Chemical Composition and Sources of Marine Aerosol over the Western North Pacific Ocean in Winter

Abstract
:1. Introduction
2. Methodology
2.1. Aerosol Collection and Chemical Analysis
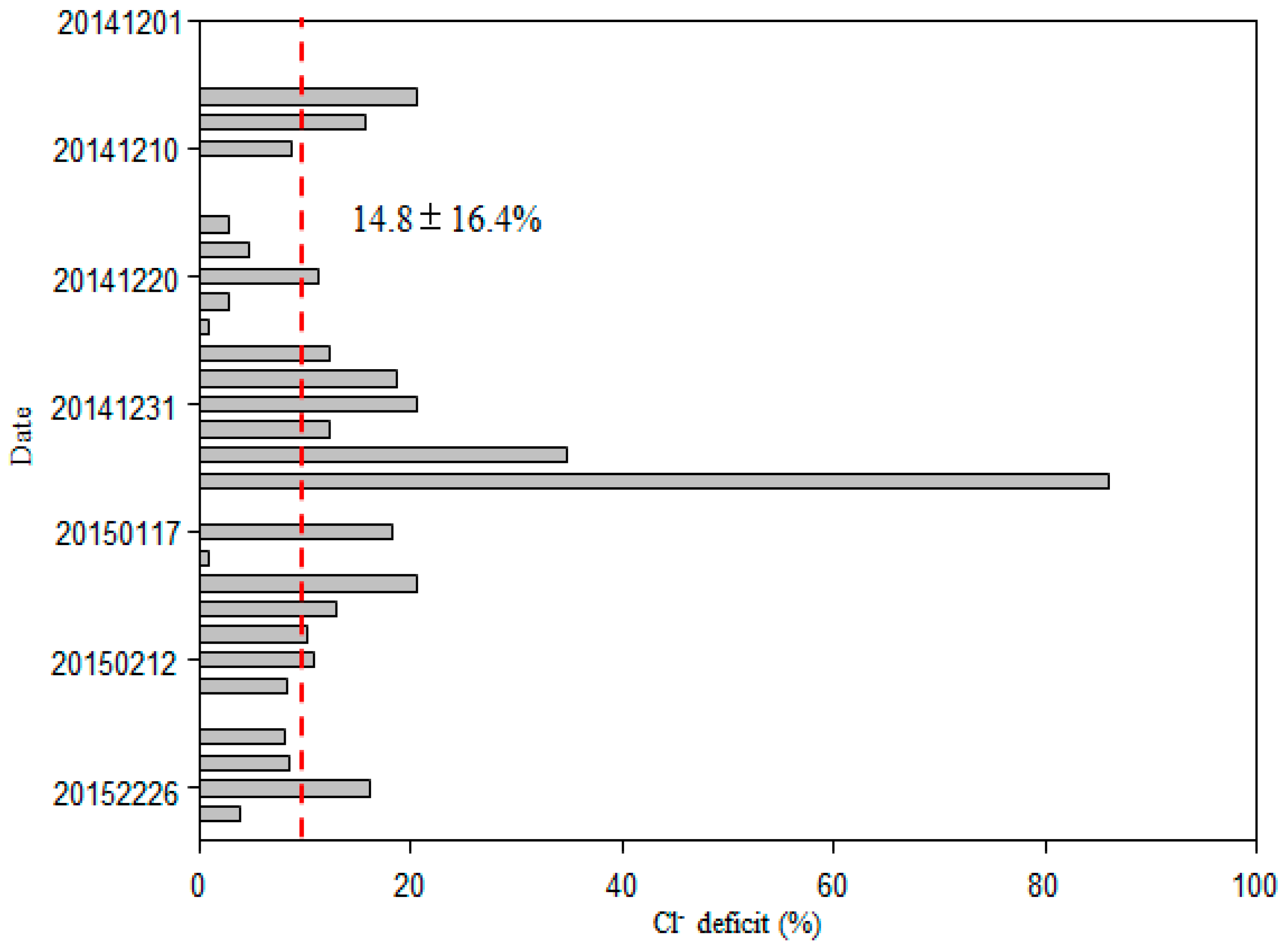
2.2. Concentrations of the Non-Sea-Salt Fraction
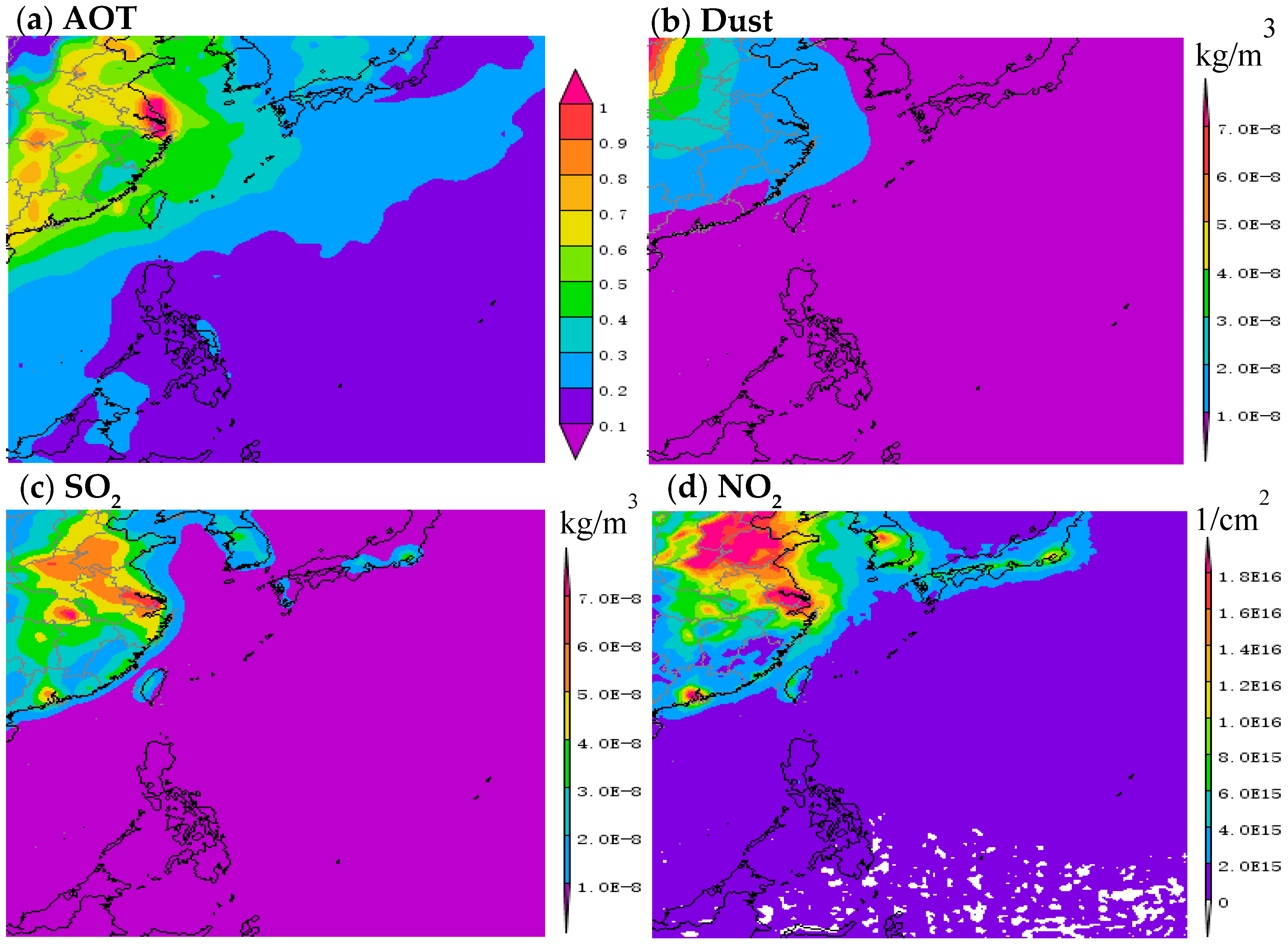
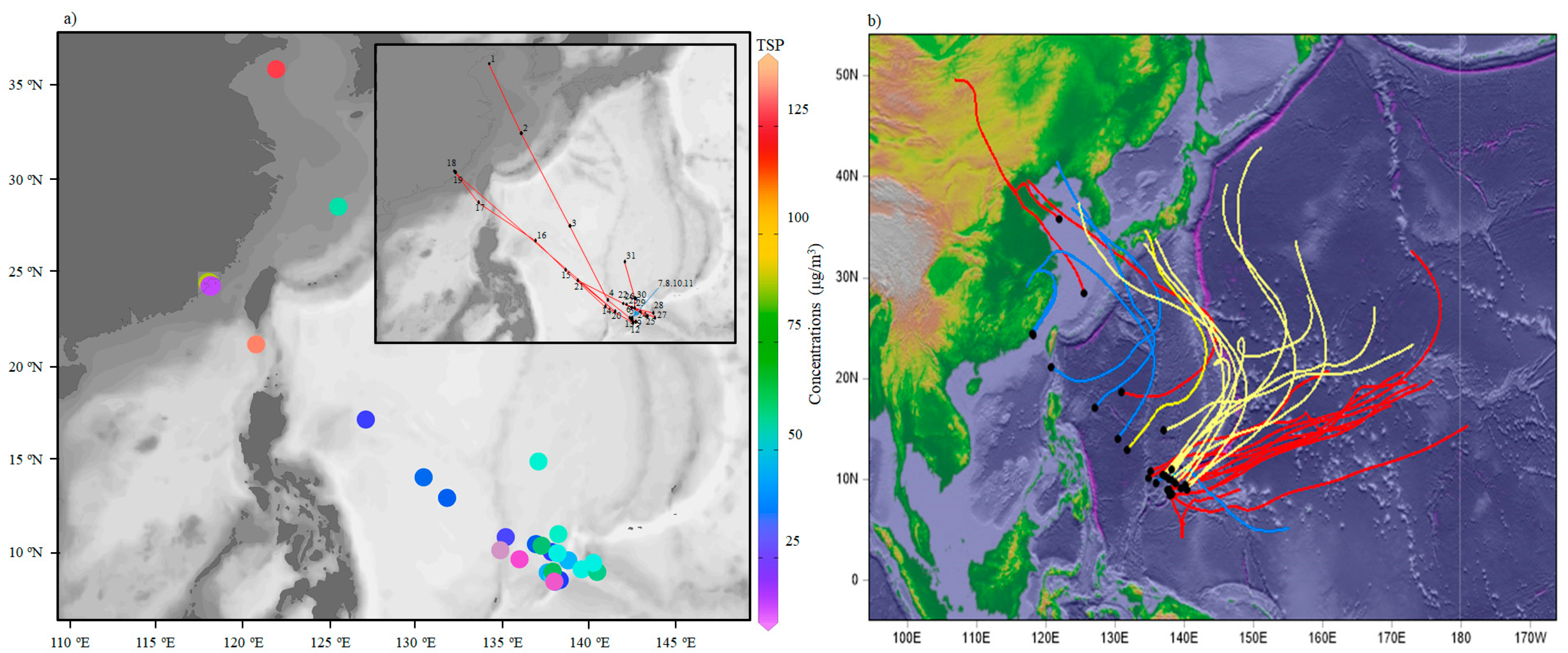
2.3. Back Trajectories and Aerosol Optical Thickness
3. Results and Discussion
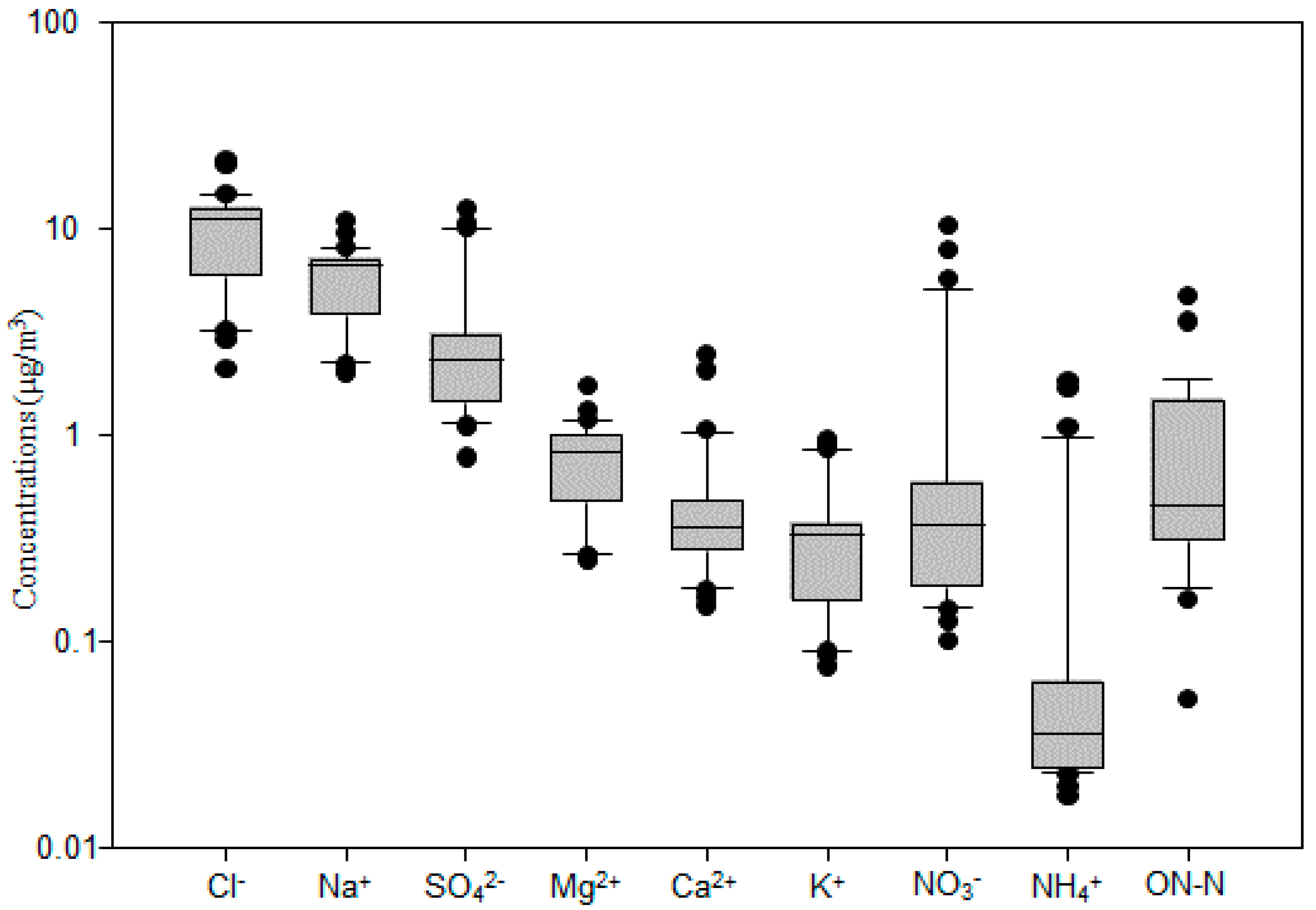
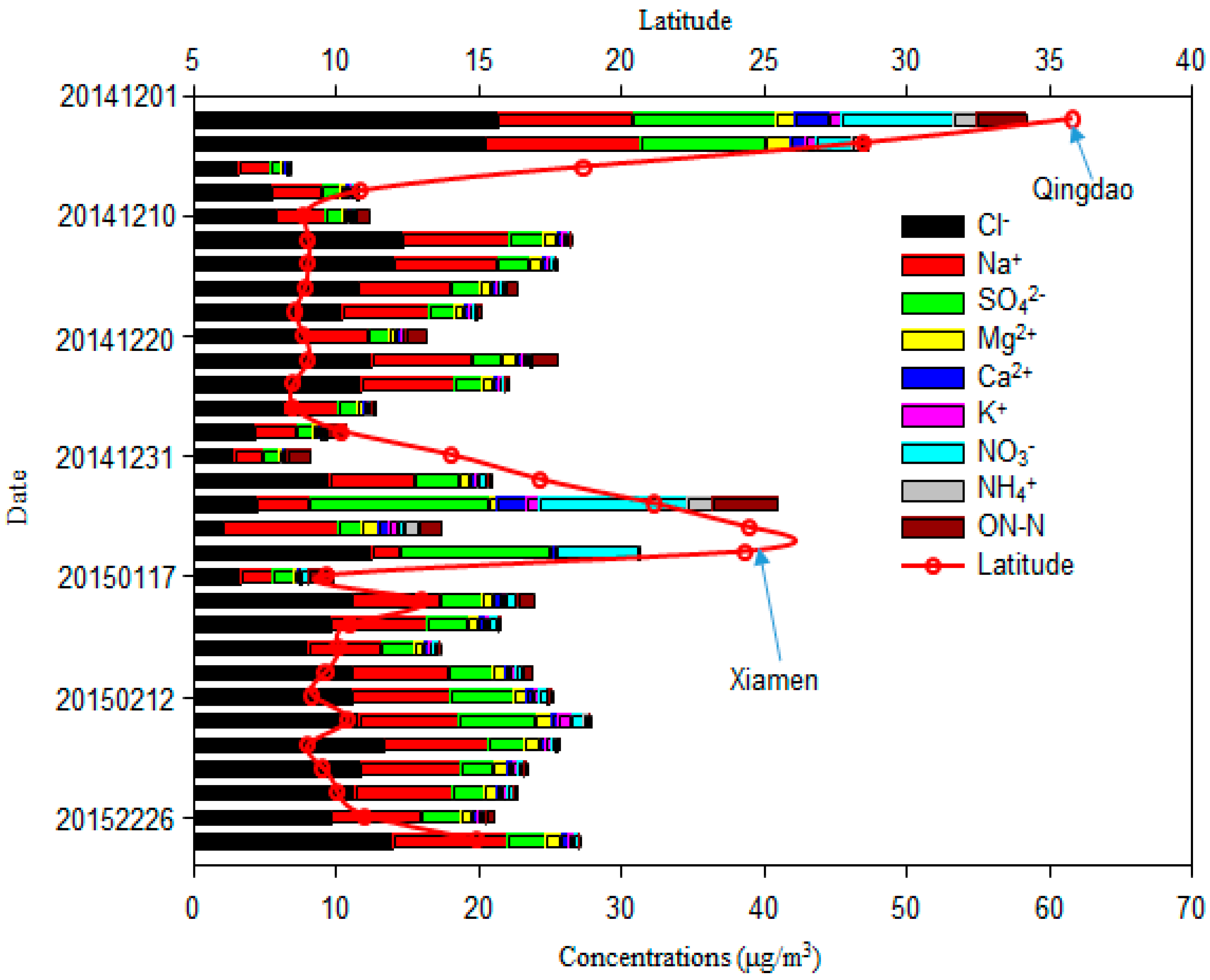
3.1. Total Suspended Particulate and Major Ion Concentrations of Marine Aerosol Samples
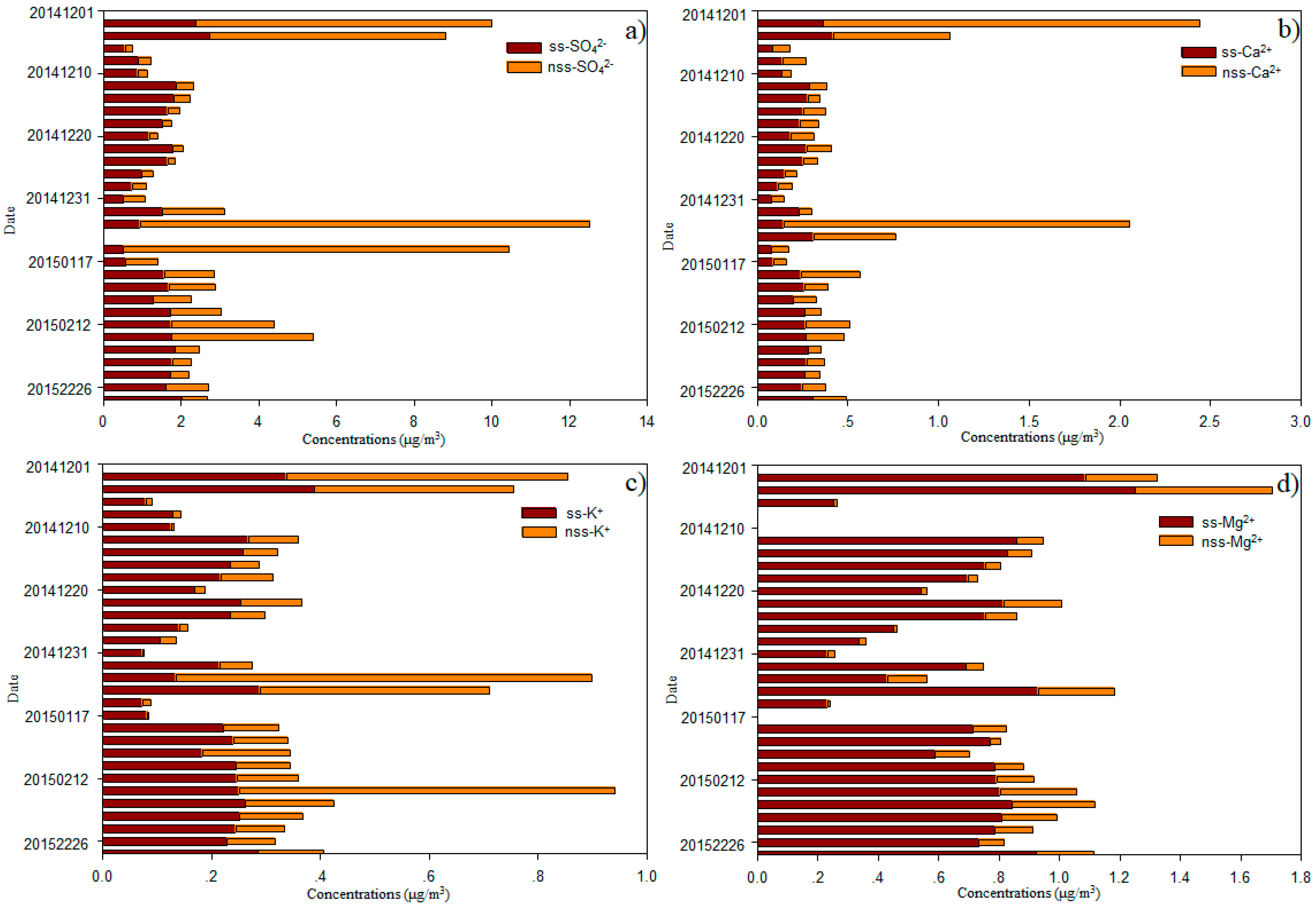
3.2. Major Ion Characteristics and Sources of Aerosol over the Western North Pacific
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wang, L.; Qi, J.H.; Shi, J.H.; Chen, X.J.; Gao, H.W. Source apportionment of particulate pollutants in the atmosphere over the Northern Yellow Sea. Atmos. Environ. 2013, 70, 425–434. [Google Scholar] [CrossRef]
- Xiao, H.W.; Xiao, H.Y.; Luo, L.; Shen, C.Y.; Long, A.M.; Chen, L.; Long, Z.H.; Li, D.N. Atmospheric aerosol compositions over the South China Sea: temporal variability and source apportionment. Atmos. Chem. Phys. 2017, 17, 3199–3214. [Google Scholar] [CrossRef]
- Zhang, M.; Chen, J.M.; Wang, T.; Cheng, T.T.; Lin, L.; Bhatia, R.S.; Havey, M. Chemical characterization of aerosols over the Atlantic Ocean and the Pacific Ocean during two cruises in 2007 and 2008. J. Geophys. Res. Atmos. 2010, 115, 1842–1851. [Google Scholar] [CrossRef]
- Miyazaki, Y.; Kawamura, K.; Sawano, M. Size distributions of organic nitrogen and carbon in remote marine aerosols: Evidence of marine biological origin based on their isotopic ratios. Geophys. Res. Lett. 2010, 37, 460–472. [Google Scholar] [CrossRef]
