Evolutionary Toxicology as a Tool to Assess the Ecotoxicological Risk in Freshwater Ecosystems




Abstract
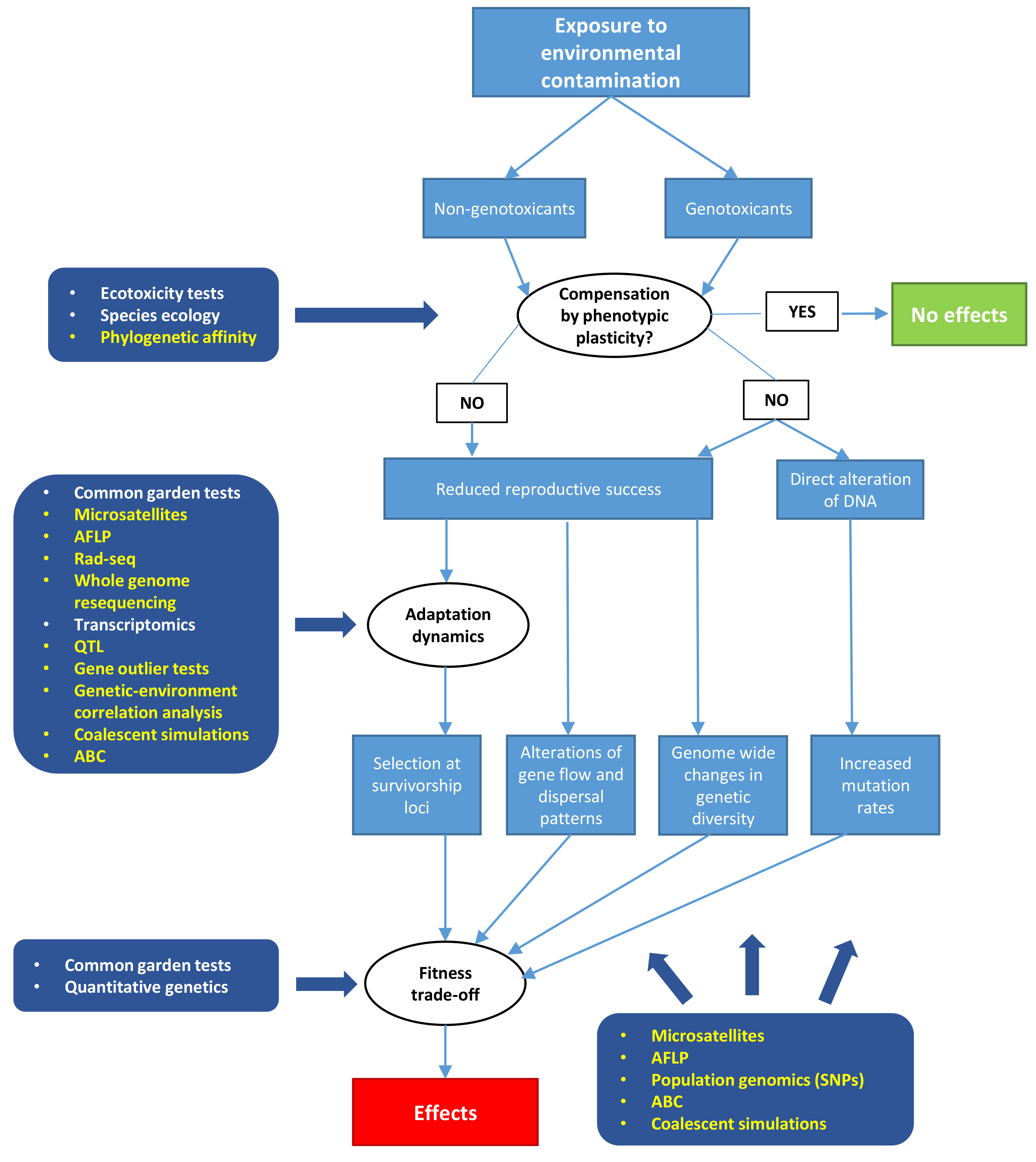
:1. Introduction
2. The Implementation of Population Genetics and Genomics in Evolutionary Toxicology: Markers, Techniques, and Data Analysis
3. Paradigmatic Case Studies
3.1. Application in the Field
3.2. Laboratory Applications
4. Potential Role of Evolutionary Toxicology in Ecological Risk Assessments
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Simmons, D.B.D.; Benskin, J.P.; Cosgrove, J.R.; Duncker, B.P.; Ekman, D.R.; Martyniuk, C.J.; Sherry, J.P. Omics for aquatic ecotoxicology: Control of extraneous variability to enhance the analysis of environmental effects. Environ. Toxicol. Chem. 2015, 34, 1693–1704. [Google Scholar] [CrossRef] [PubMed]
- Klerks, P.L.; Xie, L.; Levinton, J.S. Quantitative genetics approaches to study evolutionary processes in ecotoxicology; a perspective from research on the evolution of resistance. Ecotoxicology 2011, 20, 513–523. [Google Scholar] [CrossRef] [PubMed]
- Brown, A.R.; Hosken, D.J.; Balloux, F.; Bickley, L.K.; LePage, G.; Owen, S.F.; Hetheridge, M.J.; Tyler, C.R. Genetic variation, inbreeding and chemical exposure—Combined effects in wildlife and critical considerations for ecotoxicology. Philos. Trans. R. Soc. B Biol. Sci. 2009, 364, 3377–3390. [Google Scholar] [CrossRef] [PubMed]
- Anderson, C.; Cunha, L.; Sechi, P.; Kille, P.; Spurgeon, D. Genetic variation in populations of the earthworm, Lumbricus rubellus, across contaminated mine sites. BMC Genet. 2017, 18, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Coutellec, M.A.; Barata, C. An introduction to evolutionary processes in ecotoxicology. Ecotoxicology 2011, 20, 493–496. [Google Scholar] [CrossRef] [PubMed]
- Bickham, J.W.; Smolen, M.J. Somatic and heritable effects of environmental genotoxins and the emergence of evolutionary toxicology. Environ. Health Perspect. 1994, 102, 25–28. [Google Scholar] [CrossRef] [PubMed]
- Bickham, J.W. The four cornerstones of evolutionary toxicology. Ecotoxicology 2011, 20, 497–502. [Google Scholar] [CrossRef] [PubMed]
- Stoks, R.; Debecker, S.; Van, K.D.; Janssens, L. Integrating ecology and evolution in aquatic toxicology: Insights from damselflies. Freshw. Sci. 2015, 34, 1032–1039. [Google Scholar] [CrossRef]
- Bickham, J. Effects of chemical contaminants on genetic diversity in natural populations: Implications for biomonitoring and ecotoxicology. Mutat. Res. 2000, 463, 33–51. [Google Scholar] [CrossRef]
- Bourret, V.; Couture, P.; Campbell, P.G.C.; Bernatchez, L. Evolutionary ecotoxicology of wild yellow perch (Perca flavescens) populations chronically exposed to a polymetallic gradient. Aquat. Toxicol. 2008, 86, 76–90. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, R.; Baird, D.J.; Soares, A.M.V.M.; Lopes, I. Contaminant driven genetic erosion: A case study with Daphnia longispina. Environ. Toxicol. Chem. 2012, 31, 977–982. [Google Scholar] [CrossRef] [PubMed]
