
Atmospheric Degradation of Two Hydrofluoroketones: Theoretical Rate Constants for the Gas-Phase OH-Oxidation of HFK-447mcc and HFK-465mc


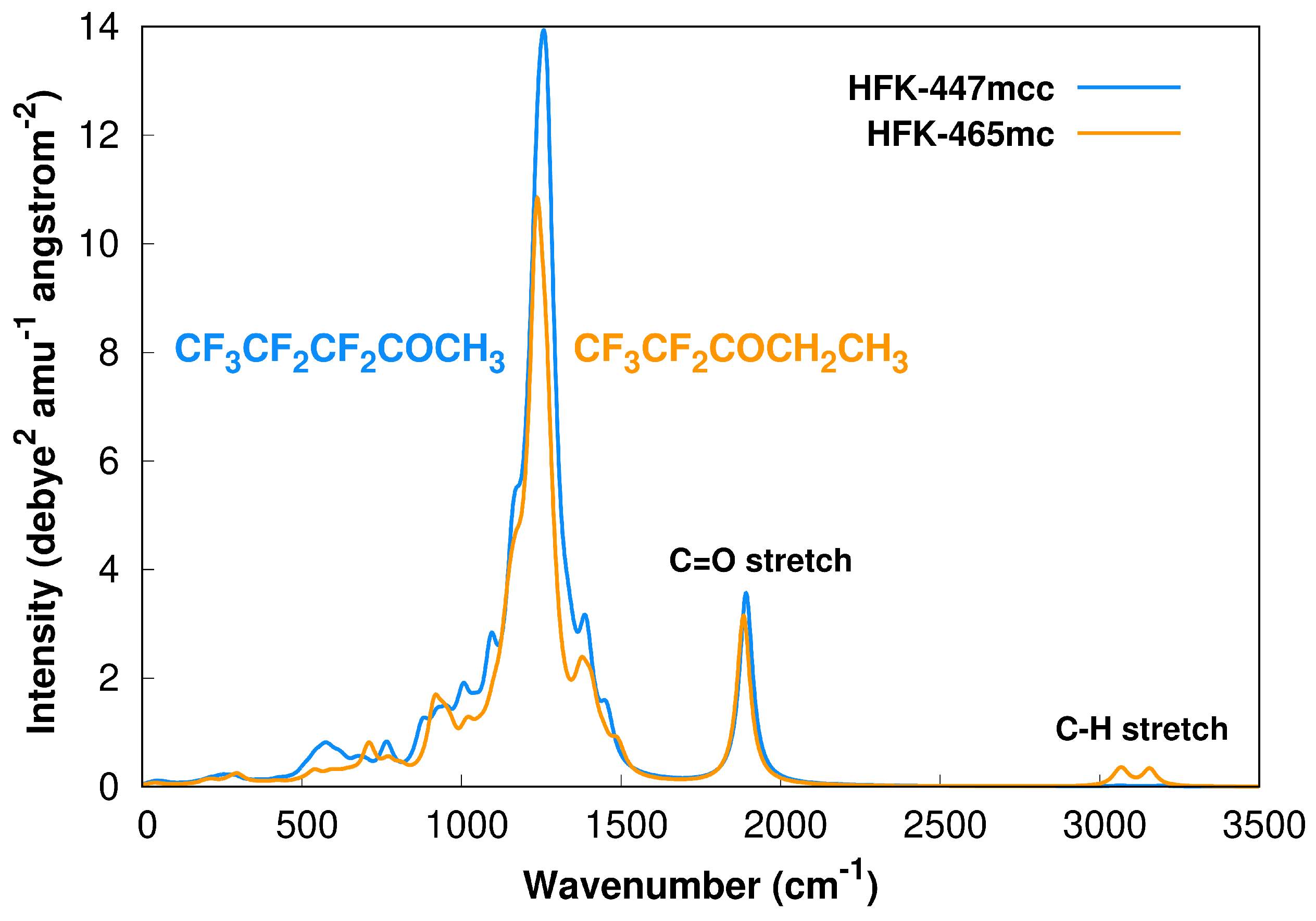{kind=link}
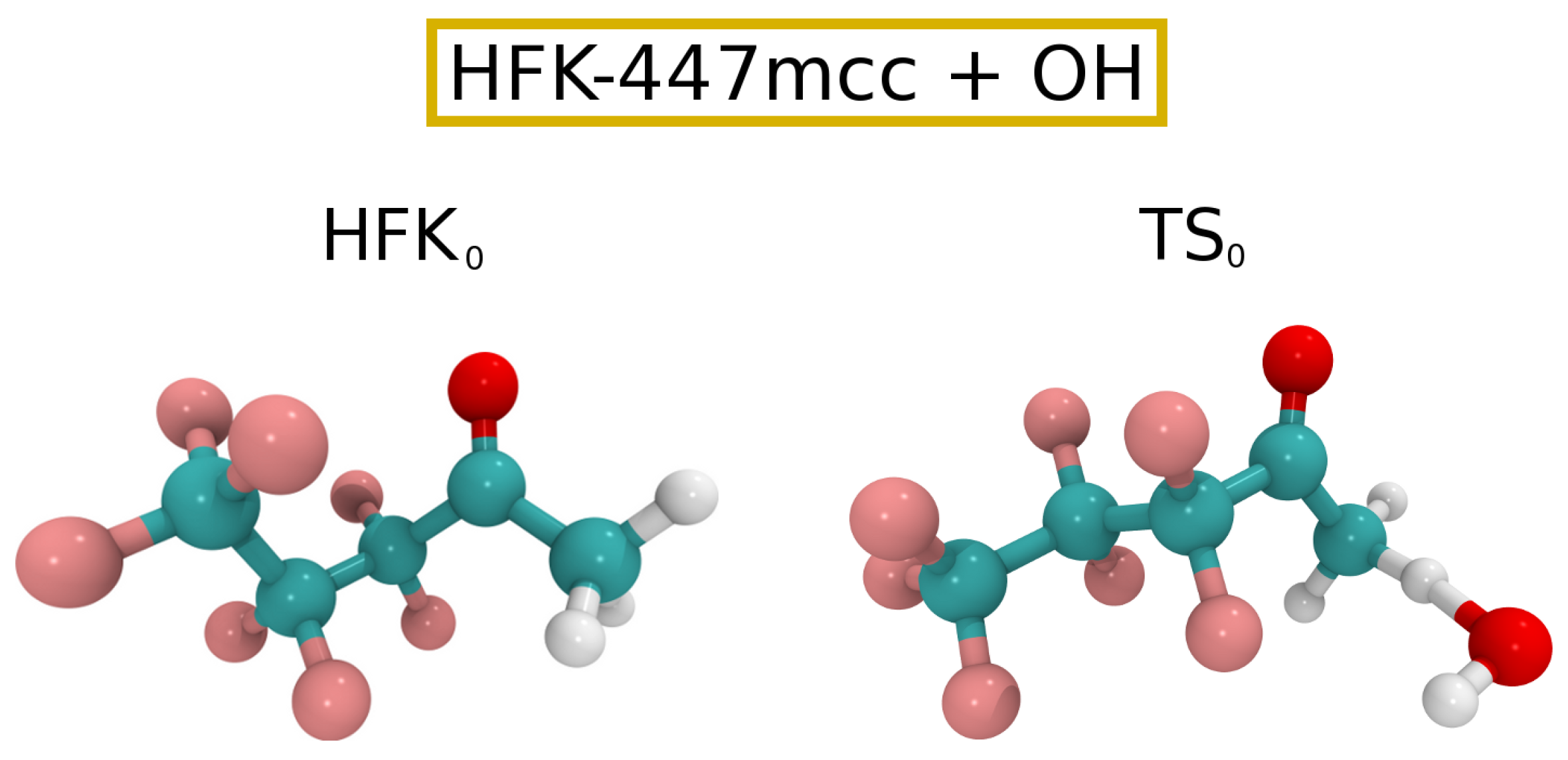{kind=link}
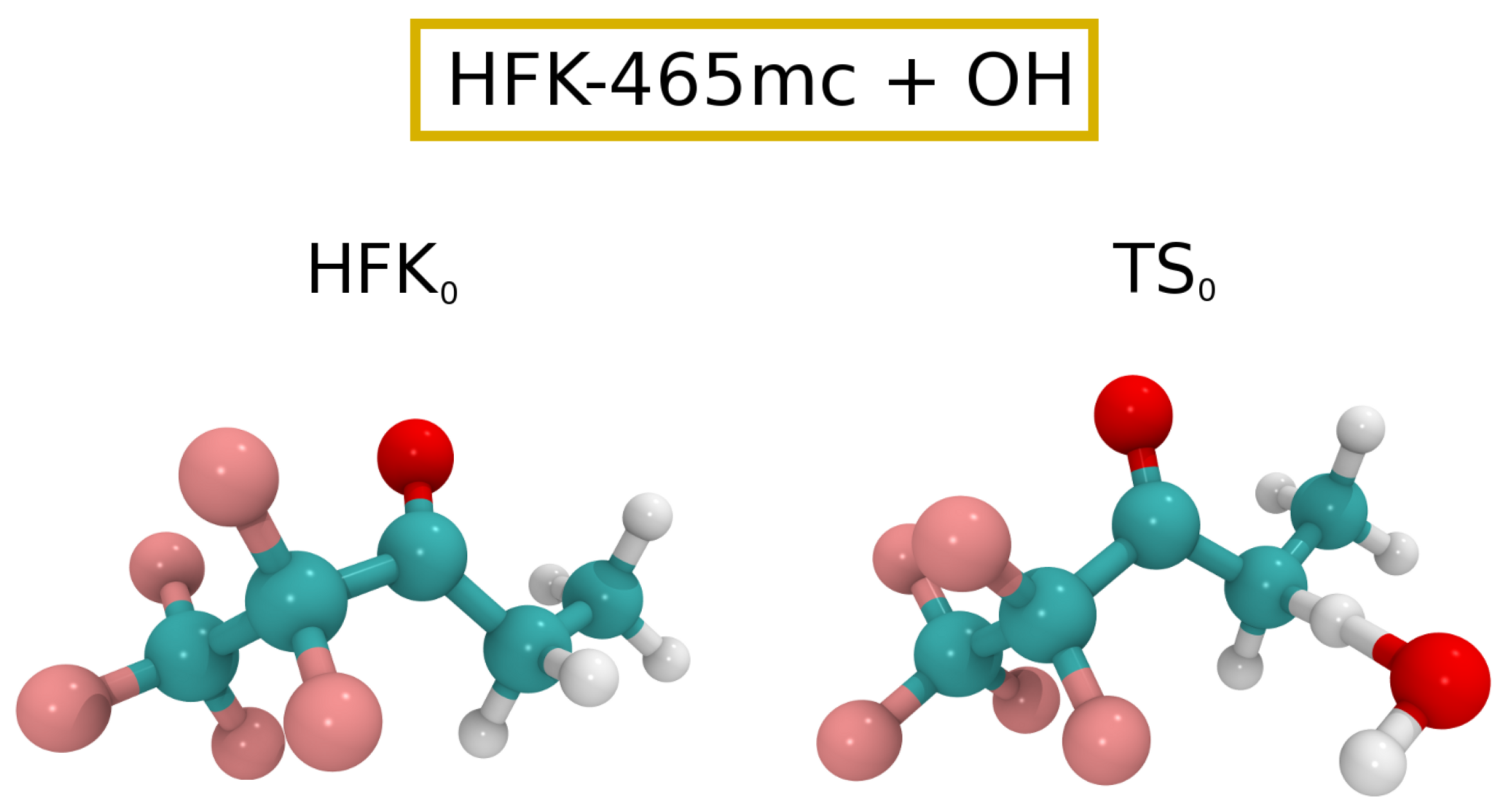{kind=link}
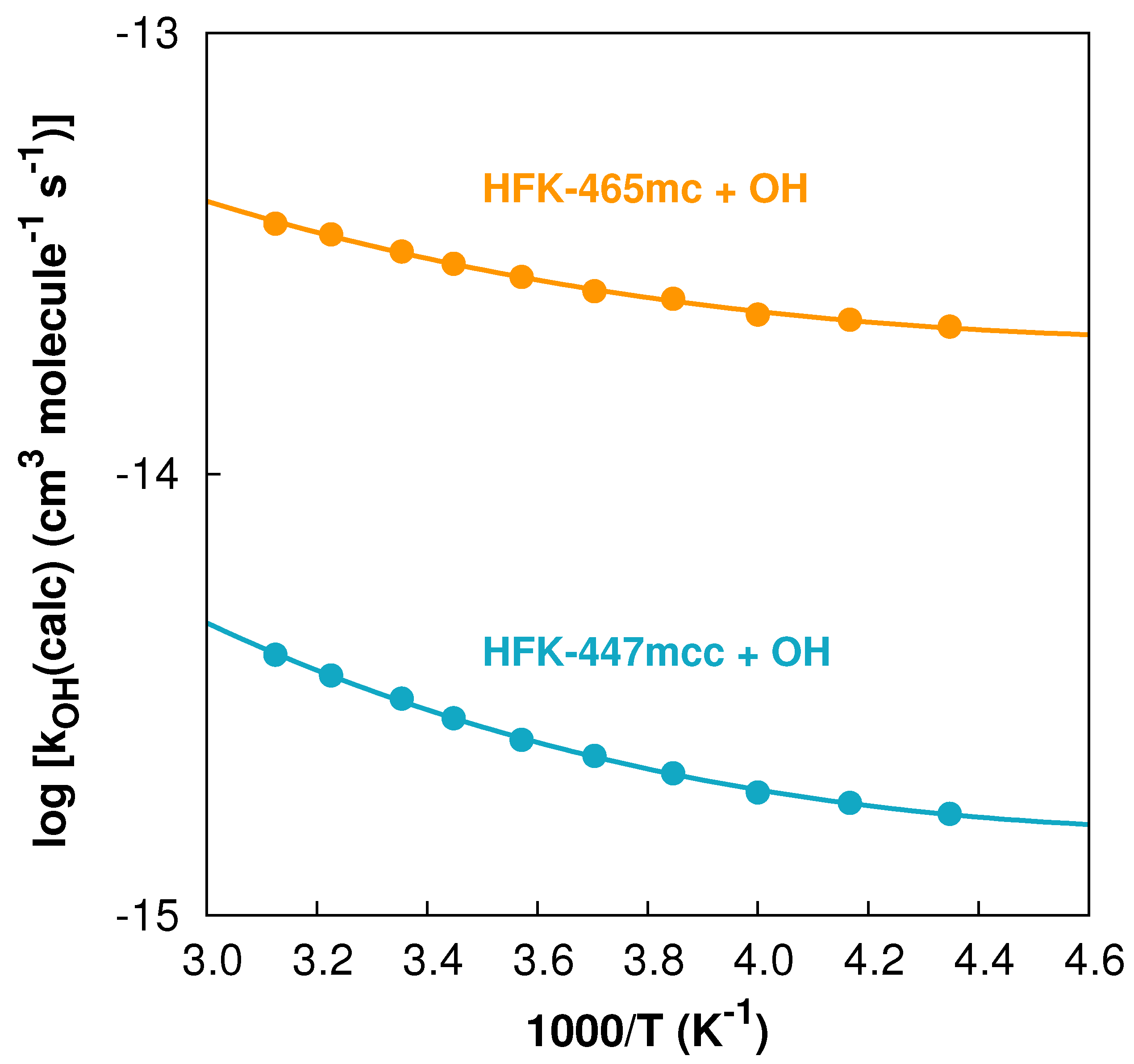{kind=link}
Abstract
:1. Introduction
2. Methodology
2.1. Theoretical Background
2.2. Computational Methods
3. Results and Discussion
