only a small portion of the total for DOC because analytical techniques are challenging. Where possible, we performed chi-squared optimization to the macromolecular measurements by varying residence times to match compilations [
3,
12].
Total class-specific dissolved carbon in micromolar was fed into the equation sequence from our earlier section with processing in the Python coding ecosystem. The primary surfactant quantities Γ, π, and ε were estimated locally. Then, the micrometeorological roughness and altered flux indicators were computed for the most important climate transfer modes. Relative reductions to gas transfer and bubble popping-efficiency were calculated by linearizing to experiments presented in Frew ([
5], Figure 9) and Modini et al. ([
11], Figure 2). Results presented in the current section all used the same main threshold value of 3 mN/m to saturate the π influence. Wind speeds were downloaded directly from an NCEP-NOAA reanalysis, as offered on the Columbia LDEO website.
The rest of this section details our core results, which are positioned together in the bottom few lines of
Table 3 and display several signs of potential import for a biological EOS concept. Fluxes for momentum, trace gases, and sea salt are defined as in [
2,
5,
14], respectively. The reader will immediately notice reductions to the critical proportionality constants distributed almost across the board, but not at all uniformly. There are clearly spatiotemporal conditions under which the food web does -or does not- exert its monolayer influence. Some losses in exchange or transfer are much greater than tens of percent for the drag coefficient (and therefore also in our simplified micrometeorology relative to heat and water vapor [
2]), the trace gas piston velocity ratio (film to clean [
5]), and the analogous quantity for bubble popping efficiency [
11]. We will now interpret more thoroughly, moving from left to right across the lower table entries in order to organize some critical comments.
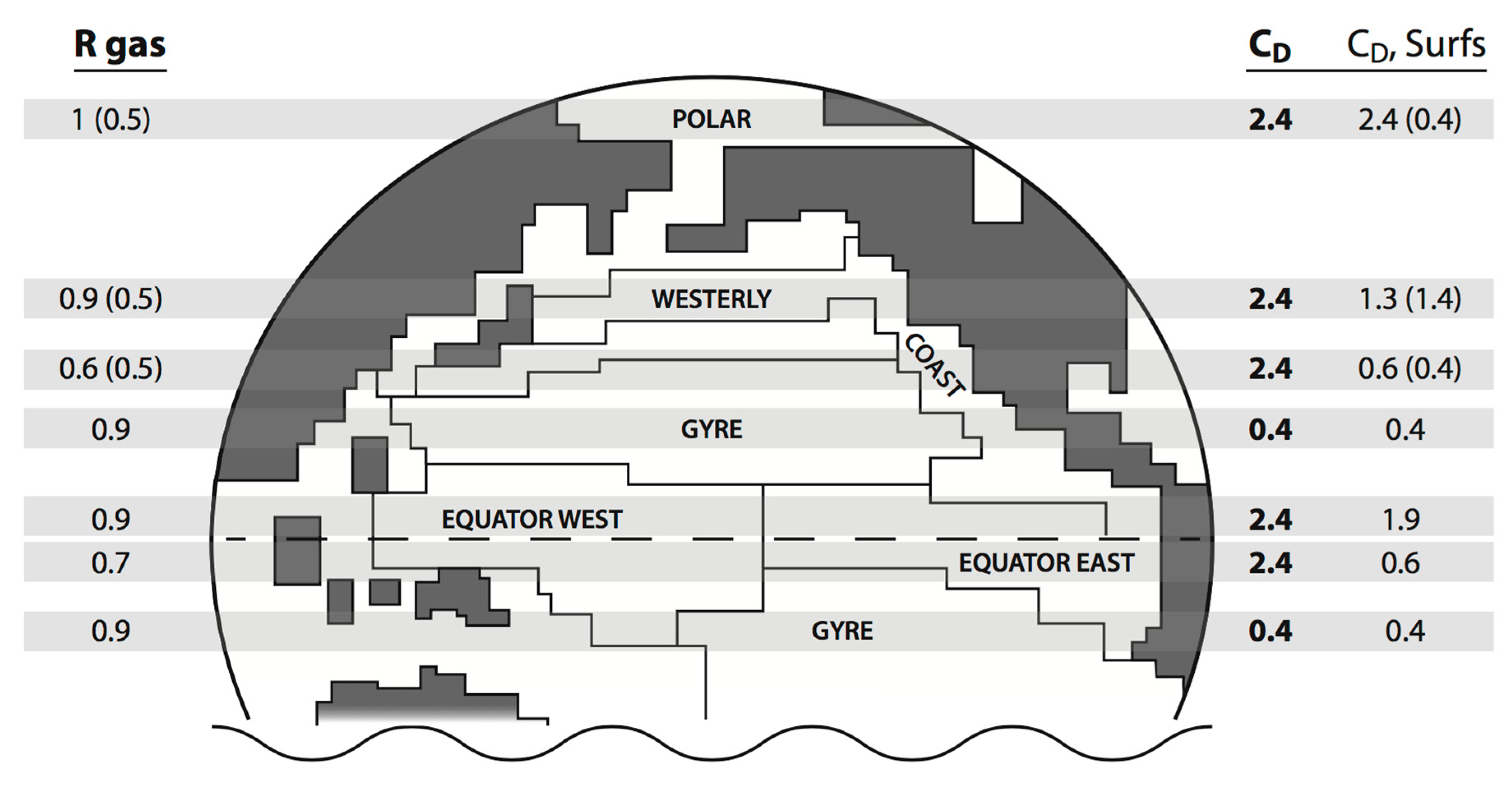
Polar transfer types are generally depressed by an ongoing bloom, while along mid-latitude coastlines or in an upwelling regime, the effects may be year-long (a-seasonal). The computed westerly influence is also continuous in our coarse interpretation, but a subtle point is raised in this part of the world—the wind regime happens to bracket the transition from low to average speeds (as indicated in