- Duce, R.A.; Laroche, J.; Altieri, K.; Arrigo, K.R.; Baker, A.R.; Capone, D.G.; Cornell, S.; Dentener, F.; Galloway, J.; Ganeshram, R.S. Impacts of atmospheric anthropogenic nitrogen on the open ocean. Science 2008, 320, 893–897. [Google Scholar] [PubMed]
- Hsu, S.C.; Liu, S.C.; Kao, S.J.; Jeng, W.L.; Huang, Y.T.; Tseng, C.M.; Tsai, F.; Tu, J.Y.; Yang, Y. Sources, solubility, and acid processing of aerosol iron and phosphorous over the South China Sea: East Asian dust and pollution outflows vs. Southeast Asian biomass burning. Atmos. Chem. Phys. Discuss. 2014, 14, 21433–21472. [Google Scholar] [CrossRef]
- Wen, L.S.; Jiann, K.T.; Santschi, P.H. Physicochemical speciation of bioactive trace metals (Cd, Cu, Fe, Ni) in the oligotrophic South China Sea. Mar. Chem. 2006, 101, 104–129. [Google Scholar] [CrossRef]
- Kim, I.N.; Lee, K.; Gruber, N.; Karl, D.M.; Bullister, J.L.; Yang, S.; Kim, T.W. Increasing anthropogenic nitrogen in the North Pacific Ocean. Science 2014, 346, 1102–1106. [Google Scholar] [CrossRef] [PubMed]
- Doney, S.C.; Mahowald, N.; Lima, I.; Feely, R.A.; Mackenzie, F.T.; Lamarque, J.F.; Rasch, P.J. Impact of anthropogenic atmospheric nitrogen and sulfur deposition on ocean acidification and the inorganic carbon system. Proc. Natl. Acad. Sci. USA 2007, 104, 14580–14585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altieri, K.E.; Fawcett, S.E.; Peters, A.J.; Sigman, D.M.; Hastings, M.G. Marine biogenic source of atmospheric organic nitrogen in the subtropical North Atlantic. Proc. Natl. Acad. Sci. USA 2016, 113, 925–930. [Google Scholar] [CrossRef] [PubMed]
- Altieri, K.E.; Hastings, M.G.; Peters, A.J.; Oleynik, S.; Sigman, D.M. Isotopic evidence for a marine ammonium source in rainwater at Bermuda. Glob. Biogeochem. Cycles 2014, 28, 1066–1080. [Google Scholar] [CrossRef] [Green Version]