3.1. OH Oxidation of HFK-447mcc
3.2. OH Oxidation of HFK-465mc
4. Conclusions
Supplementary Materials
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Molina, M.J.; Rowland, F.S. Stratospheric Sink for Chlorofluoromethanes: Chlorine Atom-Catalysed Destruction of Ozone. Nature 1974, 249, 810–812. [Google Scholar] [CrossRef]
- Farman, J.D.; Gardiner, B.G.; Shanklin, J.D. Large Losses of Total Ozone in Antarctica Reveal Seasonal ClOx/NOx Interaction. Nature 1985, 315, 207–210. [Google Scholar] [CrossRef]
- United Nations: Montreal Protocol on Substances that Deplete the Ozone Layer-Adjustments and Amendment. Int. Leg. Mater. 1993, 32, 874–887. [CrossRef]
- Lazarou, Y.G.; Papagiannakopoulos, P. Theoretical Investigation of the Thermochemistry of Hydrofluoroethers. Chem. Phys. Lett. 1999, 301, 19–28. [Google Scholar] [CrossRef]
- McGivern, B.P. Conference of the Parties to the Framework Convention on Climate Change: Kyoto Protocol. Int. Leg. Mater. 1998, 37, 22–43. [Google Scholar] [CrossRef]
- Heath, E.A. Amendment to the Montreal Protocol on Substances that Deplete the Ozone Layer (Kigali Amendment). Int. Leg. Mater. 2017, 56, 193–205. [Google Scholar] [CrossRef] [Green Version]
- UNEP. HFCs: A Critical Link in Protecting Climate and the Ozone Layer; United Nations Environment Programme (UNEP): 2011; 36p. Available online: https://www.unep.org/resources/report/hfcs-critical-link-protecting-climate-and-ozone-layer (accessed on 6 August 2022).