Table 1). The situation for the eastern-basin margins is similar at the equator, while to the west, control is attenuated because wind speeds are quite low. Subtropical gyres fall in the same category. Two columns are appended toward the right of
Table 3 containing the global average and the extreme Southern Ocean winds juxtaposed with asymptotic biology (here reflected by our macromolecular carbon chemistry). These reference points demonstrate that even at medium to high wind speed the effects may be significant.
The results are striking in many cases, with implied flux reductions sometimes as large as an order of magnitude. We conducted a series of checks to provide ourselves and the reader with some perspective on
Table 3. First of all, it should be noted that π(A) relationships derived from our EOS closely match multi-basin data published by a host of chemical oceanography groups from the 1960s onward (Jarvis et al. [
7] shown in Figure 3, plus other references in [
6]; compare with appendix plots in the latter case). The agreement may be taken as evidence that our proxy pairing of albumin and stearate is actually quite an appropriate first choice. And this is unsurprising, given the effort put into the selection process by a dedicated and pioneering community [
6,
7,
8]. More importantly, however, an averaged real (surfactant-modulated) drag coefficient from across the cells of
Table 3 is 1.2 × 10
−3. This is very close to the widely accepted, oft-cited and commonly applied global value [
2,
17]. The range of results, meanwhile, resembles that adopted in Elliott et al. [
6] as a crude ramp for generalized flux effects (π = 0.3–3 as a critical decade-span). We have not weighted our calculations by total ecozone area here, but since the coverages are roughly uniform, this should not be a large factor [
10]. The spread in trace gas piston velocities of about a factor of two (relative) strongly resembles distributions from numerous typical studies, several of which are actually shown as individual data points in our primary reference [
5]. The bubble film burst efficiency range falls in line as well. It is consistent with dozens of indications for organic surfactant sensitivity appearing in the definitive monograph Lewis and Schwartz [
14].
