
Baltic Sea Spray Emissions: In Situ Eddy Covariance Fluxes vs. Simulated Tank Sea Spray


Abstract
:1. Introduction
2. Experimental Site and Methods
2.1. Site
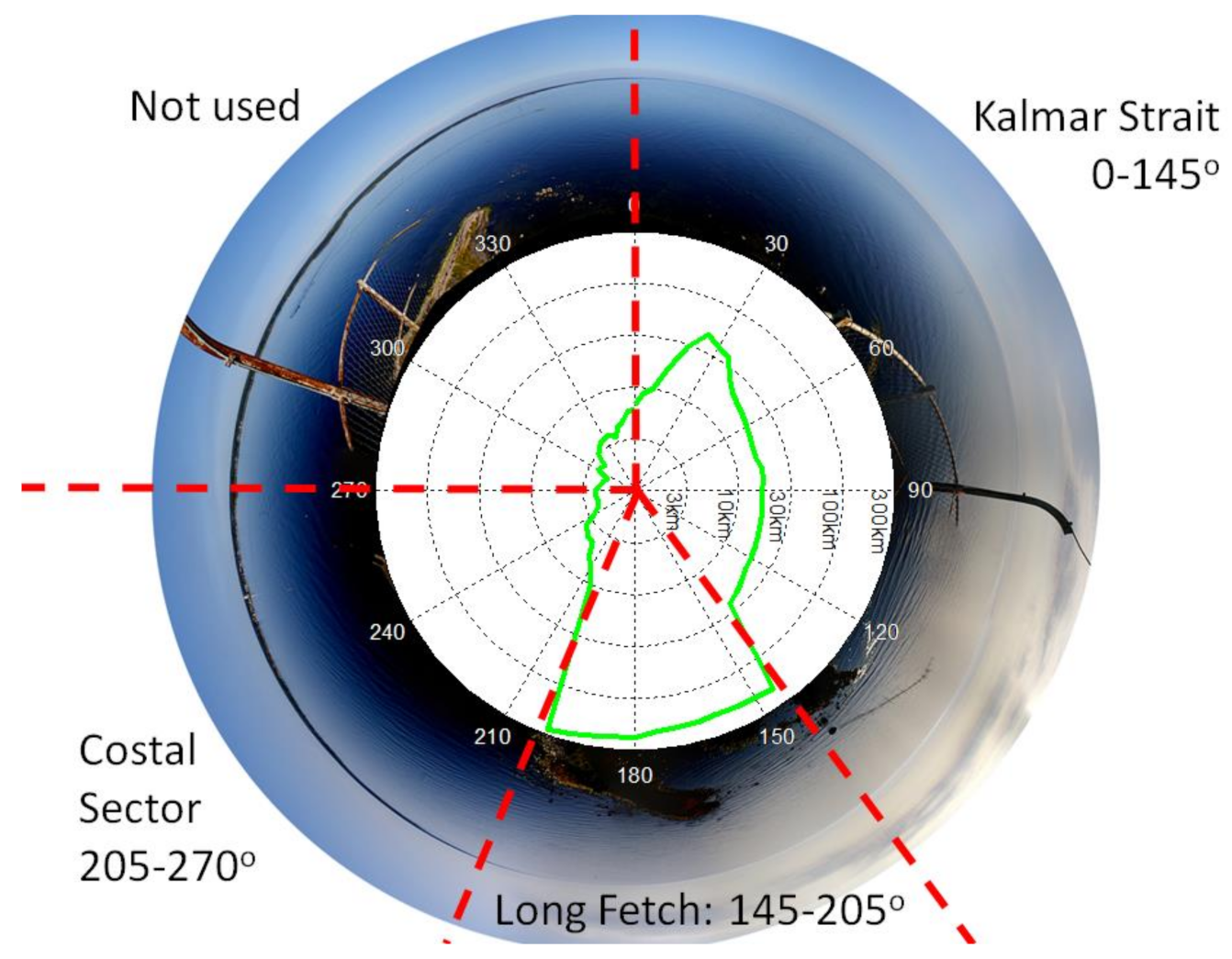
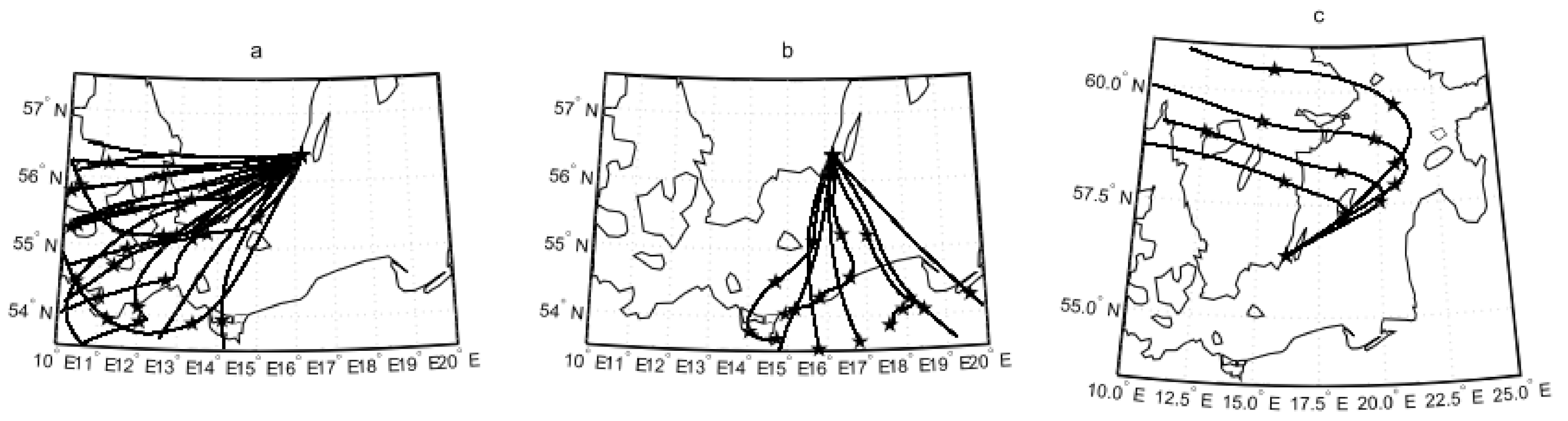
2.2. Wind Sectors, Fetch, Sea Bottom, and Footprint
2.3. Weather and Air Mass Origin
2.4. Flux Measurements
2.4.1. Instruments
2.4.2. Flux Calculations
2.4.3. Errors and Corrections
2.5. Laboratory-produced Aerosol
2.6. Sea Spray Source Parameterizations and Their Adaption to Brackish Water
3. Results and Discussion
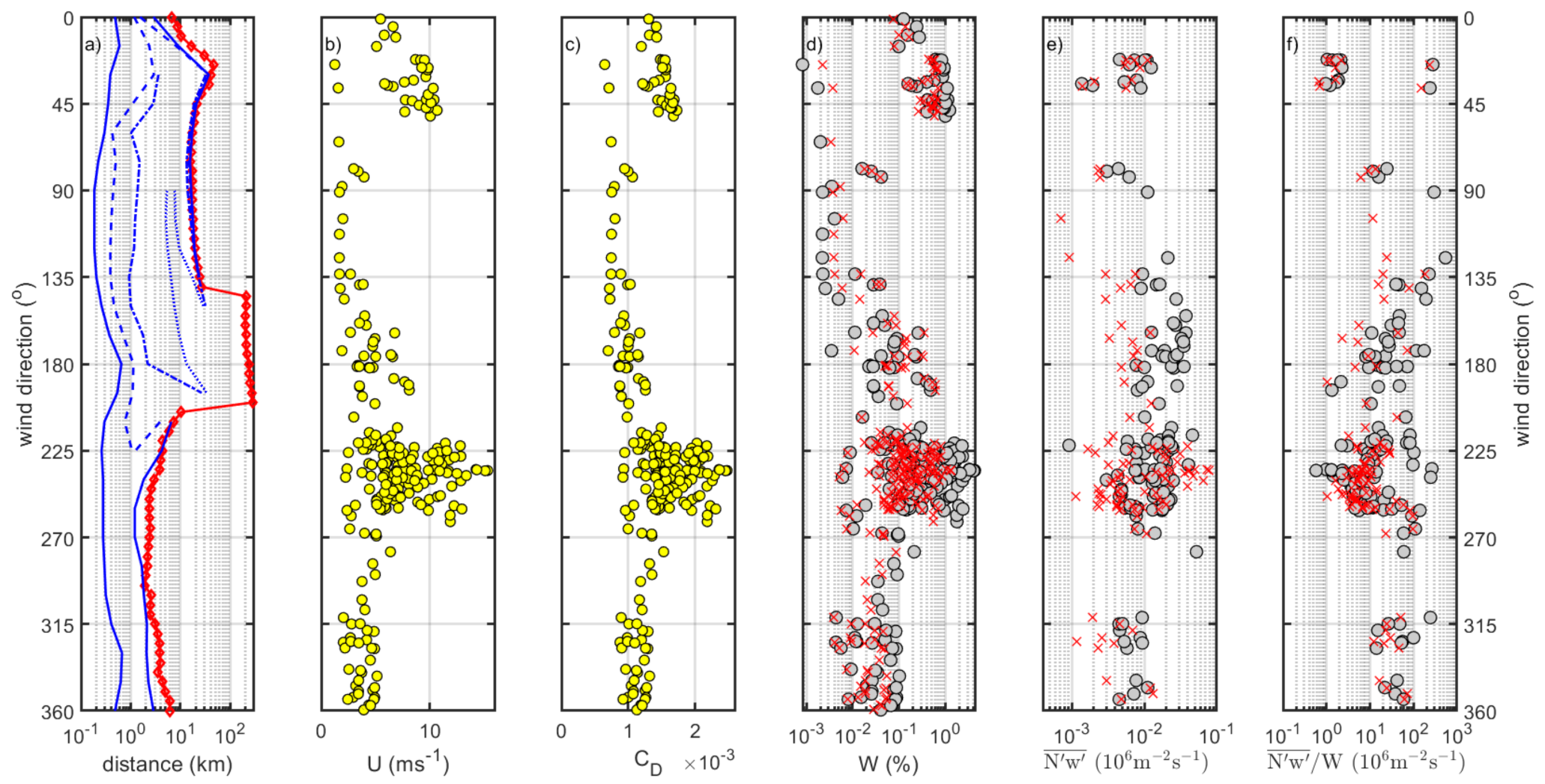
3.1. Aerosol Flux Direction
3.2. Aerosol Flux and White Cap Coverage by Wind Direction