- Hodnebrog, Ø.; Etminan, M.; Fuglestvedt, J.S.; Marston, G.; Myhre, G.; Nielsen, C.J.; Shine, K.P.; Wallington, T.J. Global Warming Potentials and Radiative Efficiencies of Halocarbons and Related Compounds: A Comprehensive Review. Rev. Geophys. 2013, 51, 300–378. [Google Scholar] [CrossRef] [Green Version]
- Wallington, T.J.; Andersen, M.P.S.; Nielsen, O.J. Atmospheric Chemistry of Halogenated Organic Compounds. In Advances in Atmospheric Chemistry; World Scientific: Singapore, 2017; Volume 1, pp. 305–402. [Google Scholar]
- Mellouki, A.; Le Bras, G.; Sidebottom, H. Kinetics and Mechanisms of the Oxidation of Oxygenated Organic Compounds in the Gas Phase. Chem. Rev. 2003, 103, 5077–5096. [Google Scholar] [CrossRef]
- Calvert, J.G.; Mellouki, A.; Orlando, J.J.; Pilling, M.J.; Wallington, T.J. The Mechanisms of Atmospheric Oxidation of the Oxygenates, 1st ed.; Oxford University Press: New York, NY, USA, 2011. [Google Scholar]
- Mellouki, A.; Wallington, T.J.; Chen, J. Atmospheric Chemistry of Oxygenated Volatile Organic Compounds: Impacts on Air Quality and Climate. Chem. Rev. 2015, 115, 3984–4014. [Google Scholar] [CrossRef]
- Vereecken, L.; Francisco, J.S. Theoretical Studies of Atmospheric Reaction Mechanisms in the Troposphere. Chem. Soc. Rev. 2012, 41, 6259–6293. [Google Scholar] [CrossRef]
- Vereecken, L.; Glowacki, D.R.; Pilling, M.J. Theoretical Chemical Kinetics in Tropospheric Chemistry: Methodologies and Applications. Chem. Rev. 2015, 115, 4063–4114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atkinson, R.; Aschmann, S.M.; Carter, W.P.L.; Pitts, J.N., Jr. Rate Constants for the Gas-Phase Reaction of OH Radicals with a Series of Ketones at 299 ± 2 K. Int. J. Chem. Kinet. 1982, 14, 839–847. [Google Scholar] [CrossRef]
- Le Calvé, S.; Hitier, D.; Le Bras, G.; Mellouki, A. Kinetic Studies of OH Reactions with a Series of Ketones. J. Phys. Chem. A 1998, 102, 4579–4584. [Google Scholar] [CrossRef]
- Otake, K.; Yasumoto, M.; Yamada, Y.; Murata, J.; Urata, S. Critical Parameters and Vapor Pressure Measurements of Potential Replacements for Chlorofluorocarbons - Four Hydrofluoroketones and a Hydrofluoroamine. J. Chem. Eng. Data 2003, 48, 1380–1383. [Google Scholar] [CrossRef]
- Anastas, P.T.; Warner, J.C. Green Chemistry: Theory and Practice; Oxford University Press: Oxford, UK, 1998. [Google Scholar]
- Erythropel, H.C.; Zimmerman, J.B.; de Winter, T.M.; Petitjean, L.; Melnikov, F.; Lam, C.H.; Lounsbury, A.W.; Mellor, K.E.; Janković, N.Z.; Tu, Q.; et al. The Green ChemisTREE: 20 Years After Taking Root With the 12 Principles. Green Chem. 2018, 20, 1929–1961. [Google Scholar] [CrossRef]
- Kurylo, M.J.; Orkin, V.L. Determination of Atmospheric Lifetimes via the Measurement of OH Radical Kinetics. Chem. Rev. 2003, 103, 5049–5076. [Google Scholar] [CrossRef]