- Jickells, T.D.; Kelly, S.D.; Baker, A.R.; Biswas, K.; Dennis, P.F.; Spokes, L.J.; Witt, M.; Yeatman, S.G. Isotopic evidence for a marine ammonia source. Geophys. Res. Lett. 2003, 30, 359–376. [Google Scholar]
- Boreddy, S.K.R.; Kawamura, K. A 12-year observation of water-soluble ions in TSP aerosols collected at a remote marine location in the WNP: An outflow region of Asian dust. Atmos. Chem. Phys. 2015, 15, 6437–6453. [Google Scholar] [CrossRef]
- Jung, J.; Furutani, H.; Uematsu, M. Atmospheric inorganic nitrogen in marine aerosol and precipitation and its deposition to the North and South Pacific Oceans. J. Atmos Chem. 2011, 68, 157–181. [Google Scholar]
- Sasakawa, M.; Uematsu, M. Chemical composition of aerosol, sea fog, and rainwater in the marine boundary layer of the northWNP and its marginal seas. J. Geophys. Res. Atmos. 2002, 107, ACH 17-1–ACH 17-9. [Google Scholar] [CrossRef]
- Kunwar, B.; Kawamura, K. One-year observations of carbonaceous and nitrogenous components and major ions in the aerosols from subtropical Okinawa Island, an outflow region of Asian dusts. Atmos. Chem. Phys. 2014, 14, 1819–1836. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.Q.; Wang, J.Z.; Niu, T.; Zhou, C.; Chen, M.; Liu, J. The Variability of Spring Sand-Dust Storm Frequency in Northeast Asia from 1980 to 2011. Acta Meteorol. Sin. 2013, 27, 119–127. [Google Scholar] [CrossRef]
- Xiao, H.; Xie, L.; Long, A.; Ye, F.; Pan, Y.; Li, D.; Long, Z.; Chen, L.; Xiao, H.; Liu, C. Use of isotopic compositions of nitrate in TSP to identify sources and chemistry in South China Sea. Atmos. Environ. 2015, 109, 70–78. [Google Scholar] [CrossRef] [Green Version]
- Xiao, H.W.; Xiao, H.Y.; Long, A.M.; Wang, Y.L.; Liu, C.Q. Chemical composition and source apportionment of rainwater at Guiyang, SW China. J. Atmos. Chem. 2013, 70, 269–281. [Google Scholar] [CrossRef] [Green Version]