- Brady, S.P.; Monosson, E.; Matson, C.W.; Bickham, J.W. Evolutionary toxicology: Toward a unified understanding of life’s response to toxic chemicals. Evolut. Appl. 2017, 10, 745–751. [Google Scholar] [CrossRef] [PubMed]
- Oziolor, E.M.; Bickham, J.W.; Matson, C.W. Evolutionary toxicology in an omics world. Evolut. Appl. 2017, 10, 752–761. [Google Scholar] [CrossRef] [PubMed]
- Shaw, J.R.; Hampton, T.H.; King, B.L.; Whitehead, A.; Galvez, F.; Gross, R.H.; Keith, N.; Notch, E.; Jung, D.; Glaholt, S.P.; et al. Natural selection canalizes expression variation of environmentally induced plasticity-enabling genes. Mol. Biol. Evolut. 2014, 31, 3002–3015. [Google Scholar] [CrossRef] [PubMed]
- Wernersson, A.S.; Carere, M.; Maggi, C.; Tusil, P.; Soldan, P.; James, A.; Sanchez, W.; Dulio, V.; Broeg, K.; Reifferscheid, G.; et al. The European technical report on aquatic effect-based monitoring tools under the water framework directive. Environ. Sci. Eur. 2015, 27, 7. [Google Scholar] [CrossRef] [Green Version]
- Belfiore, N.M.; Anderson, S.L. Effects of contaminants on genetic patterns in aquatic organisms: A review. Mutat. Res. Rev. Mutat. Res. 2001, 489, 97–122. [Google Scholar] [CrossRef]
- Gienapp, P.; Fior, S.; Guillaume, F.; Lasky, J.R.; Sork, V.L.; Csilléry, K. Genomic quantitative genetics to study evolution in the wild. Trends Ecol. Evolut. 2017, 32, 897–908. [Google Scholar] [CrossRef] [PubMed]
- Suarez-Ulloa, V.; Gonzalez-Romero, R.; Eirin-Lopez, J.M. Environmental epigenetics: A promising venue for developing next-generation pollution biomonitoring tools in marine invertebrates. Mar. Pollut. Bull. 2015, 98, 5–13. [Google Scholar] [CrossRef] [PubMed]
- Peterson, E.K.; Buchwalter, D.B.; Kerby, J.L.; Lefauve, M.K.; Varian-Ramos, C.W.; Swaddle, J.P. Integrative behavioral ecotoxicology: Bringing together fields to establish new insight to behavioral ecology, toxicology, and conservation. Curr. Zool. 2017, 63, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Brander, S.M.; Biales, A.D.; Connon, R.E. The role of epigenomics in aquatic toxicology. Environ. Toxicol. Chem. 2017, 36, 2565–2573. [Google Scholar] [CrossRef] [PubMed]
- Mussali-galante, P.; Tovar-sánchez, E.; Valverde, M.; Rojas, E. Reviews of Environmental Contamination and Toxicology; Springer: New York, NY, USA, 2014; Volume 227. [Google Scholar]
- Selkoe, K.A.; Toonen, R.J. Microsatellites for ecologists: A practical guide to using and evaluating microsatellite markers. Ecol. Lett. 2006, 9, 615–629. [Google Scholar] [CrossRef] [PubMed]
- Agnèse, J.F.; Adépo-Gourène, B.; Nyingi, D. Functional microsatellite and possible selective sweep in natural populations of the black-chinned tilapia Sarotherodon melanotheron (Teleostei, Cichlidae). Mar. Genom. 2008, 1, 103–107. [Google Scholar] [CrossRef] [PubMed]
- Rengmark, A.H.; Lingaas, F. Genomic structure of the Nile tilapia (Oreochromis niloticus) transferrin gene and a haplotype associated with saltwater tolerance. Aquaculture 2007, 272, 146–155. [Google Scholar] [CrossRef]