- Finlayson-Pitts, B.J.; Pitts, J.N., Jr. Chemistry of the Upper and Lower Atmosphere: Theory, Experiments, and Applications, 1st ed.; Academic Press: San Diego, CA, USA, 2000. [Google Scholar]
- Calvert, J.G.; Orlando, J.J.; Stockwell, W.R.; Wallington, T.J. The Mechanisms of Reactions Influencing Atmospheric Ozone, 1st ed.; Oxford University Press: New York, NY, USA, 2015. [Google Scholar]
- Díaz-de-Mera, Y.; Aranda, A.; Notario, A.; Rodríguez, A.; Rodríguez, D.; Bravo, I. Photolysis Study of Fluorinated Ketones Under Natural Sunlight Conditions. Phys. Chem. Chem. Phys. 2015, 17, 22991–22998. [Google Scholar] [CrossRef]
- Ren, Y.; Bernard, F.; Daële, V.; Mellouki, A. Atmospheric Fate and Impact of Perfluorinated Butanone and Pentanone. Environ. Sci. Technol. 2019, 53, 8862–8871. [Google Scholar] [CrossRef]
- Vereecken, L.; Peeters, J. The 1,5-H-Shift in 1-Butoxy: A Case Study in the Rigorous Implementation of Transition State Theory for a Multirotamer System. J. Chem. Phys. 2003, 119, 5159–5170. [Google Scholar] [CrossRef]
- Fernández-Ramos, A.; Ellingson, B.A.; Meana-Pañeda, R.; Marques, J.M.C.; Truhlar, D.G. Symmetry Numbers and Chemical Reaction Rates. Theor. Chem. Acc. 2007, 118, 813–826. [Google Scholar] [CrossRef] [Green Version]
- Petit, A.S.; Harvey, J.N. Atmospheric Hydrocarbon Activation by the Hydroxyl Radical: A Simple yet Accurate Computational Protocol for Calculating Rate Coefficients. Phys. Chem. Chem. Phys. 2012, 14, 184–191. [Google Scholar] [CrossRef] [PubMed]
- Rissanen, M.P.; Kurtén, T.; Sipilä, M.; Thornton, J.A.; Kangasluoma, J.; Sarnela, N.; Junninen, H.; Jørgensen, S.; Schallhart, S.; Kajos, M.K.; et al. The Formation of Highly Oxidized Multifunctional Products in the Ozonolysis of Cyclohexene. J. Am. Chem. Soc. 2014, 136, 15596–15606. [Google Scholar] [CrossRef] [PubMed]
- Møller, K.H.; Otkjaer, R.V.; Hyttinen, N.; Kurtén, T.; Kjaergaard, H.G. Cost-Effective Implementation of Multiconformer Transition State Theory for Peroxy Radical Hydrogen Shift Reactions. J. Phys. Chem. A 2016, 120, 10072–10087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Viegas, L.P. Exploring the Reactivity of Hydrofluoropolyethers toward OH Through a Cost-Effective Protocol for Calculating Multiconformer Transition State Theory Rate Constants. J. Phys. Chem. A 2018, 122, 9721–9732. [Google Scholar] [CrossRef]
- Novelli, A.; Vereecken, L.; Bohn, B.; Dorn, H.; Gkatzelis, G.L.; Hofzumahaus, A.; Holland, F.; Reimer, D.; Rohrer, F.; Rosanka, S.; et al. Importance of Isomerization Reactions for OH Radical Regeneration from the Photo-Oxidation of Isoprene Investigated in the Atmospheric Simulation Chamber SAPHIR. Atmos. Chem. Phys. 2020, 20, 3333–3355. [Google Scholar] [CrossRef] [Green Version]
- Vereecken, L.; Nozière, B. H Migration in Peroxy Radicals Under Atmospheric Conditions. Atmos. Chem. Phys. 2020, 20, 7429–7458. [Google Scholar] [CrossRef]