3.3. Aerosol Emissions by Size
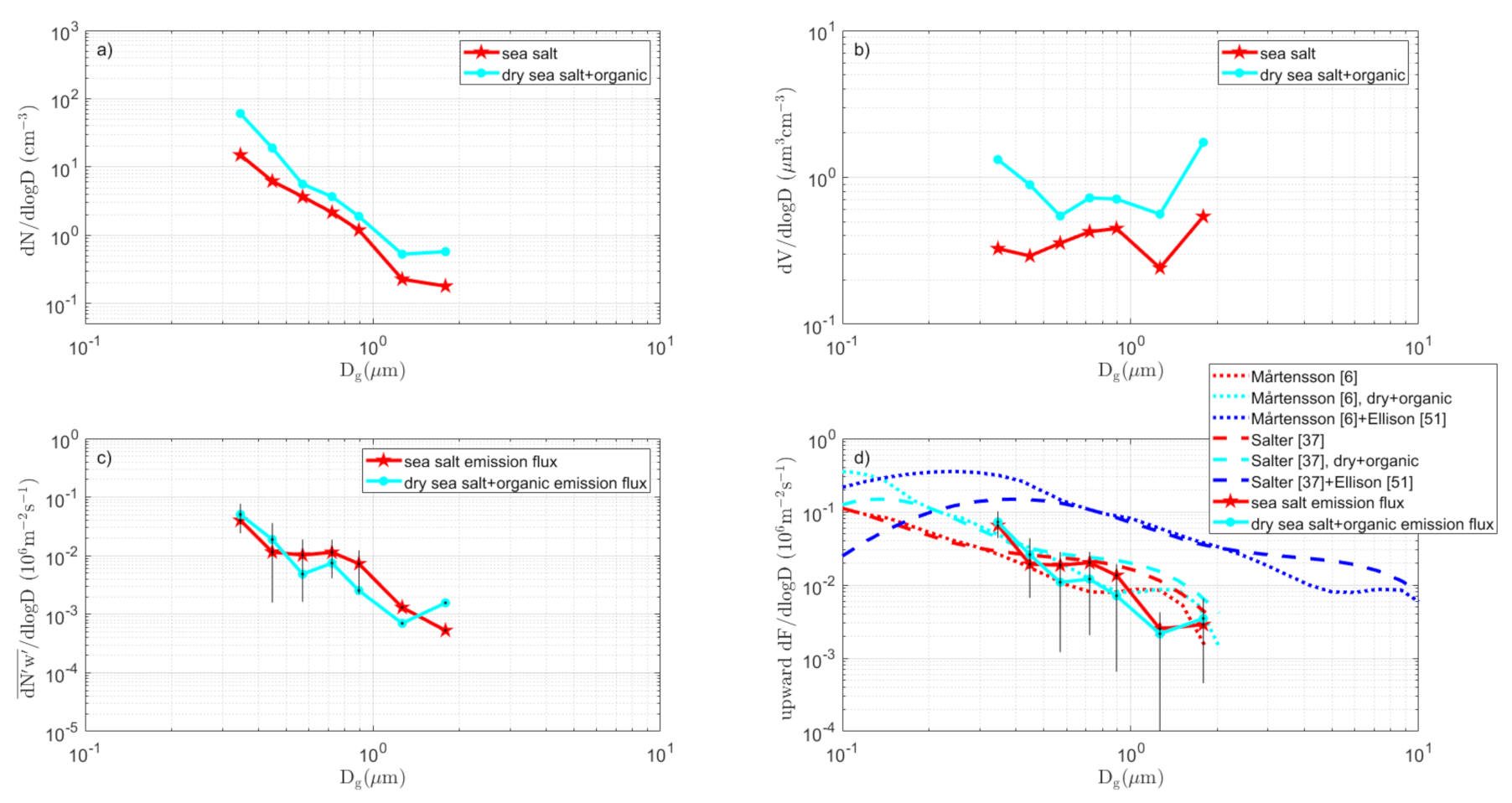
3.3.1. Coastal Sector
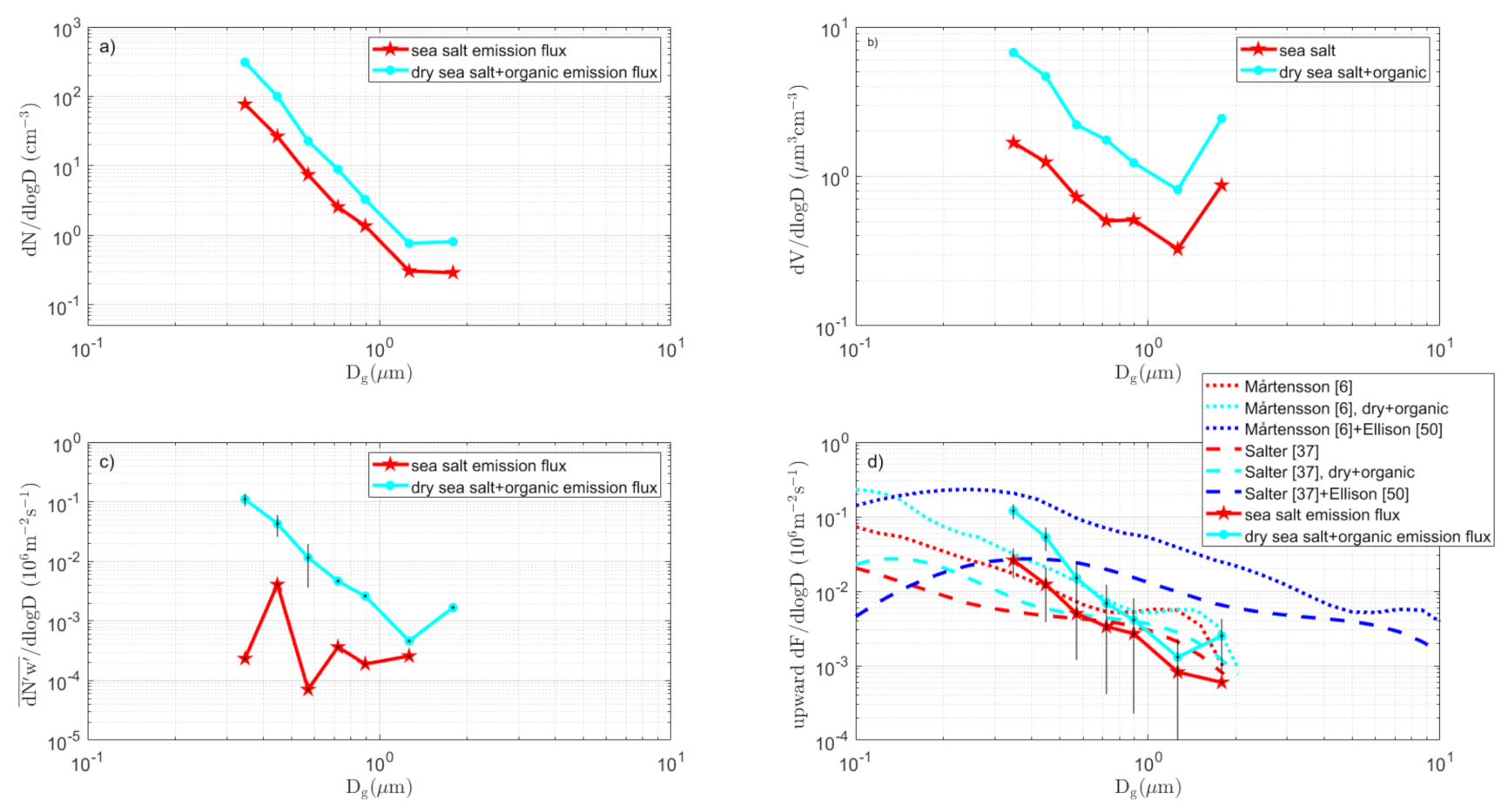
3.3.2. Long Fetch Sector
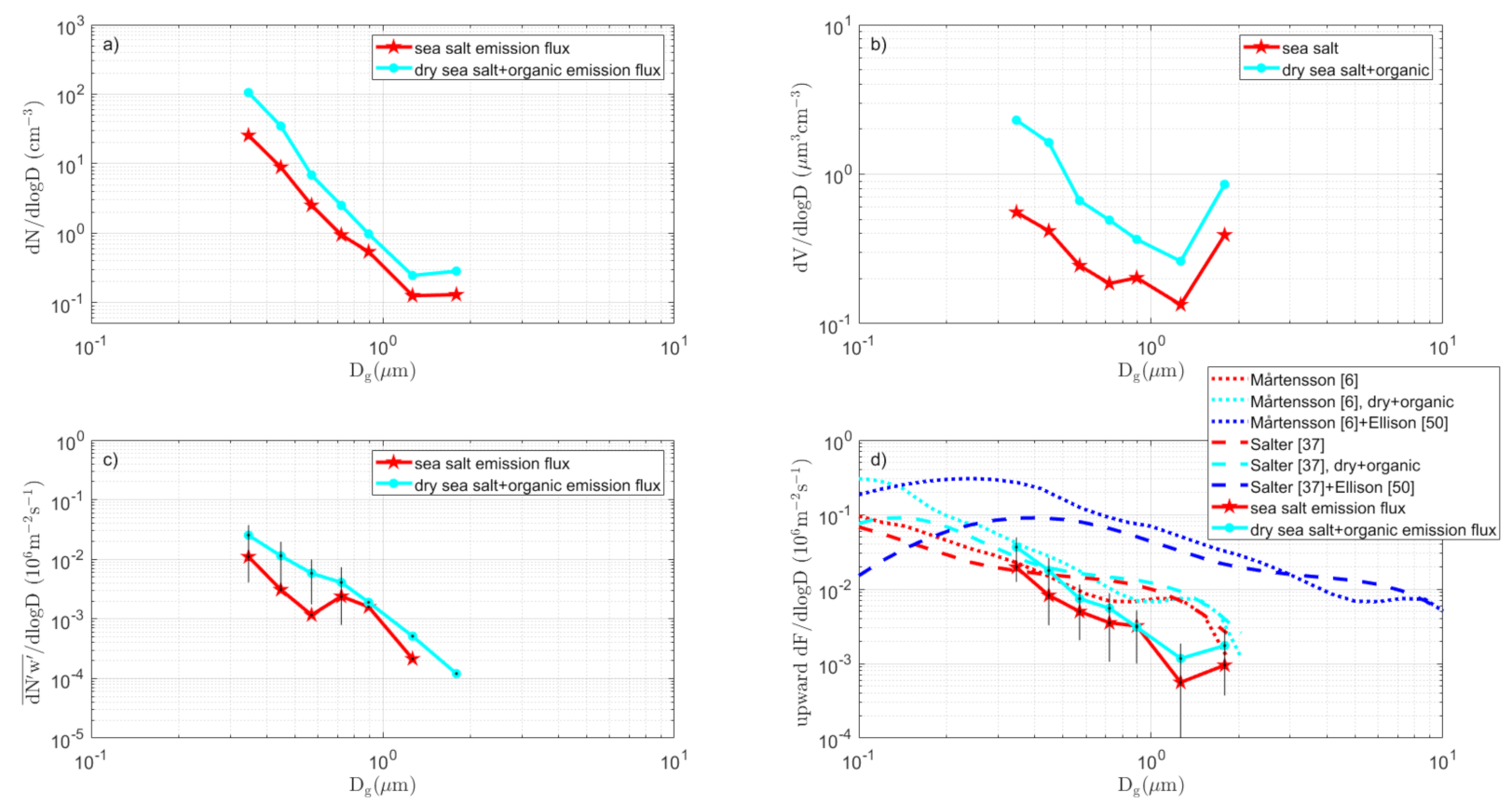
3.3.3. Kalmar Strait Sector
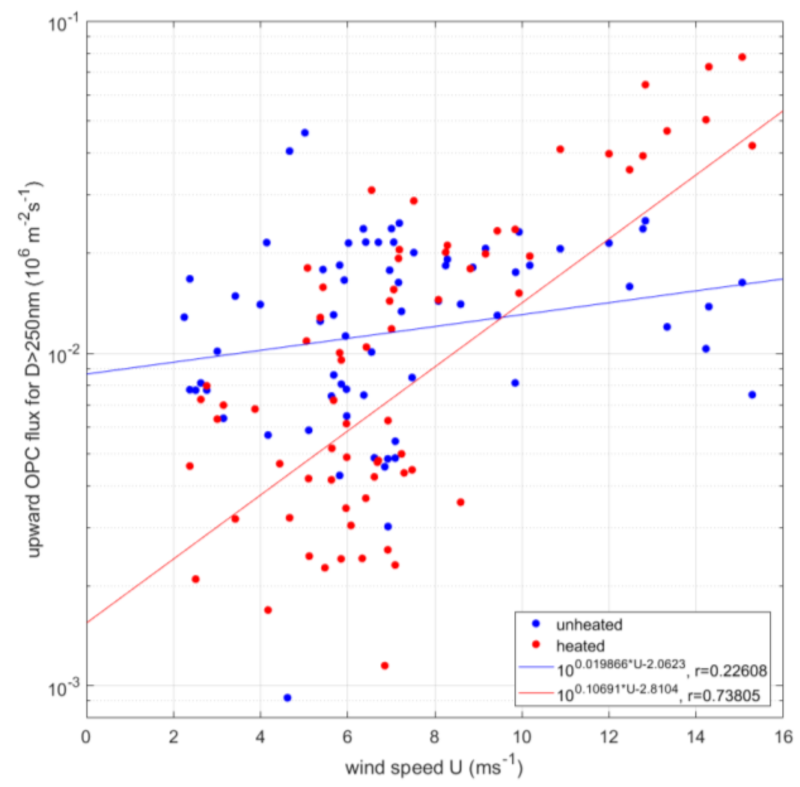
3.4. Wind Driven Sea Spray Flux
- It may be that there is an organic semi-volatile fraction along with the sea salt that had a less pronounced wind dependency. The gradient flux measurements [53] suggests that the organic mass percentage of the sea spray would decrease with increasing wind speed (at the Irish west coast facing the North-Atlantic). It is however not straightforward to derive from that study how the number of emitted sea spray particles with organic in relate to the wind speed. Will the exponential increase in sea spray emission or the decreasing percentage of organic mass win, or lose, or result in a near steady state despite increasing wind velocity? The interpretation of Figure 9 in view of the results of [53] is further complicated because we do not know if the organic sea spray fraction and the sea salt sea spray fraction was entirely internally mixed. With at least a significant part of externally mixed organic sea spray, alternatively if bubbles scavenge relatively less organic surfactants at high wind speed we cannot exclude this possibility (see discussion further down in Section 3.8).