- Vos, P.; Hogers, R.; Bleeker, M.; Reijans, M.; Lee, T.V.D.; Frijters, A.; Pot, J.; Peleman, J.; Kuiper, M.; Zabeau, M.; et al. AFLP: A new technique for DNA fingerprinting. Nucleic Acids Res. 1995, 23, 4407–4414. [Google Scholar] [CrossRef] [PubMed]
- Bouétard, A.; Côte, J.; Besnard, A.-L.; Collinet, M.; Coutellec, M.-A. Environmental versus anthropogenic effects on population adaptive divergence in the freshwater snail Lymnaea stagnalis. PLoS ONE 2014, 9, e106670. [Google Scholar] [CrossRef] [PubMed]
- Bouétard, A.; Noirot, C.; Besnard, A.L.; Bouchez, O.; Choisne, D.; Robe, E.; Klopp, C.; Lagadic, L.; Coutellec, M.A. Pyrosequencing-based transcriptomic resources in the pond snail Lymnaea stagnalis, with a focus on genes involved in molecular response to diquat-induced stress. Ecotoxicology 2012, 21, 2222–2234. [Google Scholar] [CrossRef] [PubMed]
- Bélanger-Deschênes, S.; Couture, P.; Campbell, P.G.C.; Bernatchez, L. Evolutionary change driven by metal exposure as revealed by coding SNP genome scan in wild yellow perch (Perca flavescens). Ecotoxicology 2013, 22, 938–957. [Google Scholar] [CrossRef] [PubMed]
- Hendry, A.P.; Gotanda, K.M.; Svensson, E.I. Human influences on evolution, and the ecological and societal consequences. Philos. Trans. R. Soc. B Biol. Sci. 2017, 372, 20160028. [Google Scholar] [CrossRef] [PubMed]
- Csilléry, K.; Blum, M.G.B.; Gaggiotti, O.E.; François, O. Approximate Bayesian Computation (ABC) in practice. Trends Ecol. Evolut. 2010, 25, 410–418. [Google Scholar] [CrossRef] [PubMed]
- Stefani, F.; Rusconi, M.; Valsecchi, S.; Marziali, L. Evolutionary ecotoxicology of perfluoralkyl substances (PFASs) inferred from multigenerational exposure: A case study with Chironomus riparius (Diptera, Chironomidae). Aquat. Toxicol. 2014, 156, 41–51. [Google Scholar] [CrossRef] [PubMed]
- Momigliano, P.; Jokinen, H.; Fraimout, A.; Florin, A.-B.; Norkko, A.; Merilä, J. Extraordinarily rapid speciation in a marine fish. Proc. Natl. Acad. Sci. USA 2017, 114, 6074–6079. [Google Scholar] [CrossRef] [PubMed]
- Cornuet, J.; Pudlo, P.; Veyssier, J.; Dehne-garcia, A.; Marin, J.; Estoup, A.; Gautier, M.; Cnrs, U.M.R. DIYABC v2. 0: A software to make approximate Bayesian computation inferences about population history using single nucleotide polymorphism, DNA sequence and microsatellite data. Bioinformatics 2018, 30, 1187–1189. [Google Scholar] [CrossRef] [PubMed]
- Hoban, S.; Gaggiotti, O.; Bertorelle, G. Sample Planning Optimization Tool for conservation and population Genetics (SPOTG): A software for choosing the appropriate number of markers and samples. Methods Ecol. Evolut. 2013, 4, 299–303. [Google Scholar] [CrossRef]
- Hoban, S.; Arntzen, J.A.; Bruford, M.W.; Godoy, J.A.; Rus Hoelzel, A.; Segelbacher, G.; Vilà, C.; Bertorelle, G. Comparative evaluation of potential indicators and temporal sampling protocols for monitoring genetic erosion. Evolut. Appl. 2014, 7, 984–998. [Google Scholar] [CrossRef] [PubMed]