- Viegas, L.P. Gas-Phase OH-Oxidation of 2-Butanethiol: Multiconformer Transition State Theory Rate Constant with Constrained Transition State Randomization. Chem. Phys. Lett. 2022, 803, 139829. [Google Scholar] [CrossRef]
- Hansen, J.C.; Francisco, J.S. Radical-Molecule Complexes: Changing Our Perspective on the Molecular Mechanisms of Radical-Molecule Reactions and Their Impact on Atmospheric Chemistry. ChemPhysChem 2002, 3, 833–840. [Google Scholar] [CrossRef]
- Raines, R.T.; Hansen, D.E. An Intuitive Approach to Steady-State Kinetics. J. Chem. Educ. 1988, 65, 757–759. [Google Scholar] [CrossRef]
- Singleton, D.L.; Cvetanović, R.J. Temperature Dependence of the Reaction of Oxygen Atoms with Olefins. J. Am. Chem. Soc. 1976, 98, 6812–6819. [Google Scholar] [CrossRef]
- Alvarez-Idaboy, J.R.; Mora-Diez, N.; Vivier-Bunge, A. A Quantum Chemical and Classical Transition State Theory Explanation of Negative Activation Energies in OH Addition To Substituted Ethenes. J. Am. Chem. Soc. 2000, 122, 3715–3720. [Google Scholar] [CrossRef]
- Galano, A.; Alvarez-Idaboy, J.R.; Bravo-Pérez, G.; Ruiz-Santoyo, M.E. Gas Phase Reactions of C1–C4 Alcohols with the OH Radical: A Quantum Mechanical Approach. Phys. Chem. Chem. Phys. 2002, 4, 4648–4662. [Google Scholar] [CrossRef]
- Cruz-Torres, A.; Galano, A. On the Mechanism of Gas-Phase Reaction of C1–C3 Aliphatic Thiols + OH Radicals. J. Phys. Chem. A 2007, 111, 1523–1529. [Google Scholar] [CrossRef] [PubMed]
- Xiao, F.; Sun, X.; Li, Z.; Li, X. Theoretical Study of Radical-Molecule Reactions with Negative Activation Energies in Combustion: Hydroxyl Radical Addition to Alkenes. ACS Omega 2020, 5, 12777–12788. [Google Scholar] [CrossRef]
- Viegas, L.P. Simplified Protocol for the Calculation of Multiconformer Transition State Theory Rate Constants Applied to Tropospheric OH-Initiated Oxidation Reactions. J. Phys. Chem. A 2021, 125, 4499–4512. [Google Scholar] [CrossRef]
- Smith, I.W.M. Collisional Energy Transfer, Intramolecular Vibrational Relaxation and Unimolecular Reactions. J. Chem. Soc. Faraday Trans. 1997, 93, 3741–3750. [Google Scholar] [CrossRef]
- Smith, I.W.M.; Ravishankara, A.R. Role of Hydrogen-Bonded Intermediates in the Bimolecular Reactions of the Hydroxyl Radical. J. Phys. Chem. A 2002, 106, 4798–4807. [Google Scholar] [CrossRef]
- McCabe, D.C.; Brown, S.S.; Gilles, M.K.; Talukdar, R.K.; Smith, I.W.M.; Ravishankara, A.R. Kinetics of the Removal of OH(v = 1) and OD(v = 1) by HNO3 and DNO3 from 253 to 383 K. J. Phys. Chem. A 2003, 107, 7762–7769. [Google Scholar] [CrossRef]
- Talukdar, R.K.; Gierczak, T.; McCabe, D.C.; Ravishankara, A.R. Reaction of Hydroxyl Radical with Acetone. 2. Products and Reaction Mechanism. J. Phys. Chem. A 2003, 107, 5021–5032. [Google Scholar] [CrossRef]
- Patchkovskii, S. Brute Force Symmetry Analyzer. Available online: https://github.com/nquesada/symmetry (accessed on 6 August 2022).