- Xiao, H.; Xiao, H.; Zhang, Z.; Wang, Y.; Long, A.; Liu, C. Chemical characteristics and source apportionment of atmospheric precipitation in Yongxing Island. China Environ. Sci. 2016, 36, 3237–3244, (Chinese with English Abstract). [Google Scholar]
- Piazzalunga, A.; Bernardoni, V.; Fermo, P.; Vecchi, R. Optimisation of analytical procedures for the quantification of ionic and carbonaceous fractions in the atmospheric aerosol and applications to ambient samples. Anal. Bioanal. Chem. 2013, 405, 1123–1132. [Google Scholar] [CrossRef] [PubMed]
- Jickells, T.; Baker, A.R.; Cape, J.N.; Cornell, S.E.; Nemitz, E. The cycling of organic nitrogen through the atmosphere. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2013, 368, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Keene, W.C.; Pszenny, A.A.; Galloway, J.N.; Hawley, M.E. Sea-salt corrections and interpretation of constituent ratios in marine precipitation. J. Geophys. Res. Atmos. 1986, 91, 6647–6658. [Google Scholar]
- Xiao, H.W.; Xiao, H.Y.; Long, A.M.; Wang, Y.L.; Liu, C.Q. Sources and meteorological factors that control seasonal variation of δ 34 S values in rainwater. Atmos. Res. 2014, 149, 154–165. [Google Scholar] [CrossRef]
- Zhang, X.; Zhuang, G.; Guo, J.; Yin, K.; Zhang, P. Characterization of aerosol over the Northern South China Sea during two cruises in 2003. Atmos. Environ. 2007, 41, 7821–7836. [Google Scholar] [CrossRef]
- Laskin, A.; Moffet, R.C.; Gilles, M.K.; Fast, J.D.; Zaveri, R.A.; Wang, B.; Nigge, P.; Shutthanandan, J. Tropospheric chemistry of internally mixed sea salt and organic particles: Surprising reactivity of NaCl with weak organic acids. J. Geophys. Res. Atmos. 2012, 117, 156–169. [Google Scholar] [CrossRef]