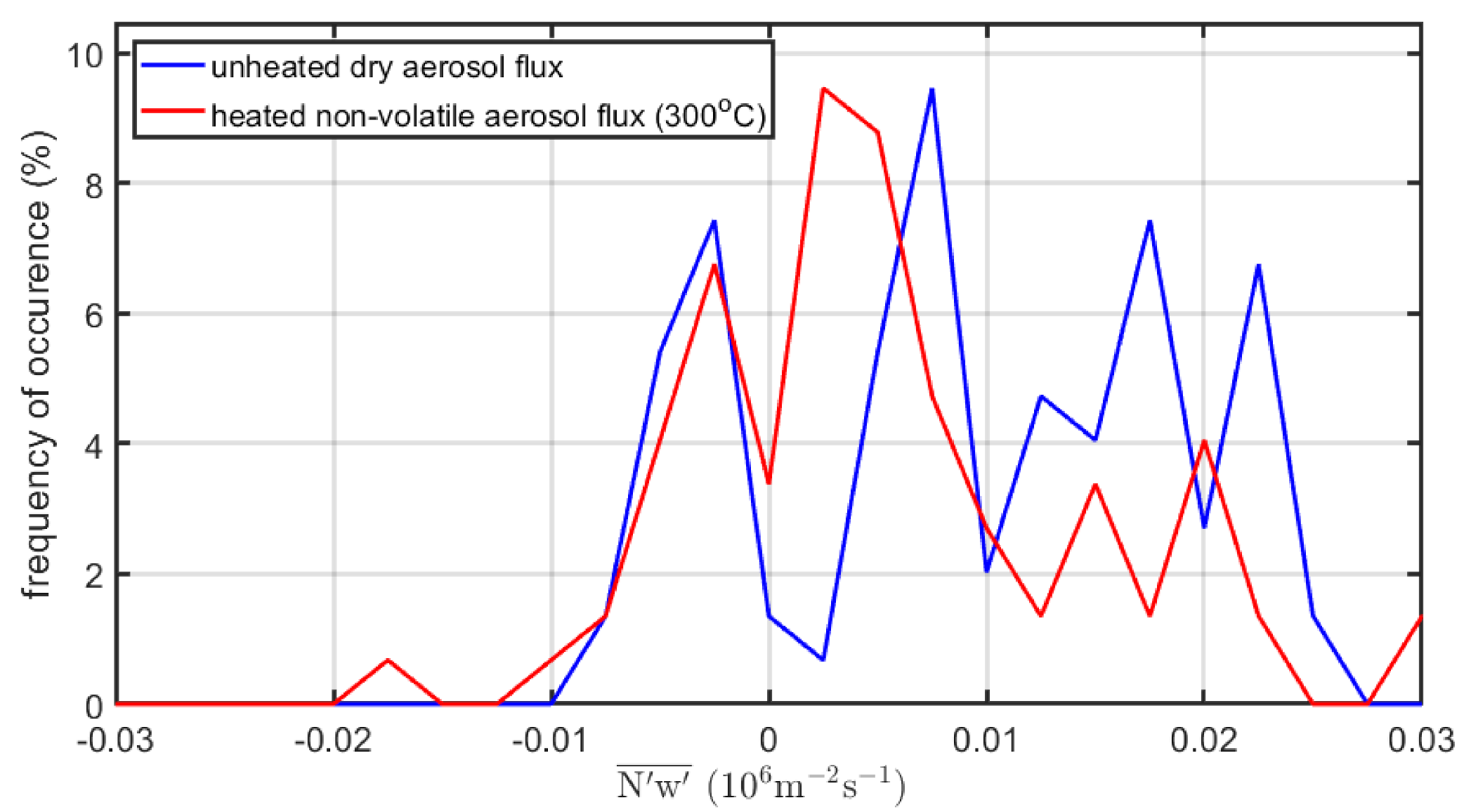
- While previous studies (in the Arctic and Atlantic Oceans) have been in fairly remote marine regions, the data of the current study originated from a region with strong anthropogenic influence on the aerosol. We should expect that the dry aerosol concentration contained significant amounts of anthropogenic non-sea-spray aerosols. These would not covariate positively with the vertical wind since they do not have a source within the footprint. They may contribute to the flux with a smaller negative covariance due to deposition at the surface, but this is usually an order of magnitude smaller than the sea spray emissions. Dry deposition is usually considered to be wind-dependent as well, e.g., [8], which could obscure any wind dependency in the sea spray source flux. This should cause more scatter (and lower correlation coefficient), less pronounced slope to the horizontal wind, and a larger zero bias, as observed in Figure 9. The heated non-volatile aerosol would most likely suffer much less from anthropogenic influence, since at 300 °C the only remaining anthropogenic aerosol component would be soot (which is likely to primarily occur at smaller sizes considering their number).