- Laporte, M.; Pavey, S.A.; Rougeux, C.; Pierron, F.; Lauzent, M.; Budzinski, H.; Labadie, P.; Geneste, E.; Couture, P.; Baudrimont, M.; et al. RAD sequencing reveals within-generation polygenic selection in response to anthropogenic organic and metal contamination in North Atlantic Eels. Mol. Ecol. 2016, 25, 219–237. [Google Scholar] [CrossRef] [PubMed]
- Bach, L.; Dahllöf, I. Local contamination in relation to population genetic diversity and resilience of an arctic marine amphipod. Aquat. Toxicol. 2012, 114–115, 58–66. [Google Scholar] [CrossRef] [PubMed]
- Boyer, B. Genome scan in the mosquito Aedes rusticus: Population structure and detection of positive selection after insecticide treatment. Mol. Ecol. 2010, 325–337. [Google Scholar] [CrossRef]
- Vega-Retter, C.; Vila, I.; Véliz, D. Signatures of directional and balancing selection in the silverside Basilichthys microlepidotus (Teleostei: Atherinopsidae) inhabiting a polluted river. Evolut. Biol. 2015, 42, 156–168. [Google Scholar] [CrossRef]
- Bank, C.; Ewing, G.B.; Ferrer-Admettla, A.; Foll, M.; Jensen, J.D. Thinking too positive? Revisiting current methods of population genetic selection inference. Trends Genet. 2014, 30, 540–546. [Google Scholar] [CrossRef] [PubMed]
- Hoban, S.; Kelley, J.L.; Lotterhos, K.E.; Antolin, M.F.; Bradburd, G.; Lowry, D.B.; Poss, M.L.; Reed, L.K.; Storfer, A.; Whitlock, M.C. Finding the genomic basis of local adaptation: Pitfalls, practical solutions, and future directions. Am. Nat. 2016, 188, 379–397. [Google Scholar] [CrossRef] [PubMed]
- Hohenlohe, P.A.; Phillips, P.C.; Cresko, W.A. Using population genomics to detect selection in natural populations: Key concepts and methodological considerations. Int. J. Plant Sci. 2011, 171, 1059–1071. [Google Scholar] [CrossRef] [PubMed]
- Tiffin, P.; Ross-Ibarra, J. Advances and limits of using population genetics to understand local adaptation. Trends Ecol. Evolut. 2014, 29, 673–680. [Google Scholar] [CrossRef] [PubMed]
- Merilä, J.; Hendry, A.P. Climate change, adaptation, and phenotypic plasticity: The problem and the evidence. Evolut. Appl. 2014, 7, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Whitehead, A.; Galvez, F.; Zhang, S.; Williams, L.M.; Oleksiak, M.F. Functional genomics of physiological plasticity and local adaptation in killifish. J. Hered. 2011, 102, 499–511. [Google Scholar] [CrossRef] [PubMed]
- Comeron, J.M. Background selection as null hypothesis in population genomics: Insights and challenges from Drosophila studies. Philos. Trans. R. Soc. B Biol. Sci. 2017, 372, 20160471. [Google Scholar] [CrossRef] [PubMed]
- Proestou, D.A.; Flight, P.; Champlin, D.; Nacci, D. Targeted approach to identify genetic loci associated with evolved dioxin tolerance in Atlantic Killifish (Fundulus heteroclitus). BMC Evolut. Biol. 2014, 14. [Google Scholar] [CrossRef] [PubMed]