- Thornton, J.A.; Kercher, J.P.; Riedel, T.P.; Wagner, N.L.; Cozic, J.; Holloway, J.S.; Dube´, W.P.; Wolfe, G.M.; Quinn, P.K.; Middlebrook, A.M.; et al. A large atomic chlorine source inferred from mid-continental reactive nitrogen chemistry. Nature 2010, 464, 271–274. [Google Scholar] [CrossRef] [PubMed]
- Boreddy, S.K.R.; Kawamura, K. A review of dicarboxylic acids and related compounds in atmospheric aerosols: Molecular distributions, sources and transformation. Atmos. Res. 2016, 170, 140–160. [Google Scholar]
- Dueñas, C.; Fernández, M.C.; Gordo, E.; Cañete, S.; Pérez, M. Chemical and radioactive composition of bulk deposition in Málaga (Spain). Atmos. Environ. 2012, 62, 1–8. [Google Scholar] [CrossRef]
- Taylor, S.R. Abundance of chemical elements in the continental crust: a new table. Geochim. Cosmochim. Acta 1964, 28, 1273–1285. [Google Scholar] [CrossRef]
- Deng, C.; Zhuang, G.; Huang, K.; Li, J.; Zhang, R.; Wang, Q.; Liu, T.; Sun, Y.; Guo, Z.; Fu, J.S. Chemical characterization of aerosols at the summit of Mountain Tai in Central East China. Atmos. Chem. Phys. 2011, 11, 7319–7332. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.H.; Yang, G.P.; Liu, C.Y.; Su, L.P. Chemical Characteristics of Aerosol Composition over the Yellow Sea and the East China Sea in Autumn. J. Atmos. Sci. 2013, 70, 1784–1794. [Google Scholar] [CrossRef]
- Choi, Y.; Rhee, T.S.; Collett, J.L., Jr.; Park, T.; Park, S.-M.; Seo, B.-K.; Park, G.; Park, K.; Lee, T. Aerosol concentrations and composition in the North Pacific marine boundary layer. Atmos. Environ. 2017, 171, 165–172. [Google Scholar] [CrossRef]
- Martino, M.; Hamliton, D.; Baker, A.R.; Jickells, T.D.; Bromley, T.; Nojiri, Y.; Quack, B.; Boyd, P.W. Western Pacific atmospheric nutrient deposition fluxes, their impact on surface ocean productivity. Glob. Biogeochem. Cycles 2014, 28, 712–728. [Google Scholar] [Green Version]