- Eckart, C. The Penetration of a Potential Barrier by Electrons. Phys. Rev. 1930, 35, 1303–1309. [Google Scholar] [CrossRef]
- O’Boyle, N.M.; Vandermeersch, T.; Flynn, C.J.; Maguire, A.R.; Hutchison, G.R. Confab-Systematic Generation of Diverse Low-Energy Conformers. J. Cheminf. 2011, 3, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halgren, T.A. Merck Molecular Force Field. I. Basis, Form, Scope, Parameterization, and Performance of MMFF94. J. Comput. Chem. 1996, 17, 490–519. [Google Scholar] [CrossRef]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An Open Chemical Toolbox. J. Cheminf. 2011, 3, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Truhlar, D.G. Exploring the Limit of Accuracy of the Global Hybrid Meta Density Functional for Main-Group Thermochemistry, Kinetics, and Noncovalent Interactions. J. Chem. Theory Comput. 2008, 4, 1849–1868. [Google Scholar] [CrossRef] [PubMed]
- Jensen, F. Unifying General and Segmented Contracted Basis Sets. Segmented Polarization Consistent Basis Sets. J. Chem. Theory Comput. 2014, 10, 1074–1085. [Google Scholar] [CrossRef]
- Marques, J.M.C.; Llanio-Trujillo, J.L.; Abreu, P.E.; Pereira, F.B. How Different Are Two Chemical Structures? J. Chem. Inf. Model. 2010, 50, 2129–2140. [Google Scholar] [CrossRef]
- Viegas, L.P.; Jensen, F. Reactivity of α,ω-Dihydrofluoropolyethers toward OH Predicted by Multiconformer Transition State Theory and the Interacting Quantum Atoms Approach. J. Phys. Chem. A 2020, 124, 3460–3470. [Google Scholar] [CrossRef]
- Schmidt, M.W.; Baldridge, K.K.; Boatz, J.A.; Elbert, S.T.; Gordon, M.S.; Jensen, J.H.; Koseki, S.; Matsunaga, N.; Nguyen, K.A.; Su, S.; et al. General Atomic and Molecular Electronic Structure System. J. Comput. Chem. 1993, 14, 1347–1363. [Google Scholar] [CrossRef]
- Viegas, L.P. Assessment of Model Chemistries for Hydrofluoropolyethers: A DFT/M08-HX Benchmark Study. Int. J. Quantum Chem. 2017, 117, e25381. [Google Scholar] [CrossRef]
- Palafox, M.A. DFT Computations on Vibrational Spectra: Scaling Procedures to Improve the Wavenumbers. Phys. Sci. Rev. 2018, 3, 0184. [Google Scholar] [CrossRef]
- Prinn, R.G.; Huang, J.; Weiss, R.F.; Cunnold, D.M.; Fraser, P.J.; Simmonds, P.G.; McCulloch, A.; Harth, C.; Salameh, P.; O’Doherty, S.; et al. Evidence for Substantial Variations of Atmospheric Hydroxyl Radicals in the Past Two Decades. Science 2001, 292, 1882–1888. [Google Scholar] [CrossRef] [PubMed]
- McGillen, M.R.; Carter, W.P.L.; Mellouki, A.; Orlando, J.J.; Picquet-Varrault, B.; Wallington, T.J. Database for the Kinetics of the Gas-Phase Atmospheric Reactions of Organic Compounds. Earth Syst. Sci. Data 2020, 12, 1203–1216. [Google Scholar] [CrossRef]
- Altarawneh, M. A Closer Look Into the Contribution of Atmospheric Gas-Phase Pathways in the Formation of Perfluorocarboxylic Acids. Atmos. Pol. Res. 2021, 12, 101255. [Google Scholar] [CrossRef]
- Kooij, D.M. Über die Zersetzung des Gasförmigen Phosphorwasserstoffs. Z. Phys. Chem. 1893, 12, 155–161. [Google Scholar] [CrossRef]
- Laidler, K.J. The Development of the Arrhenius Equation. J. Chem. Ed. 1984, 61, 494–498. [Google Scholar] [CrossRef]
- Ren, Y.; Baramoussi, E.M.E.; Daële, V.; Mellouki, A. Atmospheric Chemistry of Ketones: Reaction of OH Radicals with 2-Methyl-3-Pentanone, 3-Methyl-2-Pentanone and 4-Methyl-2-Pentanone. Sci. Total Environ. 2021, 780, 146249. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Viegas, L.P. Atmospheric Degradation of Two Hydrofluoroketones: Theoretical Rate Constants for the Gas-Phase OH-Oxidation of HFK-447mcc and HFK-465mc. Atmosphere 2022, 13, 1256. https://doi.org/10.3390/atmos13081256
Viegas LP. Atmospheric Degradation of Two Hydrofluoroketones: Theoretical Rate Constants for the Gas-Phase OH-Oxidation of HFK-447mcc and HFK-465mc. Atmosphere. 2022; 13(8):1256. https://doi.org/10.3390/atmos13081256
Chicago/Turabian StyleViegas, Luís Pedro. 2022. "Atmospheric Degradation of Two Hydrofluoroketones: Theoretical Rate Constants for the Gas-Phase OH-Oxidation of HFK-447mcc and HFK-465mc" Atmosphere 13, no. 8: 1256. https://doi.org/10.3390/atmos13081256
APA StyleViegas, L. P. (2022). Atmospheric Degradation of Two Hydrofluoroketones: Theoretical Rate Constants for the Gas-Phase OH-Oxidation of HFK-447mcc and HFK-465mc. Atmosphere, 13(8), 1256. https://doi.org/10.3390/atmos13081256