- Foll, M.; Gaggiotti, O. A genome-scan method to identify selected loci appropriate for both dominant and codominant markers: A Bayesian perspective. Genetics 2008, 180, 977–993. [Google Scholar] [CrossRef] [PubMed]
- Vitalis, R.; Gautier, M.; Dawson, K.J.; Beaumont, M.A. Detecting and measuring selection from gene frequency data. Genetics 2014, 196, 799–817. [Google Scholar] [CrossRef] [PubMed]
- Günther, T.; Coop, G. Robust identification of local adaptation from allele frequencies. Genetics 2013, 195, 205–220. [Google Scholar] [CrossRef] [PubMed]
- Gautier, M. Genome-wide scan for adaptive divergence and association with population-specific covariates. Genetics 2015, 201, 1555–1579. [Google Scholar] [CrossRef] [PubMed]
- Messer, P.W. SLiM: Simulating evolution with selection and linkage. Genetics 2013, 194, 1037–1039. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, R.D. A flexible forward simulator for populations subject to selection and demography. Bioinformatics 2008, 24, 2786–2787. [Google Scholar] [CrossRef] [PubMed]
- Jensen, J.D.; Thornton, K.R.; Andolfatto, P. An approximate bayesian estimator suggests strong, recurrent selective sweeps in drosophila. PLoS Genet. 2008, 4. [Google Scholar] [CrossRef] [PubMed]
- Bazin, E.; Dawson, K.J.; Beaumont, M.A. Likelihood-free inference of population structure and local adaptation in a Bayesian hierarchical model. Genetics 2010, 185, 587–602. [Google Scholar] [CrossRef] [PubMed]
- Haasl, R.J.; Payseur, B.A. Fifteen years of genomewide scans for selection: Trends, lessons and unaddressed genetic sources of complication. Mol. Ecol. 2016, 5–23. [Google Scholar] [CrossRef] [PubMed]
- Nei, M.; Maruyama, T.; Chakraborty, R. The bottleneck effect and genetic variability in populations. Evolution 1975, 29, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Hartl, D.L. A Primer of Population Genetics; Sinauer Associates, Inc.: Sunderland, MA, USA, 2001; p. 221. [Google Scholar]
- De Meeûs, T.; Michalakis, Y.; Renaud, F.; Olivieri, I. Polymorphism in heterogeneous environments, evolution of habitat selection and sympatric speciation: Soft and hard selection models. Evolut. Ecol. 1993, 7, 175–198. [Google Scholar] [CrossRef]
- Bougas, B.; Normandeau, E.; Grasset, J.; Defo, M.A.; Campbell, P.G.C.; Couture, P.; Bernatchez, L. Transcriptional response of yellow perch to changes in ambient metal concentrations-A reciprocal field transplantation experiment. Aquat. Toxicol. 2016, 173, 132–142. [Google Scholar] [CrossRef] [PubMed]
- Guinand, B.; Fustier, M.A.; Labonne, M.; Jourdain, E.; Calvès, I.; Quiniou, L.; Cerqueira, F.; Laroche, J. Genetic structure and heterozygosity-fitness correlation in young-of-the-year sole (Solea solea L.) inhabiting three contaminated West-European estuaries. J. Sea Res. 2013, 80, 35–49. [Google Scholar] [CrossRef]
- Whitehead, A.; Clark, B.W.; Reid, N.M.; Hahn, M.E.; Nacci, D. When evolution is the solution to pollution: Key principles, and lessons from rapid repeated adaptation of killifish (Fundulus heteroclitus) populations. Evolut. Appl. 2017, 10, 762–783. [Google Scholar] [CrossRef] [PubMed]