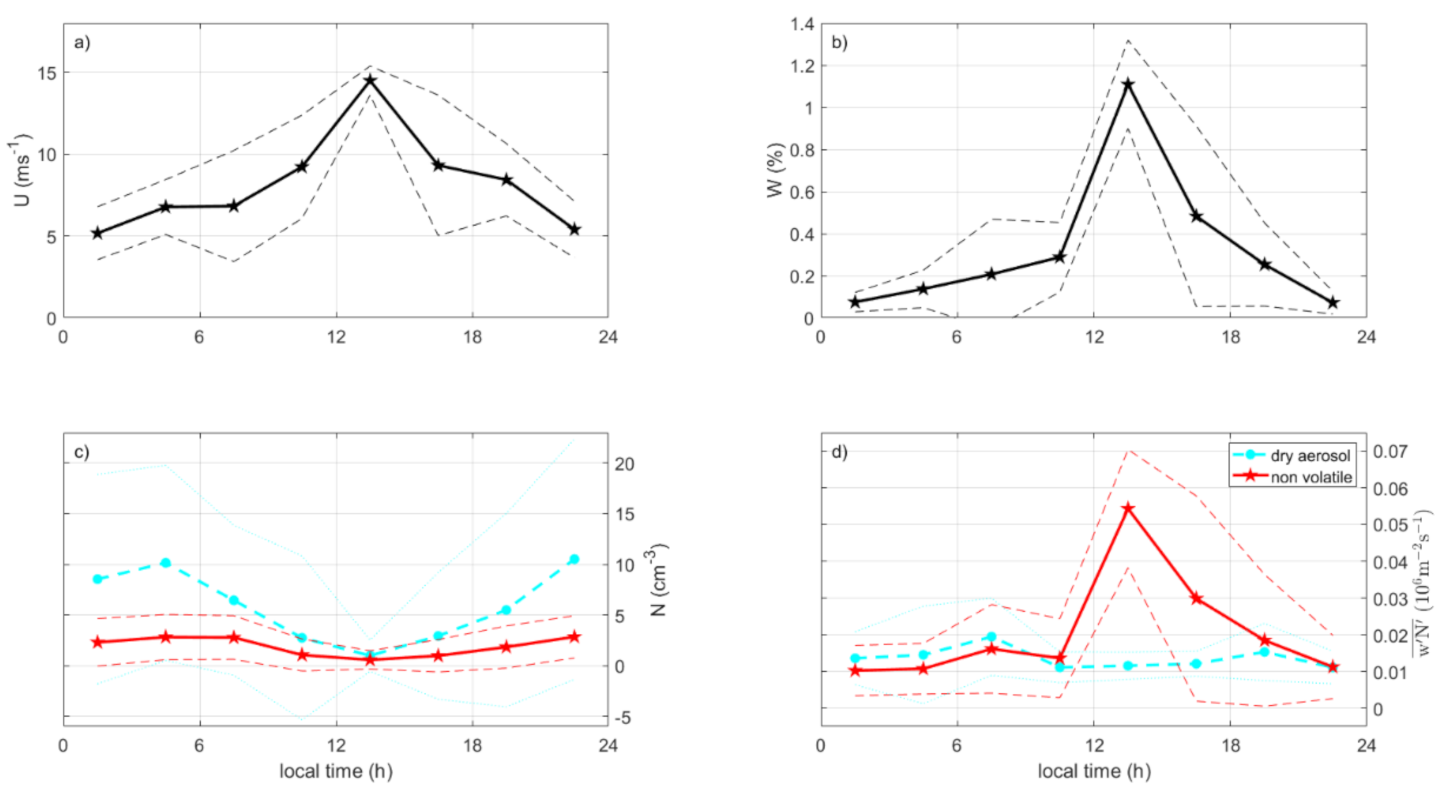
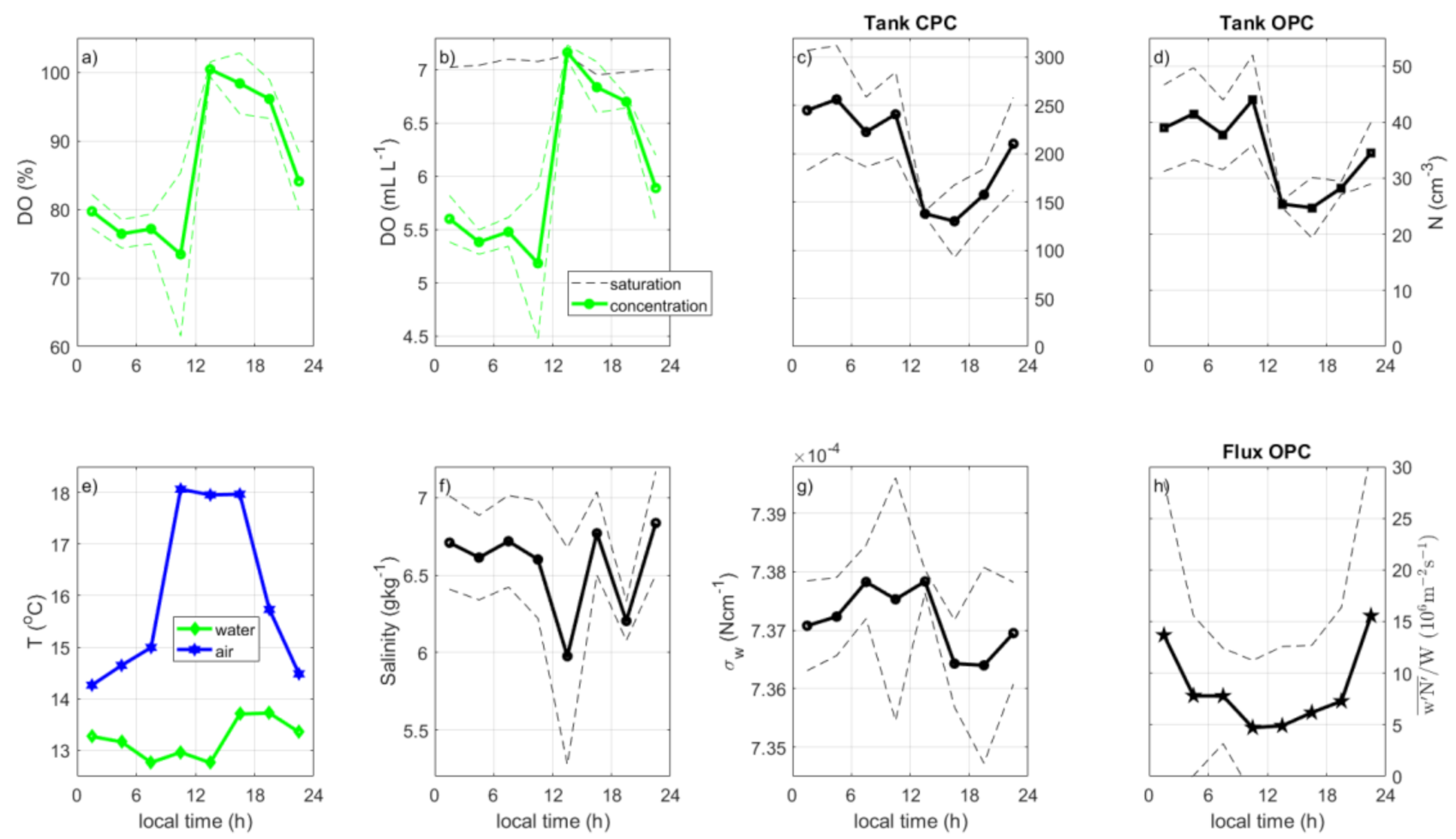
3.5. Ambient Diurnal Cycles
3.6. Diurnal Cycles According to in situ Fluxes vs. Tank Sea Spray Production
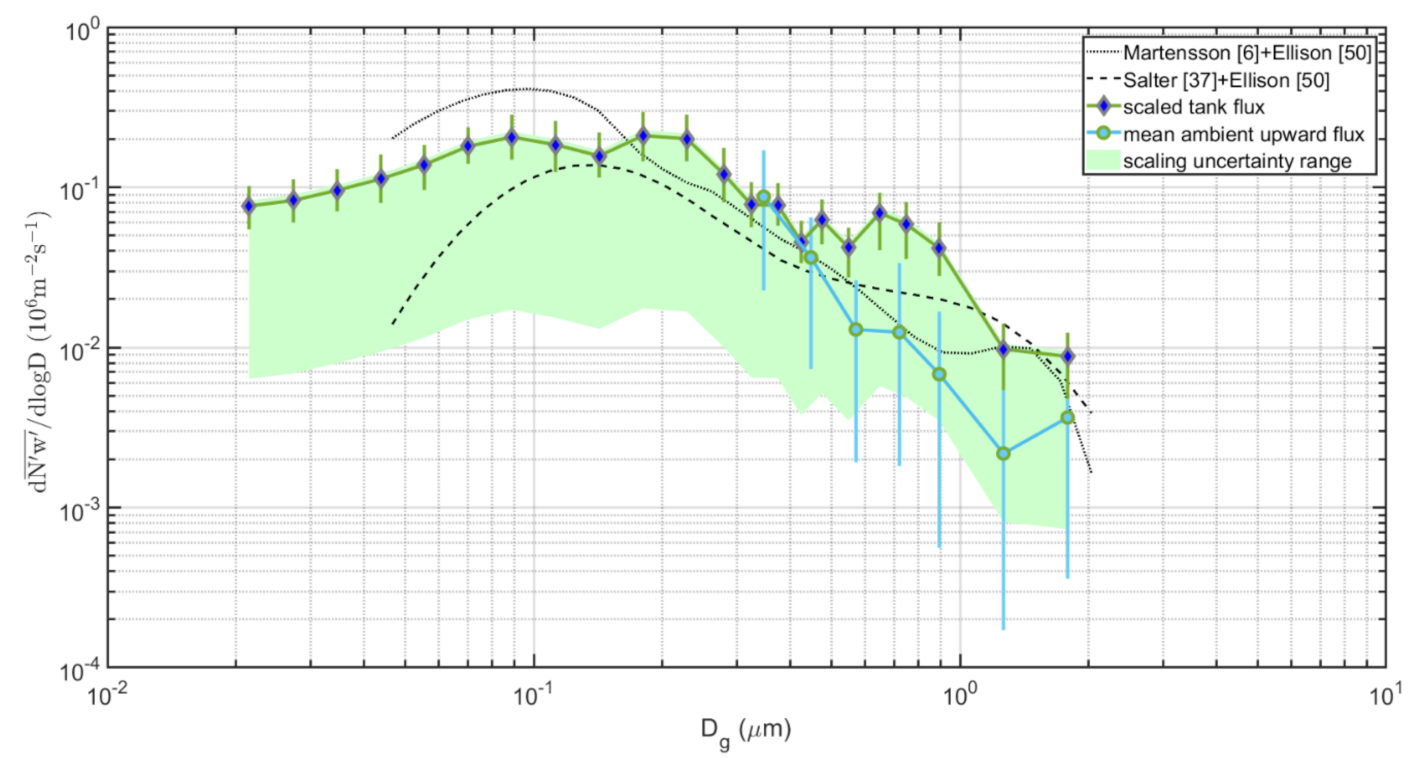
3.7. The Complete Sea Spray Emission Size Spectra vs. Source Parameterizations
- (i)
- The difference in slope over aerosol size between the tank sea spray simulation and the direct in situ EC fluxes remains.
- (ii)
- The conclusion that sea spray is produced in a wide sub micrometer range, centered and peaking at about 0.09–0.3 µm dry diameter is not affected by the scaling.
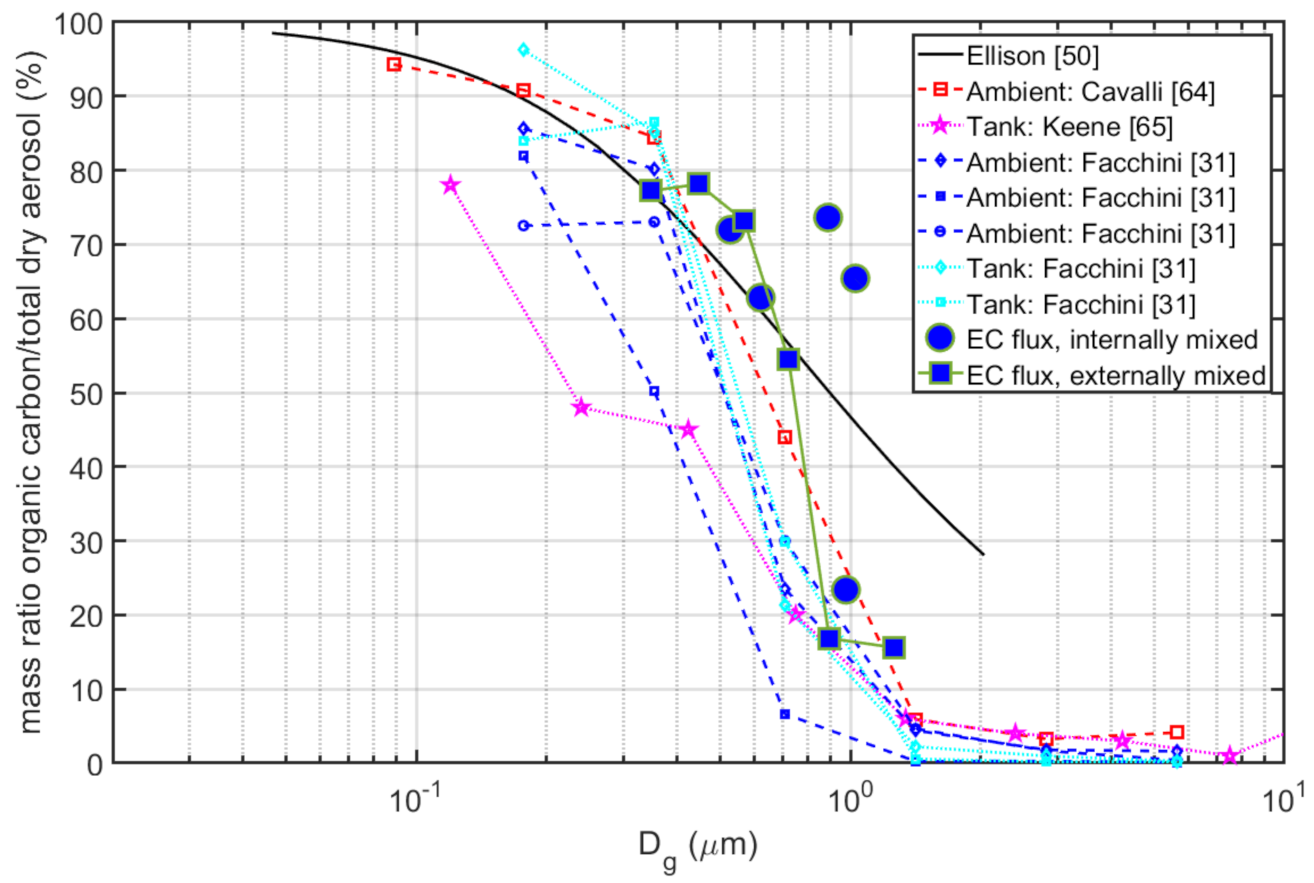
3.8. Chemical Interpretation of The Semi-Volatile/Non-Volatile EC Flux Fractions
4. Summary and Conclusions
- We have presented the first ever sea spray aerosol EC fluxes at near-coastal conditions and with limited fetch, and the first over a water surface with low salinity. Under these conditions, increased wind speed produces more sea spray than usual, and the emissions approximately follow the wind power law, that is, these exhibit a strong non-linear response to increase in wind speed. This compares reasonably well with previous studies from remote open oceans with normal salinity.