- Zhao, Y.; Zhang, L.; Pan, Y.; Wang, Y.; Paulot, F.; Henze, D.K. Atmospheric nitrogen deposition to the northwestern Pacific: Seasonal variation and source attribution. Atmos. Chem. Phys. 2015, 15, 13657–13703. [Google Scholar] [CrossRef]
- Kundu, S.; Kawamura, K.; Lee, M.; Andreae, T.W.; Hoffer, A.; Andreae, M.O. Comparison of Amazonian biomass burning and East Asian marine aerosols: Bulk organics, diacids and related compounds, water-soluble inorganic ions, stable carbon and nitrogen isotope ratios. Low Temp. Sci. 2010, 68, 89–100. [Google Scholar]
- Kunwar, B.; Kawamura, K.; Zhu, C. Stable carbon and nitrogen isotopic compositions of ambient aerosols collected from Okinawa Island in the WNP Rim, an outflow region of Asian dusts and pollutants. Atmos. Environ. 2016, 131, 243–253. [Google Scholar] [CrossRef]
- Pavuluri, C.M.; Kawamura, K.; Swaminathan, T. Time-resolved distributions of bulk parameters, diacids, ketoacids and α-dicarbonyls and stable carbon and nitrogen isotope ratios of TC and TN in tropical Indian aerosols: Influence of land/sea breeze and secondary processes. Atmos. Res. 2015, 153, 188–199. [Google Scholar] [CrossRef]
- Pavuluri, C.M.; Kawamura, K.; Tachibana, E.; Swaminathan, T. Elevated nitrogen isotope ratios of tropical Indian aerosols from Chennai: Implication for the origins of aerosol nitrogen in South and Southeast Asia. Atmos. Environ. 2010, 44, 3597–3604. [Google Scholar] [CrossRef] [Green Version]
- Shi, J.; Gao, H.; Qi, J.; Zhang, J.; Yao, X. Sources, compositions, and distributions of water-soluble organic nitrogen in aerosols over the China Sea. J. Geophys, Res. Atmos. 2010, 115, 1383–1392. [Google Scholar] [CrossRef]