- Inostroza, P.A.; Vera-Escalona, I.; Wicht, A.J.; Krauss, M.; Brack, W.; Norf, H. Anthropogenic stressors shape genetic structure: Insights from a model freshwater population along a land use gradient. Environ. Sci. Technol. 2016, 50, 11346–11356. [Google Scholar] [CrossRef] [PubMed]
- Athrey, N.R.G.; Leberg, P.L.; Klerks, P.L. Laboratory culturing and selection for increased resistance to cadmium reduce genetic variation in the least killifish, Heterandria formosa. Environ. Toxicol. Chem. 2007, 26, 1916–1921. [Google Scholar] [CrossRef] [PubMed]
- Nowak, C.; Vogt, C.; Pfenninger, M.; Schwenk, K.; Oehlmann, J.; Streit, B.; Oetken, M. Rapid genetic erosion in pollutant-exposed experimental chironomid populations. Environ. Pollut. 2009, 157, 881–886. [Google Scholar] [CrossRef] [PubMed]
- Vogt, C.; Nowak, C.; Barateiro, J.; Oetken, M.; Schwenk, K. Multi-generation studies with Chironomus riparius—Effects of low tributyltin concentrations on life history parameters and genetic diversity. Chemosphere 2007, 67, 2192–2200. [Google Scholar] [CrossRef] [PubMed]
- Couture, P.; Busby, P.; Gauthier, C.; Rajotte, J.W.; Pyle, G.G. Seasonal and regional variations of metal contamination and condition indicators in yellow perch (Perca flavescens) along two polymetallic gradients. I. Factors influencing tissue metal concentrations. Hum. Ecol. Risk Assess. 2008, 14, 97–125. [Google Scholar] [CrossRef]
- Couture, P.; Rajotte, J.W.; Pyle, G.G. Seasonal and regional variations in metal contamination and condition indicators in yellow perch (Perca flavescens) along two polymetallic gradients. III. Energetic and physiological indicators. Hum. Ecol. Risk Assess. 2008, 14, 146–165. [Google Scholar] [CrossRef]
- Pyle, G.; Busby, P.; Gauthier, C.; Rajotte, J.; Couture, P. Seasonal and regional variations in metal contamination and condition indicators in yellow perch (Perca flavescens) along two polymetallic gradients. II. Growth patterns, longevity, and condition. Hum. Ecol. Risk Assess. 2008, 14, 126–145. [Google Scholar] [CrossRef]
- Reid, N.M.; Proestou, D.A.; Clark, B.W.; Warren, W.C.; Colbourne, J.K.; Shaw, J.R.; Karchner, S.I.; Hahn, M.E.; Nacci, D.; Oleksiak, M.F.; et al. The genomic landscape of rapid repeated evolutionary adaptation to toxic pollution in wild fish. Science 2016, 354, 1305–1308. [Google Scholar] [CrossRef] [PubMed]
- Sobral, O.; Marin-Morales, M.A.; Ribeiro, R. Could contaminant induced mutations lead to a genetic diversity overestimation? Ecotoxicology 2013, 22, 838–846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Josephs, E.B.; Stinchcombe, J.R.; Wright, S.I. What can genome-wide association studies tell us about the evolutionary forces maintaining genetic variation for quantitative traits? New Phytol. 2017, 214, 21–33. [Google Scholar] [CrossRef] [PubMed]
- The IUCN Red list of Threatened Species. Version 2016-3; Version 20; IUCN (International Union for Conservation of Nature): Gland, Switzerland, 2016.