- It is, however, only the heated non-volatile (sea salt) aerosol fluxes that show a highly significant increase in wind speed, similar to what we have come to expect from sea spray EC fluxes. Considering that this is a much more polluted region compared to the remote oceans where previous studies had taken place, the dry unheated aerosol fluxes probably include too much semi-volatile anthropogenic aerosols (which contribute negative deposition fluxes that may mask the emission fluxes, not to mention increasing the random errors). This emphasizes the importance of including a volatility system, a thermodenuder, in EC aerosol flux systems when studying sea spray emissions in polluted regions.
- It was not trivial to combine tank and EC flux data into a continuous sea spray emission size spectrum from 0.01 µm to 2 µm diameter. We are unable to be conclusive regarding the scaling factor between the fluxes derived from the tank measurements and EC fluxes. The primary reason is that it appears both tank-derived fluxes and EC fluxes have different slopes over the aerosol size. Future work is needed to study if this is a feature related to the different methods used, and if it can be overcome.
- Differences between various sectors (Table 1) in the particle production per white cap area were relatively small (2.2 – 2.7 × 107 m−2s−1), which suggests that the approach by [6] and many others (to parameterize the sea spray emissions per white cap surface separately from the white cap cover wind-function) is a reasonable simplification. This was contradicted by the scaling of our tank-derived fluxes to our EC fluxes (Figure 12), which resulted in a range of scaling factors that cannot be explained by the white cap fraction vs. tank bubble surface. There were most likely other complications that may add to the uncertainty in the scaling between tank experiments and in situ fluxes. We had hoped that this problem would have been circumnavigated if one scales the tank sea spray simulations to actual in situ EC fluxes, as we have done in this study. However, as noted above, because the two methods resulted in sea spray emission fluxes with different slopes over aerosol size, we still had problems to resolve.
- Diurnal cycles for in situ particle production were closely related to wind speed and white cap coverage, with maxima at around noon. Tank sea spray production had an afternoon minimum in their diurnal cycle. In situ sea spray production changed from a peak at noon to a minimum at noon when normalized to the production per white cap area. Although with somewhat different timing, the diurnal cycles supported the existence of a biologically driven diurnal cycle in the sea spray production as suggested by [13], where the dissolved oxygen indicated a connection to photosynthesis or respiration.
- Surprisingly, there appeared to be less organic sea spray production from the shallow waters in the coastal zone and Kalmar Strait zone than in the long fetch zone. It may well be that this is an artefact due to negative deposition fluxes of anthropogenic particles embedded in the upward net fluxes.
- In the long fetch zone, we were able to distinguish between observed aerosol emission fluxes of dry aerosol (unheated, both sea spray and organics) and non-volatile aerosols (heated, sea salt only) in the smallest size bins of the OPC. This did not apply to the fluxes from shallow and coastal waters. The long fetch zone aerosol flux size distributions and organic fraction were in agreement with previous studies, but we were unable to conclude if the organic and sea salt sea spray in the OPC range occurred as internal or external mixtures. Both are equally plausible.
- In our study, our estimates of the relative organic sea spray distribution over size using the EC method in combination with a thermodenuder demonstrated that this combination of methods was able to distinguish between organic and sea salt sea spray. It also confirms the conclusion by [31] that the shift from a predominantly sea salt mass in the super micrometer to a predominantly organic mass at ≈0.1 µm diameter is a signature of the sea spray aerosol.
- As a first guess, over sea spray emissions from a brackish water such as the Baltic Sea, in both [6] and [37], sea salt source parameterization appeared to work, as long as the fluxes were modified for the actual salinity by shifting the particle diameters proportionally to the cubic root of the salinity, as suggested by [6].
- The Ellison [50] organic mono-layer model appears to be able to explain most of the differences we observed between the dry aerosol and heated non-volatile aerosol. However, the rate of change in organic fraction over aerosol size, as suggested by [50], appeared somewhat smaller than most observations, including this study (see Figure 13).
- The observed particle production by size was somewhere in the vicinity of both pure sea salt models [6,37], when these were modified for the salinity and simple surfactant monolayer model, as in [50]. In some sectors and size intervals, the emissions were even larger, and water soluble organics or liquid colloidal micelles within the droplets/particles could have contributed to the organic fraction. In most cases, we were not able to be conclusive due to uncertainty in the flux measurements.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Notations
| N | aerosol number concentration, cm−3 |
| half hourly average aerosol number concentration, cm−3 | |
| N’ | turbulent fluctuation in aerosol number concentration, cm−3 |
| w | vertical wind speed, ms−1 |
| half hourly average vertical wind speed, ms−1 | |
| w’ | turbulent fluctuation in vertical wind speed, ms−1 |
| turbulent aerosol number flux, eddy covariance flux, m−2s−1 | |
| F | aerosol emission flux, m−2s−1 |
| Fm | measured turbulent aerosol flux limited by the sensor response time, m−2s−1 |
| Fc | aerosol flux corrected for limited sensor response, or aerosol emission flux, m−2s−1 |
| τc | frequency first order response time constant, s |
| nm | normalized frequency |
| z | measurement height, m |
| L | Monin–Obukov length, m |
| mean horizontal wind speed, ms−1 | |
| U | mean horizontal wind speed at 10 m, ms−1 |
| Q | sampling flow, m3s−1 |
| σw | vertical wind standard deviation, ms−1 |
| t | sampling period, s |
| W | white cap coverage, % |
| Xf | fetch, m |
| CD | drag coefficient |
| S | salinity, ppt |
| Dg | geometric mean of the upper and lower diameters of each OPC (optical particle counter) size bin, used as the center value for the size bin when plotted with a logarithmic diameter axis, µm |
| D35 | sea salt sea spray particle dry diameter at S = 35ppt, µm |
| DS | sea salt sea spray particle dry diameter at salinity S, µm |
| DO | dissolved oxygen, % or mL L−1 |
| σw | surface tension, Nm−1 |
References
- Blanchard, D.C. The Production, Distribution, and Bacterial Enrichment of the Sea-Salt Aerosol. C. Mathematical and Physical Sciences No. 108, NATO ASI Series; Reidel Publishing Company: Dordrecht, The Netherlands, 1983; pp. 407–454. [Google Scholar]
- Blanchard, D.C.; Syzdek, L.D. Film drop production as a function of bubble size. J. Geophys. Res. Space Phys. 1988, 93, 3649–3654. [Google Scholar] [CrossRef]
- Resch, F.; Afeti, G. Submicron film drop production by bubbles in seawater. J. Geophys. Res. Space Phys. 1992, 97, 3679–3683. [Google Scholar] [CrossRef]
- Spiel, D.E. On the births of film drops from bubbles bursting on seawater surfaces. J. Geophys. Res. Space Phys. 1998, 103, 24907–24918. [Google Scholar] [CrossRef]
- Cipriano, R.J.; Blanchard, D.C. Bubble and Aerosol Spectra Produced by a Laboratory ‘Breaking Wave’. J. Geophys. Res. 1981, 86, 8085–8092. [Google Scholar] [CrossRef]