- Wang, G.; Xie, M.; Hu, S.; Gao, S.; Tachibana, E.; Kawamura, K. Dicarboxylic acids, metals and isotopic compositions of C and N in atmospheric aerosols from inland China: implications for dust and coal burning emission and secondary aerosol formation. Atmos. Chem. Phys. 2010, 10, 6087–6096. [Google Scholar] [CrossRef] [Green Version]
- Ogunro, O.O.; Burrows, S.M.; Elliott, S.; Frossard, A.A.; Hoffman, F.; Frossard, A.A.; Hoffman, F.; Letscher, R.T.; Moore, J.K.; Russell, L.M.; et al. Global distribution and surface activity of macromolecules in offline simulations of marine organic chemistry. Biogeochemistry 2015, 126, 25–56. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Na+ | Cl− | SO42− | nss-SO42− | K+ | nss-K+ | Mg2+ | nss-Mg2+ | Ca2+ | nss-Ca2+ | NH4+ | NO3− | ON | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| (a) | |||||||||||||
| Na+ | 1 | 0.65 | 0.21 | 0.17 | 0.73 * | 0.49 | 0.99 ** | 0.92 ** | 0.52 | 0.40 | 0.44 | 0.05 | −0.26 |
| Cl− | 1 | 0.59 | 0.37 | 0.38 | 0.19 | 0.64 | 0.56 | 0.45 | 0.38 | 0.18 | 0.36 | −0.08 | |
| SO42− | 1 | 0.98 ** | 0.55 | 0.61 | 0.24 | 0.29 | 0.67 | 0.69 | 0.57 | 0.92 ** | 0.66 | ||
| nss-SO42− | 1 | 0.62 | 0.71 | 0.20 | 0.29 | 0.63 | 0.65 | 0.68 | 0.93 ** | 0.78 | |||
| K+ | 1 | 0.95 ** | 0.75 * | 0.77 * | 0.89 ** | 0.83 * | 0.91 ** | 0.57 | 0.56 | ||||
| nss-K+ | 1 | 0.52 | 0.57 | 0.90 ** | 0.89 ** | 0.96 ** | 071 * | 0.77 | |||||
| Mg2+ | 1 | 0.96 ** | 0.52 | 0.40 | 0.45 | 0.07 | −0.24 | ||||||
| nss-Mg2+ | 1 | 0.50 | 0.39 | 0.45 | 0.10 | −0.16 | |||||||
| Ca2+ | 1 | 0.99 ** | 0.94 ** | 0.79 * | 0.80 | ||||||||
| nss-Ca2+ | 1 | 0.94 ** | 0.84 ** | 0.87 * | |||||||||
| NH4+ | 1 | 0.74 * | 0.86 * | ||||||||||
| NO3− | 1 | 0.89 * | |||||||||||
| ON | 1 | ||||||||||||
| (b) | |||||||||||||
| Na+ | 1 | 0.97 ** | 0.61 ** | 0.25 | 0.67 ** | 0.38 | 0.98 ** | 0.60 ** | 0.80 ** | 0.30 | 0.15 | 0.13 | −0.44 * |