- Baer, C.F. Does mutation rate depend on itself. PLoS Biol. 2008, 6, e52. [Google Scholar] [CrossRef] [PubMed]
- Rusconi, M.; Marziali, L.; Stefani, F.; Valsecchi, S.; Bettinetti, R.; Mazzoni, M.; Rosignoli, F.; Polesello, S. Evaluating the impact of a fluoropolymer plant on a river macrobenthic community by a combined chemical, ecological and genetic approach. Sci. Total Environ. 2015, 538, 654–663. [Google Scholar] [CrossRef] [PubMed]
- Ashley, M.V.; Willson, M.F.; Pergams, O.R.W.; O’Dowd, D.J.; Gende, S.M.; Brown, J.S. Evolutionarily enlightened management. Biol. Conserv. 2003, 111, 115–123. [Google Scholar] [CrossRef]
- Laval, G.; Excoffier, L. SIMCOAL 2.0: A program to simulate genomic diversity over large recombining regions in a subdivided population with a complex history. Bioinformatics 2004, 20, 2485–2487. [Google Scholar] [CrossRef] [PubMed]
- Mussali-Galante, P.; Tovar-Sánchez, E.; Valverde, M.; Rojas, E. Genetic Structure and Diversity of Animal Populations Exposed to Metal Pollution BT—Reviews of Environmental Contamination and Toxicology; Whitacre, D.M., Ed.; Springer International Publishing: Cham, Germany, 2014; Volume 227, pp. 79–106. ISBN 978-3-319-01327-5. [Google Scholar]
- Smith, T.B.; Bernatchez, L. Evolutionary change in human-altered environments. Mol. Ecol. 2008, 17, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Bergland, A.O.; Behrman, E.L.; O’Brien, K.R.; Schmidt, P.S.; Petrov, D.A. Genomic evidence of rapid and stable adaptive oscillations over seasonal time scales in Drosophila. PLoS Genet. 2014, 10. [Google Scholar] [CrossRef] [PubMed]
- Acerenza, L. Constraints, trade-offs and the currency of fitness. J. Mol. Evolut. 2016, 82, 117–127. [Google Scholar] [CrossRef] [PubMed]
- Dutilleul, M.; Réale, D.; Goussen, B.; Lecomte, C.; Galas, S.; Bonzom, J.M. Adaptation costs to constant and alternating polluted environments. Evolut. Appl. 2017, 10, 839–851. [Google Scholar] [CrossRef] [PubMed]
- Shirley, M.D.F.; Sibly, R.M. Genetic basis of a between-environment trade-off involving resistance to Cadmium in Drosophila melanogaster. Evolution 1999, 53, 826–836. [Google Scholar] [CrossRef] [PubMed]
- Harrison, P.A.; Berry, P.M.; Simpson, G.; Haslett, J.R.; Blicharska, M.; Bucur, M.; Dunford, R.; Egoh, B.; Garcia-Llorente, M.; Geamănă, N.; et al. Linkages between biodiversity attributes and ecosystem services: A systematic review. Ecosyst. Serv. 2014, 9, 191–203. [Google Scholar] [CrossRef]
- Brady, S.P.; Richardson, J.L.; Kunz, B.K. Incorporating evolutionary insights to improve ecotoxicology for freshwater species. Evolut. Appl. 2017, 10, 829–838. [Google Scholar] [CrossRef] [PubMed]
- Szamecz, B.; Boross, G.; Kalapis, D.; Kovács, K.; Fekete, G.; Farkas, Z.; Lázár, V.; Hrtyan, M.; Kemmeren, P.; Groot Koerkamp, M.J.A.; Rutkai, E.; Holstege, F.C.P.; et al. The genomic landscape of compensatory evolution. PLoS Biol. 2014, 12. [Google Scholar] [CrossRef] [PubMed]
- Wan, J.S.H.; Pang, C.K.; Bonser, S.P. Does the cost of adaptation to extremely stressful environments diminish over time? A literature synthesis on how plants adapt to heavy metals and pesticides. Evolut. Biol. 2017, 44, 411–426. [Google Scholar] [CrossRef]
- Palmgren, M.; Engström, K.; Hallström, B.M.; Wahlberg, K.; Søndergaard, D.A.; Sall, T.; Vahter, M.; Broberg, K. AS3MT-mediated tolerance to arsenic evolved by multiple independent horizontal gene transfers from bacteria to eukaryotes. PLoS ONE 2017, 12, e0175422. [Google Scholar] [CrossRef] [PubMed]
- Hendriks, A.J. How to deal with 100,000+ substances, sites, and species: Overarching principles in environmental risk assessment. Environ. Sci. Technol. 2013, 47, 3546–3547. [Google Scholar] [CrossRef] [PubMed]
- Hua, J.; Wuerthner, V.P.; Jones, D.K.; Mattes, B.; Cothran, R.D.; Relyea, R.A.; Hoverman, J.T. Evolved pesticide tolerance influences susceptibility to parasites in amphibians. Evolut. Appl. 2017, 10, 802–812. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Markers | Errors | ||||
|---|---|---|---|---|---|
| Low Bottleneck | High Bottleneck | ||||
| Microsatellites | 20 loci | Type I | 0.36 | Type I | 0.43 |
| Type II | 0.36 | Type II | 0.34 | ||
| 50 loci | Type I | 0.43 | Type I | 0.35 | |