- Mårtensson, E.M.; Nilsson, E.D.; de Leeuw, G.; Cohen, L.H.; Hansson, H.-C. Laboratory simulations and parameterization of the primary marine aerosol production. J. Geophys. Res. 2003, 108, 4297. [Google Scholar]
- Monahan, E.C.; Ó’Muircheartaigh, I. Optimal Power-Law Description of Oceanic Whitecap Coverage Dependence on Wind Speed. J. Phys. Oceanogr. 1980, 10, 2094–2099. [Google Scholar] [CrossRef] [Green Version]
- Slinn, A.A.; Slinn, W.G.N. Predictions for particle deposition on natural waters. Atmos. Environ. 1980, 14, 1013–1016. [Google Scholar] [CrossRef]
- Zufall, M.J.; Davidson, C.I.; Caffrey, P.F.; Ondov, J.M. Airborne Concentrations and Dry Deposition Fluxes of Particluate Species to Surrogate Surfaces Deployed in Southern Lake Michigan. Environ. Sci. Technol. 1998, 32, 1623–1628. [Google Scholar] [CrossRef]
- Nair, P.R.; Parameswaran, K.; Abraham, A.; Jacob, S. Wind-dependence of sea-salt and non-sea-salt aerosols over the oceanic environment. J. Atmos. Solar-Terr. Phys. 2005, 67, 884–898. [Google Scholar] [CrossRef]
- Sellegri, K.; O’Dowd, C.D.; Yoon, Y.J.; Jennings, S.G.; de Leeuw, G. Surfactants and submicron sea spray generation. J. Geophys. Res. 2006, 111, D22215. [Google Scholar] [CrossRef] [Green Version]
- Tyree, C.A.; Hellion, V.M.; Alexandrova, O.A.; Allen, J.O. Foam droplets generated from natural and artificial seawaters. J. Geophys. Res. Space Phys. 2007, 112. [Google Scholar] [CrossRef] [Green Version]
- Hultin, K.A.H.; Nilsson, E.D.; Krejci, R.; Mårtensson, E.M.; Ehn, M.; Hagström, Å.; De Leeuw, G. In situ laboratory sea spray production during the Marine Aerosol Production 2006 cruise on the northeastern Atlantic Ocean. J. Geophys. Res. Space Phys. 2010, 115. [Google Scholar] [CrossRef]
- Hultin, K.A.; Krejci, R.; Pinhassi, J.; Gómez-Consarnau, L.; Mårtensson, E.M.; Hagström, Å.; Nilsson, E.D. Aerosol and bacterial emissions from Baltic Seawater. Atmos. Res. 2011, 99, 1–14. [Google Scholar] [CrossRef]
- Unger, I.; Saak, C.-M.; Salter, M.; Zieger, P.; Patanen, M.; Bjorneholm, O. Influence of Organic Acids on the Surface Composition of Sea Spray Aerosol. J. Phys. Chem. A 2020, 124, 422–429. [Google Scholar] [CrossRef]
- Cochran, R.E.; Laskina, O.; Trueblood, J.V.; Estillore, A.D.; Morris, H.S.; Jayarathne, T.; Sultana, C.M.; Lee, C.; Lin, P.; Laskin, J.; et al. Molecular Diversity of Sea Spray Aerosol Particles: Impact of Ocean Biology on Particle Composition and Hygroscopicity. Chem 2017, 2, 655–667. [Google Scholar] [CrossRef] [Green Version]
- Schiffer, J.M.; Mael, L.E.; Prather, K.A.; Amaro, R.E.; Grassian, V.H. Sea Spray Aerosol: Where Marine Biology Meets Atmospheric Chemistry. ACS Central Sci. 2018, 4, 1617–1623. [Google Scholar] [CrossRef]
- Bird, J.C.; De Ruiter, R.; Courbin, L.; Stone, H.A. Daughter bubble cascades produced by folding of ruptured thin films. Nature 2010, 465, 759–762. [Google Scholar] [CrossRef]
- Brasz, C.F.; Bartlett, C.T.; Walls, P.L.L.; Flynn, E.G.; Yu, Y.E.; Bird, J.C. Minimum size for the top jet drop from a bursting bubble. Phys. Rev. Fluids 2018, 3, 074001. [Google Scholar] [CrossRef]
- Leifer, I.; de Leeuw, G.; Cohen, L.H. Secondary bubble production from breaking waves: The bubble burst mechanism. Geophys. Res. Lett. 2000, 27, 4077–4080. [Google Scholar] [CrossRef]
- Nilsson, E.D.; Rannik, Ü.; Buzorius, G.; Kulmala, M.; O’Dowd, C.D. Effects of continental boundary layer evolution, convection, turbulence and entrainment on aerosol formation. Tellus 2001, 53B, 441–461. [Google Scholar] [CrossRef]
- Vogt, M.; Nilsson, E.D.; Ahlm, L.; MÕrtensson, E.M.; Johansson, C. Seasonal and diurnal cycles of 0.25–2.5 µm aerosol fluxes over urban Stockholm, Sweden. Tellus B Chem. Phys. Meteorol. 2011, 63, 935–951. [Google Scholar] [CrossRef] [Green Version]
- Ahlm, L.; Krejci, R.; Nilsson, E.D.; Mårtensson, E.M.; Vogt, M.; Artaxo, P. Emission and dry deposition of accumulation mode particles in the Amazon Basin. Atmos. Chem. Phys. Discuss. 2010, 10, 10237–10253. [Google Scholar] [CrossRef] [Green Version]
- Nilsson, E.D.; Rannik, Ü.; Swietlicki, E.; Leck, C.; Aalto, P.P.; Zhou, J.; Norman, M. Turbulent aerosol fluxes over the Arctic Ocean 2. Wind-driven sources from the sea. J. Geophys. Res. 2001, 106, 32139–32154. [Google Scholar] [CrossRef]
- Geever, M.; O’Dowd, C.D.; van Ekeren, S.; Flanagan, R.; Nilsson, E.D.; de Leeuw, G.; Rannik, Ü. Submicron sea spray fluxes. Geophys. Res. Lett. 2005, 32, L15810. [Google Scholar] [CrossRef] [Green Version]
- Nilsson, E.D.; Mårtensson, E.M.; Van Ekeren, J.S.; de Leeuw, G.; Moerman, M.; O’Dowd, C. Primary marine aerosol emissions: Size resolved eddy covariance measurements with estimates of sea salt and organic carbon fractions. Atmos. Chem. Phys. Disc. 2007, 7, 13345–13400. [Google Scholar]
- Norris, S.J.; Brooks, I.M.; De Leeuw, G.; Smith, M.H.; Moerman, M.; Lingard, J.J.N. Eddy covariance measurements of sea spray particles over the Atlantic Ocean. Atmospheric Chem. Phys. Discuss. 2008, 8, 555–563. [Google Scholar] [CrossRef] [Green Version]
- Gong, S.L.; Barrie, L.A.; Prospero, J.M.; Savoie, D.L.; Ayers, G.P.; Blanchet, J.-P.; Spacek, L. Modeling sea-salt aerosols in the atmosphere 2. Atmospheric concentrations and fluxes. J. Geophys. Res. 1997, 102, 3819–3830. [Google Scholar] [CrossRef] [Green Version]
- Grini, A.; Myhre, G.; Sundet, J.K.; Isaksen, I.S.A. Modeling the Annual Cycle of Sea Salt in the Global 3D Model Oslo CTM2: Concentrations, Fluxes, and Radiative Impact. J. Clim. 2002, 15, 1717–1730. [Google Scholar] [CrossRef]
- Middlebrook, A.M.; Murphy, D.M.; Thomson, D.S. Observations of organic material in individual marine particles at Cape Grim during First Aerosol Characterization Experiment (ACE 1). J. Geophys. Res. 1998, 103, 16475–16483. [Google Scholar] [CrossRef]
- Facchini, M.C.; Rinaldi, M.; Decesari, S.; Carbone, C.; Finessi, E.; Mircea, M.; Fuzzi, S.; Ceburnis, D.; Flanagan, R.; Nilsson, E.D.; et al. Primary submicron marine aerosol dominated by insoluble organic colloids and aggregates. Geophys. Res. Lett. 2008, 35. [Google Scholar] [CrossRef]
- O’Dowd, C.D.; Langmann, B.; Varghese, S.; Scannell, C.; Ceburnis, D.; Facchini, M.C. A combined organic--inorganic sea--spray source function. Geophys. Res. Lett. 2008, 35. [Google Scholar] [CrossRef] [Green Version]
- Russell, L.M.; Hawkins, L.N.; Frossard, A.A.; Quinn, P.K.; Bates, T.S. Carbohydrate-like composition of submicron atmospheric particles and their production from ocean bubble bursting. Proc. Natl. Acad. Sci. USA 2009, 107, 6652–6657. [Google Scholar] [CrossRef] [Green Version]
- Murphy, D.M.; Anderson, J.R.; Quinn, P.K.; McInnes, L.M.; Brechtel, F.J.; Kreidenweis, S.; Middlebrook, A.M.; Pósfai, M.; Thomson, D.S.; Buseck, P.R. Influence of sea-salt on aerosol radiative properties in the Southern Ocean marine boundary layer. Nature 1998, 392, 62–65. [Google Scholar] [CrossRef]
- O’Dowd, C.D.; Smith, M.H.; Consterdine, I.E.; Lowe, J.A. Marine aerosol, sea-salt, and the marine sulphur cycle: A short review. Atmos. Environ. 1997, 31, 73–80. [Google Scholar]
- Monahan, E.C.; Spiel, D.E.; Davidson, K.L. A model of marine aerosol generation via whitecaps and wave disruption. In Oceanic Whitecaps; Monahan, E.C., Mac Niocaill, G., Eds.; Reidel Publishing Company: Dordrecht, The Netherlands, 1986; 167p. [Google Scholar]
- Salter, M.E.; Zieger, P.; Navarro, J.C.A.; Grythe, H.; Kirkevåg, A.; Rosati, B.; Riipinen, I.; Nilsson, E.D. An empirically derived inorganic sea spray source function incorporating sea surface temperature. Atmos. Chem. Phys. Discuss. 2015, 15, 11047–11066. [Google Scholar] [CrossRef] [Green Version]
- Pierce, J.R.; Adams, P.J. Global evaluation of CCN formation by direct emission of sea salt and growth of ultrafine sea salt. J. Geophys. Res. Space Phys. 2006, 111. [Google Scholar] [CrossRef]