Overall the clear trend is toward biological slow-down of selected energy-mass fluxes. Some of the actual geographic patterns are called out schematically in
Figure 1 for quick visual examination. Taken together,
Table 1,
Table 2 and
Table 3 and our figure suggest a risky and speculative “Gaian” guess (which is considered in more detail below). Stabilization of the local water column may well be favored at the marine systems level since nutrients are thus retained for recycling. Feedbacks from reduced momentum input or heat output work in this direction to zeroth order. The salt spray (loop) influence can be simulated at the level of a thought experiment by mimicking Charlson Lovelock Andreae and Warren (CLAW) hypothesis logic. Fewer cloud condensation nuclei translate through the regional stratus cloud reflectivity to enhanced sunlight penetration, and thus to the warming of upper layers. Finally, we feel compelled to remark that biological regulation of the wind-driven horizontal circulation cannot be easily excluded.
7. Uncertainties in the EOS
For the sake of brevity we deal with process-specific sensitivities in tabular form and then summarize, all in the present section. Parameters from
Table 1,
Table 2 and
Table 3 have provided for us what were very literally the first answers generated during the research effort. Even an initial cut at the surfactant-climate connection has therefore yielded a strong suggestion that there may be regional regulation by the marine biota. However, this general outcome could be fortuitous to some degree; we must now address the distributed inherent uncertainties. They are both numerous and large, appearing prominently at every stage. The reader is encouraged to scan
Table 5 to get a feeling for the severity of problems remaining, but several of the entries deserve particular comment.
Sources, Sinks: Here we refer to water column kinetics dictating the global macromolecular steady state patterns from which we draw the concentrations
C. Production and loss terms configured as in [
3] necessarily average over complex multistep mechanisms at both reactive ends. We suspect that the best way to narrow down such fundamental uncertainties will be through further in situ measurements (oceanographic, analytical chemical) conducted selectively across ecogeographic provinces of interest. The small heteroatomic (organosulfur) species dimethyl sulfide might be taken as an example and inspiration. It is maintained at somewhat greater than nanomolar levels across the entire upper ocean but is constantly under tight and variable food web constraints. Due to the familiar CLAW hypothesis, the molecule has by now been quantified tens of thousands of times. Additionally, sophisticated extrapolation techniques are at hand to fill in large geographical gaps that inevitably remain.
Temperature, salt, and acid-base dependencies: Experiments suggest that these are only minor relative to the equilibrium constants called upon in our Power Langmuir. Graham and Phillips, for example [
18], report albumin, casein, and lysozyme adsorption data from room temperature down to nearly the freezing point, and differences in net π-coverage were much less than a factor of two. Our primary lipid-behavior references [
19,
20] document values over wide ranges of both ionic strength and acidity. Still, all such shifts are traditionally accountable in the theoretical framework of thermochemistry [
4] and they could be explored as such given time and resources. It is also the case that skin temperature may apply in the monolayer, and would be somewhat cooler than bulk.
The mixing state: As in the bulk, mixtures operating in two dimensions must experience altered activity coefficients due to neighboring solute-solute interactions. Such relationships have been linearized out of our analysis for demonstration purposes, but should be reinstated as soon as possible. Planar liquids and solids are known to reinforce their respective tension behaviors if excesses are similar [
4]. Both theoretical and experimental approaches should be valuable here. One can envision a distinctive tangential thermochemistry with Gibbsian form, but spread tightly over the planetary envelope and dominated by chemical potentials from the DOC.
Molecular surrogate selection: Another issue that is likely best handled in the laboratory, with the caveat that standard commercial surfactants should be avoided in many instances [
6]. We maintain, for example, that common off-the-shelf choices such as Triton X-100 or SDS should be given careful consideration prior to application, and perhaps they should even be disfavored since true marine biopolymers are not well represented (in the sense of our background section [
7]). A corollary is that environmental surfactant work has always relied heavily on the availability of certain reagents. Thus, many high quality experimental outcomes may be less than directly transferrable to the real ocean. In fact, one of our crucial pivot references falls squarely in this category ([
8], see Table 1). To be consistent, we are forced to recommend more appropriate proxy identification followed by reinvestigation.
Heteropolycondensates: Some overlap exists with catch-all categories of marine humics and fulvics, but the point to be made with regard to uncertainty is similar. Benner [
12] has characterized unknown, random condensed structures as pervasive. Re-constituted variable organic functional types are the rule of the sea rather than the exception. Stretches of bio-detrital protein or strands of lipidic hydrophobe could well be preserved during processing, going on to act as effective adsorbers of enriched concentration with somewhat different equilibrium parameters. The main issue is that our Ogunro-style calculations [
3] for pure substance could represent significant underestimates.