| Cl− | 1 | 0.51 ** | 0.18 | 0.61 ** | 0.33 | 0.95 ** | 0.56 * | 0.74 ** | 0.23 | 0.09 | 0.05 | −0.41 * | |
| SO42− | 1 | 0.92 ** | 0.86 ** | 0.80 ** | 0.66 ** | 0.56 * | 0.76 ** | 0.61 ** | 0.75 ** | 0.71 ** | −0.35 | ||
| nss-SO42− | 1 | 0.72 ** | 0.79 ** | 0.33 | 0.42 | 0.53 ** | 0.60 ** | 0.84 ** | 0.80 ** | −0.21 | |||
| K+ | 1 | 0.95 ** | 0.75 ** | 0.72 ** | 0.66 ** | 0.41 | 0.78 ** | 0.50 * | −0.39 | ||||
| nss-K+ | 1 | 0.50 * | 0.62 ** | 0.48 * | 0.38 | 0.91 ** | 0.57 ** | −0.30 | |||||
| Mg2+ | 1 | 0.81 ** | 0.80 ** | 0.31 | 0.25 | 0.21 | −0.41 | ||||||
| nss-Mg2+ | 1 | 0.49 * | 0.22 | 0.45 * | 0.46 * | −0.22 | |||||||
| Ca2+ | 1 | 0.81 ** | 0.34 | 0.50 * | −0.26 | ||||||||
| nss-Ca2+ | 1 | 0.39 | 0.68 ** | 0.02 | |||||||||
| NH4+ | 1 | 0.61 ** | −0.13 | ||||||||||
| NO3− | 1 | −0.10 | |||||||||||
| ON | 1 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xiao, H.-W.; Xiao, H.-Y.; Shen, C.-Y.; Zhang, Z.-Y.; Long, A.-M. Chemical Composition and Sources of Marine Aerosol over the Western North Pacific Ocean in Winter. Atmosphere 2018, 9, 298. https://doi.org/10.3390/atmos9080298
Xiao H-W, Xiao H-Y, Shen C-Y, Zhang Z-Y, Long A-M. Chemical Composition and Sources of Marine Aerosol over the Western North Pacific Ocean in Winter. Atmosphere. 2018; 9(8):298. https://doi.org/10.3390/atmos9080298
Chicago/Turabian StyleXiao, Hong-Wei, Hua-Yun Xiao, Chun-Yan Shen, Zhong-Yi Zhang, and Ai-Min Long. 2018. "Chemical Composition and Sources of Marine Aerosol over the Western North Pacific Ocean in Winter" Atmosphere 9, no. 8: 298. https://doi.org/10.3390/atmos9080298
APA StyleXiao, H. -W., Xiao, H. -Y., Shen, C. -Y., Zhang, Z. -Y., & Long, A. -M. (2018). Chemical Composition and Sources of Marine Aerosol over the Western North Pacific Ocean in Winter. Atmosphere, 9(8), 298. https://doi.org/10.3390/atmos9080298