- Korhonen, H.; Carslaw, K.S.; Spracklen, D.V.; Mann, G.W.; Woodhouse, M.T. Influence of oceanic dimethyl sulfide emissions on cloud condensation nuclei concentrations and seasonality over the remote Southern Hemisphere oceans: A global model study. J. Geophys. Res. Space Phys. 2008, 113. [Google Scholar] [CrossRef]
- Heslin-Rees, D.; Burgos, M.; Hansson, H.C.; Krejci, R.; Ström, J.; Tunved, P.; Zieger, P. From a polar to a marine environment: Has the changing Arctic led to a shift in aerosol light scattering properties? Atmos. Chem. Phys. 2020, 20, 13671–13686. [Google Scholar] [CrossRef]
- Gaman, A.; Rannik, Ü.; Aalto, P.; Pohja, T.; Siivola, E.; Kulmala, M.; Vesala, T. Relaxed Eddy Accumulation System for Size-Resolved Aerosol Particle Flux Measurements. J. Atmos. Ocean. Technol. 2004, 21, 933–943. [Google Scholar] [CrossRef]
- Rheinheimer, G. Pollution in the Baltic Sea. Naturwissenschaften 1998, 85, 318–329. [Google Scholar] [CrossRef] [PubMed]
- Kljun, N.; Calanca, P.; Rotach, M.W.; Schmid, H.P. A Simple Parameterisation for Flux Footprint Predictions. Boundary-Layer Meteorol. 2004, 112, 503–523. [Google Scholar] [CrossRef]
- Kaimal, J.C.; Finnigan, J.J. Atmospheric Boundary Layer Flows: Their Structure and Measurement; Oxford University Press: Oxford, UK, 1994. [Google Scholar]
- Buzorius, G.; Rannik, Ü.; Mäkelä, J.M.; Keronen, P.; Vesala, T.; Kulmala, M. Vertical aerosol fluxes measured by the eddy covariance method and deposition of nucleation mode particles above a Scots pine forest in southern Finland. J. Geophys. Res. Space Phys. 2000, 105, 19905–19916. [Google Scholar] [CrossRef]
- Ahlm, L.; Nilsson, E.D.; Krejci, R.; Mrtensson, E.M.; Vogt, M.; Artaxo, P. Aerosol number fluxes over the Amazon rain forest during the wet season. Atmos. Chem. Phys. Discuss. 2009, 9, 9381–9400. [Google Scholar] [CrossRef] [Green Version]
- Rannik, Ü.; Vesala, T.; Keskinen, R. On the damping of temperature fluctuations in a circular tube relevant to the eddy covariance measurement technique. J. Geophys. Res. Space Phys. 1997, 102, 12789–12794. [Google Scholar] [CrossRef]
- Buzorius, G.; Rannik, Ü.; Nilsson, E.; Vesala, T.; Kulmala, M. Analysis of measurement techniques to determine dry deposition velocities of aerosol particles with diameters less than 100 nm. J. Aerosol Sci. 2003, 34, 747–764. [Google Scholar] [CrossRef]
- Jokinen, V.; Mäkelä, J.M. Closed-loop arrangement with critical orifice for DMA sheath/excess flow system. J. Aerosol Sci. 1997, 28, 643–648. [Google Scholar] [CrossRef]
- Ellison, G.B.; Tuck, A.F.; Vaida, V. Atmospheric processing of organic aerosols. J. Geophys. Res. Space Phys. 1999, 104, 11633–11641. [Google Scholar] [CrossRef]
- Zábori, J.; Matisans, M.; Krejci, R.; Nilsson, E.D.; Ström, J. Artificial primary marine aerosol production: A laboratory study with varying water temperature, salinity, and succinic acid concentration. Atmos. Chem. Phys. 2012, 12, 10709. [Google Scholar] [CrossRef] [Green Version]
- Piazzola, J.; Forget, P.; Despiau, S. A sea spray generation function for fetch-limited conditions. Ann. Geophys. 2002, 20, 121–131. [Google Scholar] [CrossRef]
- Ceburnis, D.; Rinaldi, M.; Ovadnevaite, J.; Martucci, G.; Giulianelli, L.; O’Dowd, C.D. Marine submicron aerosol gradients, sources and sinks. Atmos. Chem. Phys. 2016, 16, 12425–12439. [Google Scholar] [CrossRef] [Green Version]
- O’Dowd, C.D.; Smith, M.H. Physicochemical properties of aerosols over the northeast Atlantic: Evidence for wind-speed-related submicron sea-salt aerosol production. J. Geophys. Res. Space Phys. 1993, 98, 1137–1149. [Google Scholar] [CrossRef]
- Siedler, G.; Peters, H. Physical properties (general) of sea water. In LANDOLT-BÖRNSTEIN, Numerical Data and Functional Relationships in Science and Technology; New Series; Oceanography; Teilband: Berlin, Germany, 1986; Volume V/3a, pp. 233–264. [Google Scholar]
- Marty, J.C.; Saliot, A.; Buat-Menard, P.; Chesselet, R.; Hunter, K.A. Relationship between the lipid compositions of marine aerosols, the sea surface microlayer, and subsurface water. J. Geophys. Res. Space Phys. 1979, 84, 5707. [Google Scholar] [CrossRef]
- Tervahattu, H.; Juhanoja, J.; Kupiainen, K. Identification of an organic coating on marine aerosol particles by TOF-SIMS. J. Geophys. Res. Space Phys. 2002, 107, ACH-18. [Google Scholar] [CrossRef]
- Damay, P.; Maro, E.; Coppalle, D.; Lamaud, A.; Connan, E.; Hébert, O.; Talbaut, D. Size-resolved eddy covariance measurements of fine particle vertical fluxes. J. Aerosol Sci. 2009, 40, 1050–1058. [Google Scholar] [CrossRef]
- Clarke, A.D.; Owens, S.R.; Zhou, J. An ultrafine sea-salt flux from breaking waves: Implications for cloud condensation nuclei in the remote marine atmosphere. J. Geophys. Res. 2006, 111, D06202. [Google Scholar] [CrossRef] [Green Version]
- Petelski, T.; Piskozub, J. Vertical coarse aerosol fluxes in the atmospheric surface layer over the North Polar Waters of the Atlantic. J. Geophys. Res. Ocean. 2006, 111, C6. [Google Scholar] [CrossRef] [Green Version]
- Savelyev, I.B.; Anguelova, M.D.; Frick, G.M.; Dowgiallo, D.J.; Hwang, P.A.; Caffrey, P.F.; Bobak, J.P. On direct passive microwave remote sensing of sea spray aerosol production. Atmos. Chem. Phys. 2014, 14, 11611. [Google Scholar] [CrossRef] [Green Version]
- Markuszewski, P.; Klusek, Z.; Nilsson, E.D.; Petelski, T. Observations on relations between marine aerosol fluxes and surface-generated noise in the southern Baltic Sea. Oceanologia 2020, 62, 413–427. [Google Scholar] [CrossRef]
- Hwang, P.; Setten, M. Energy dissipation of wind-generated waves and whitecap coverage. J. Geophys. Res. 2008, 113. [Google Scholar] [CrossRef]
- Cavalli, F.; Facchini, M.C.; Decesari, S.; Mircea, M.; Emblico, L.; Fuzzi, S.; Ceburnis, D.; Yoon, Y.J.; O’Dowd, C.D.; Putaud, J.-P.; et al. Advances in characterization of size-resolved organic matter in marine aerosol over the North Atlantic. J. Geophys. Res. 2004, 109, D24215. [Google Scholar] [CrossRef]
- Keene, W.C.; Maring, H.; Maben, J.R.; Kieber, D.J.; Pszenny, A.A.P.; Dahl, E.E.; Izaguirre, M.A.; Davis, A.J.; Long, M.S.; Zhou, X.; et al. Chemical and physical characteristics of nascent aerosols produced by bursting bubbles at a model air-sea interface. J. Geophys. Res. Space Phys. 2007, 112. [Google Scholar] [CrossRef] [Green Version]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| n | Xf Range (km) | Mean CD | Mean U (ms−1) | Mean W (%) | Mean (m−2s−1) | Mean /W (m−2s−1) |
|---|---|---|---|---|---|---|
| Coastal sector, 205–270° | ||||||
| 148 | 2–10 | 1.6 × 10−3 | 7.6 (15.3) | 0.23 (1.3) | 1.4 (1.6) × 104 | 6.4 (7.0) × 106 |
| Long fetch sector, 145–205° | ||||||
| 29 | 200–300 | 9.7 × 10−4 | 4.6 (8.1) | 0.17 (0.61) | 2.2 (0.7) × 104 | 1.7 (0.6) × 107 |
| Kalmar Strait sector, 0–145° | ||||||
| 45 | ≈20 | 1.3 × 10−3 | 6.5 (10.7) | 0.31 (0.70) | 8.6 (4.7) × 103 | 4.5 (2.6) × 106 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nilsson, E.D.; Hultin, K.A.H.; Mårtensson, E.M.; Markuszewski, P.; Rosman, K.; Krejci, R. Baltic Sea Spray Emissions: In Situ Eddy Covariance Fluxes vs. Simulated Tank Sea Spray. Atmosphere 2021, 12, 274. https://doi.org/10.3390/atmos12020274
Nilsson ED, Hultin KAH, Mårtensson EM, Markuszewski P, Rosman K, Krejci R. Baltic Sea Spray Emissions: In Situ Eddy Covariance Fluxes vs. Simulated Tank Sea Spray. Atmosphere. 2021; 12(2):274. https://doi.org/10.3390/atmos12020274
Chicago/Turabian StyleNilsson, Ernst Douglas, Kim A. H. Hultin, Eva Monica Mårtensson, Piotr Markuszewski, Kai Rosman, and Radovan Krejci. 2021. "Baltic Sea Spray Emissions: In Situ Eddy Covariance Fluxes vs. Simulated Tank Sea Spray" Atmosphere 12, no. 2: 274. https://doi.org/10.3390/atmos12020274
APA StyleNilsson, E. D., Hultin, K. A. H., Mårtensson, E. M., Markuszewski, P., Rosman, K., & Krejci, R. (2021). Baltic Sea Spray Emissions: In Situ Eddy Covariance Fluxes vs. Simulated Tank Sea Spray. Atmosphere, 12(2), 274. https://doi.org/10.3390/atmos12020274