Cross-phase kinetics: These have often been explored by the marine aerosol community in conjunction with primary organic emission studies. Delays of order seconds are sometimes reported in the attainment of open ocean surfactant equilibria [
6]. It will be of interest to monitor detailed rates in the case of the several key proxies for swings in surface tension. However as a startup expedient, we have postulated that on the gravity wave time scales of the most relevance here, the adsorptive steady state is established rapidly.
Collapse of the monolayer: This behavior can be extra-thermodynamic, hysteretic, and mechanically extraordinarily complex [
4]. See the Graham and Phillips data [
18] for examples varying across a family of commercial proteins. Layering, wrapping, bunching, balling, and even the reformation of partial enzymes may be expected (primary-secondary-tertiary), positioned at and over the interface. Our current view is that these are structural pathologies that can ultimately be represented simply and directly as loss of surface-active carbon relative to the monolayer.
Alternate phases: Some authors have addressed the potential for whole ecosystems to exist locally within the so-called sea-surface microlayer, which itself spans the chemistry of concern here. We have recently reviewed a representative subset for this type of work [
6]. Detrital slimes, bacteria and even protozoans have all been documented residing very near the top of the water column (within micrometers), and they may well occupy net fractional interfacial area. This would definitely indicate displacement and disruption of the monolayer. The sacrifice to available area, however, could be parameterized at the regional scale based on appropriate observations.
Stirring: At high wind speeds the monolayer is expected to turn over rapidly in the vertical—to “churn” on itself by folding back regularly into the upper column [
5]. This is in common with other surface-confined objects, structures, or substances. One instance is sea foam, which is supported by the surfactant concepts investigated here. Another example would be the volumes of low-level air trapped into bubble plumes by wave breaking. If turbulence and the kinetics involved in reaching the interface have to be questioned under mild conditions (see just above), then adsorptive re-equilibration may be even less competitive when conditions are rough. But this dynamic is partially handled in the tabled Barger et al. data [
8], which already form a conceptual basis for our equations.
Similarity: Monin Obukhov considerations have been set aside in favor of the neutral micrometeorological regime in order to communicate our main ideas to the surface chemistry audience at an early stage. However, boundary layer neutrality is far from universal over the remote ocean. Buoyant or stably stratified air columns could lead to swamping or cancellation of surfactant dependencies over large stretches. The neutral circumstance, however, has long served the environmental community as an accessible baseline [
2,
17]. We certainly welcome those with the appropriate skills to dig deeper into the implications of similarity theory.
8. Summary and Discussion
We have argued briefly that (1) interfacial chemistry may be coherent over much of the ocean-atmosphere and, moreover, it may be comprehensible (calculable or map-able) given just a small handful of bio-macromolecular proxies, (2) the attendant adjustments to energy-mass transfer can be parameterized through surface tension differences (specifically expressed as the tangent pressure), (3) required dissolved organic distributions are derivable from global systems chemistry-transport, and finally, (4) items 1–3 imply—and actually drive—strong geographic variability in the better known climate-critical fluxes. A short list of susceptible transfer modes would include momentum, heat, water vapor, trace gases, and salt spray. Along the way, as we developed this circuit of surface science relationships, analogies have been drawn upon liberally with bulk force-volume mechanics. Hence the exercise distills to the construction of a chemically functional-operational but globally unified two-dimensional equation of state. The result can be thought of as a wrapper on the liquid water exterior of Earth. Detailed forms are dictated by laboratory data for specific surrogates representing real surface-active detrital carbon. Surfactant chemical outcomes plus flux influences therefore depend significantly on upper ocean food web dynamics, i.e., on marine ecology. Not surprisingly, uncertainties in our development are numerous. But most appear manageable, as shown in the sensitivity and uncertainty sections.