| Type II | 0.31 | Type II | 0.09 | ||
| 50 loci (rep. samples) | Type I | 0.20 | Type I | 0.18 | |
| Type II | 0.13 | Type II | 0.12 | ||
| Single nucleotide polymorphisms (SNPs) | 2000 loci | Type I | 0.38 | Type I | 0.46 |
| Type II | 0.17 | Type II | 0.14 | ||
| Organism | Contaminant | Genetic Assay | Strengths | Opportunities | Reference | |
|---|---|---|---|---|---|---|
| Field case studies | Perca flavescens | Metals (Cu, Cd) | Microsatellites, de novo transcriptome scan, microarrays | Genetic erosion and selection at unconventional targets were both detected | Test for adaptation costs toward other stressors could be performed | [10,28,60] |
| Daphnia longispina | Acidity, metals | AFLP microsatellites, Comet test | Tight implementation of quantitative, population genetics, and phenotype responses | Only traditional genetics approaches were employed, no Next Generation Sequencing (NGS) data | [11] | |
| Solea solea | Complex mixture of pesticides and organic pollutants derived from agricultural land drainage | Microsatellites | One of the few studies finding a heterozygosity-fitness correlation, although at a single locus | Low number of samples and markers, inferences at polygenic level would have likely benefitted from NGS | [61] | |
| Fundulus heteroclitus | Dioxin-like contaminants | Transcriptomics, genomics, quantitative genetics | Many different approaches converge in demonstrating adaptation to contaminants and pathways affected | Long-term effects (i.e., genetic erosion, recovery from adaptation) are still uncertain and not predictable | [62] | |
| Gammarus pulex | Effluent of wastewater treatment plants and man-made barriers | Microsatellites | Synergic effects of habitat fragmentation, mutagenic compounds, and contaminants exposure were identified | The role of de novo mutations vs. standing genetic variation in adaptation to contaminants (e.g., with genomic approaches) could be evaluated | [63] | |
| Anguilla anguilla and Anguilla rostrata | PCBs, DDTs, metals | SNPs generated by restriction site associated DNA sequencing (RAD-seq) | Polygenic adaptation was demonstrated using RAD-seq polymorphisms analyzed by the Random Forest technique | The hypothesis of long-term genetic erosion due to contaminants could be tested | [36] | |
| Laboratory case studies | Heterandria formosa | Cadmium | Microsatellites | A link between adaptation and overall genetic erosion was found; limits of closed multigeneration tests were undiscovered | Genes underpinning rapid adaptation cannot be investigated by using neutral markers | [64] |
| Chironomus riparius | Tributyltin (TBT) | Microsatellites (multigenerational approach) | Effects on genetic variability were diversified at different concentrations of TBT; repeated temporal samples | Low number of neutral markers, the complex of genes adapted in relation to TBT concentration could be unveiled by integrating NGS | [65,66] | |
| Chironomus riparius | Perfluorinated compounds | Microsatellites (multigenerational approach) | Integration of coalescent simulations approach in evolutionary toxicology demonstrating mutation rate increase; repeated temporal samples | Low number of markers, selection could not be tested or identified, NGS approaches could be implemented at this scale | [31] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rusconi, M.; Bettinetti, R.; Polesello, S.; Stefani, F. Evolutionary Toxicology as a Tool to Assess the Ecotoxicological Risk in Freshwater Ecosystems. Water 2018, 10, 490. https://doi.org/10.3390/w10040490
Rusconi M, Bettinetti R, Polesello S, Stefani F. Evolutionary Toxicology as a Tool to Assess the Ecotoxicological Risk in Freshwater Ecosystems. Water. 2018; 10(4):490. https://doi.org/10.3390/w10040490
Chicago/Turabian StyleRusconi, Marianna, Roberta Bettinetti, Stefano Polesello, and Fabrizio Stefani. 2018. "Evolutionary Toxicology as a Tool to Assess the Ecotoxicological Risk in Freshwater Ecosystems" Water 10, no. 4: 490. https://doi.org/10.3390/w10040490
APA StyleRusconi, M., Bettinetti, R., Polesello, S., & Stefani, F. (2018). Evolutionary Toxicology as a Tool to Assess the Ecotoxicological Risk in Freshwater Ecosystems. Water, 10(4), 490. https://doi.org/10.3390/w10040490