Inexpensive early (next generation) experiments can be imagined to scope the possibilities opened by our biogeochemical EOS concept. Interfacial pressure versus excess molecular area curves could be reproduced for proxies over artificial seawater in the laboratory. Attempts could be made to improve the effective—but classic—surrogate vector of two compounds (our protein and lipid choices). Field expeditions might be organized station-wise (in the oceanographic sense) to determine whether prospects for bio-flux control could be a reality, at least relative to certain familiar and well-instrumented locations. Our personal feeling is that the original threshold (limiting) tangential pressure 3 mN/m can and should be verified. This might entail a judicious combination of micrometeorological and analytical chemistry observations organized from a campaign perspective. Ship transect, aircraft platform, and satellite data will no doubt all make powerful contributions.
One broad context for appreciating a potential global bio-EOS is provided in the planetary sciences [
21]. Equations of state permeate all discussion across the solar system, whether in time or space. Force–density relationships range from crushed ideal gases composed mainly of electrons to quasi-quantized metallic hydrogen moduli, often maintained at megabars. But from among members of this vast and diverse menagerie, none are simultaneously as tenuous and efficacious as the macromolecular film version presented here. The physicochemical and informational complexity embodied in Earth’s global monolayer are unparalleled. Materials science offers further comparisons—in general, forces underlying the surface tension of a bulk liquid are notoriously strong since they imply multi-molecular bond breaking [
1]. Our notions raise the tantalizing possibility that here on Earth, life has stumbled onto a means for tuning the global hydrogen bond while also lining the entire ocean interface with Coulombic and Van der Waals regulators. Yet another standpoint is that of raw mass balance. Imagine a vertical section through the marine and atmospheric boundary layers with diverse, bio-maintained macromolecules forever riding wave tops and exerting their peculiar filtering action on world-wide energy flow. For the price of roughly a single carbon atom of thickness—all of energy, fluid and salt-gas transfer are kept in check. The last few arguments have verged on the anthropomorphic of course, and so we have stepped blithely from mere speculation into the Gaian realm. This is always a dangerous enterprise, but perhaps not inappropriate to close.
We take the liberty of concluding within a rich historical and anecdotal framework provided recently in the short paper by Cox et al. [
9]. These authors describe a dramatic sea rescue taking place in the late nineteenth century, in which several divergent cargo-chemical surfactants were fortuitously tested against one another under intense and well-documented scrutiny for their respective wave calming capabilities. First, some common petroleum products, and then later, a set of fish oils were intentionally pulled from a ship’s hold and dumped into coastal Atlantic waters. The former are mainly insoluble and simply bead up toward the atmosphere, while the latter possess a crucial monomolecular layer structure—multiple double bonds that cause 2D liquid behavior (per our background section). The petroleum failed to provide any relief of wave motion at all, while in stark contrast, natural marine oils quickly succeeded in eliminating breakers over square kilometers, thus permitting life-boat rowing activity and life-saving to commence.
Implications for chemistry of the planetary interface should be obvious, but they can only be explicitly captured, extrapolated, and studied through the construction of some sort of parameterized EOS. The fact is that detailed surfactant composition matters greatly, while the influence can be both targeted and surprising. Cox and company go on to provide a world history of slick experiments, informal and otherwise. Marine surfactant effects turn out to be mere common knowledge for those who have worked the sea. There has long been consideration, for example, of chemically engineering harbor entrances. The authors conclude by mentioning the option for regional geoengineering of storms via surface films. One idea would be to reduce heat sourcing from the tropical ocean to mitigate the severity of hurricanes.
Our main point in the present work takes on special significance in light of the Cox et al. publication. It appears that global phytoplankton may already perform some of these functions in the natural background, at least at the (ecological) provincial scale. If this is the case, biodynamic flux modulation has been ongoing for epochs. The possibilities cannot presently be excluded that marine food web dynamics have partly dictated latitudes and even locations of hurricane genesis, the energy-direction of surface ocean currents, the depth of the local mixed layer which determines nutrient supplies, and more. We feel that this list is sufficiently compelling to merit well-designed experiments at multiple scales beginning in the laboratory.

 ,
, 


{kind=link}