Experimental Analysis and Modeling of Nitrate Removal through Zero-Valent Magnesium Particles




Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Denitrification Batch Tests
2.3. Analytical Methods and the Presentation of Results
3. Results and Discussion
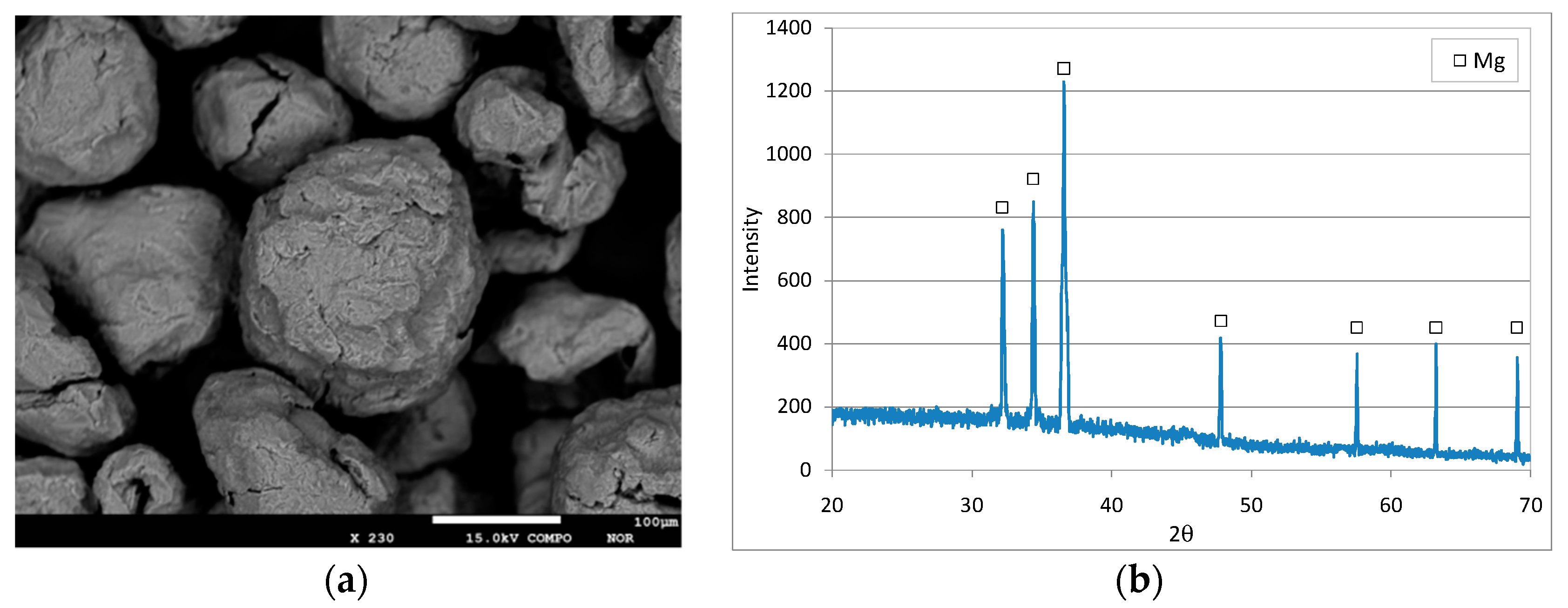
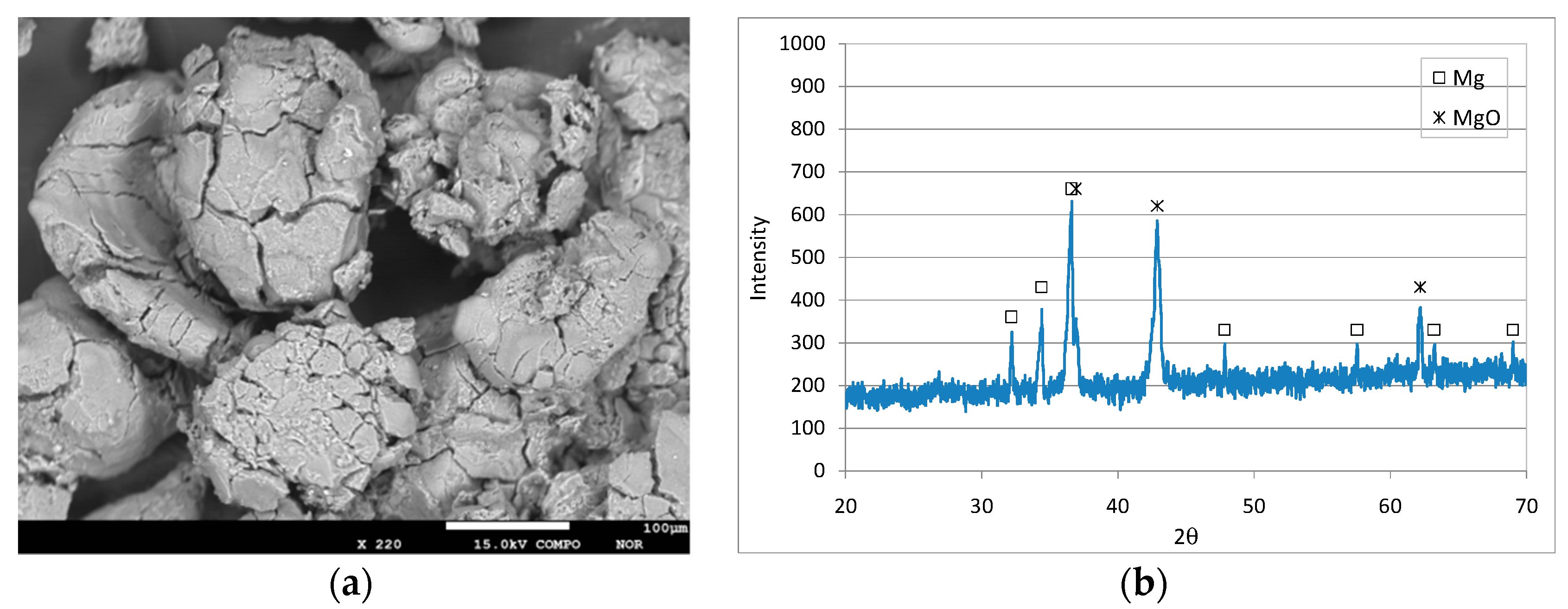
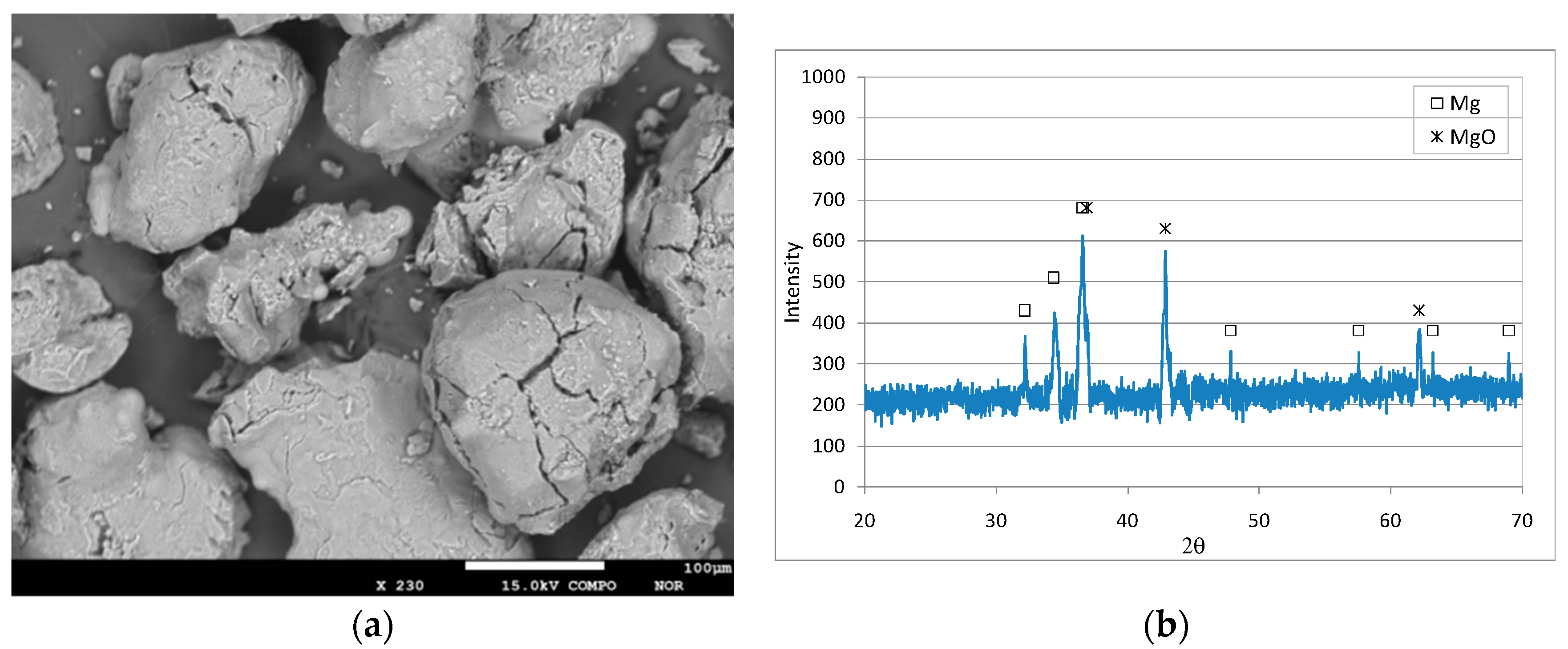
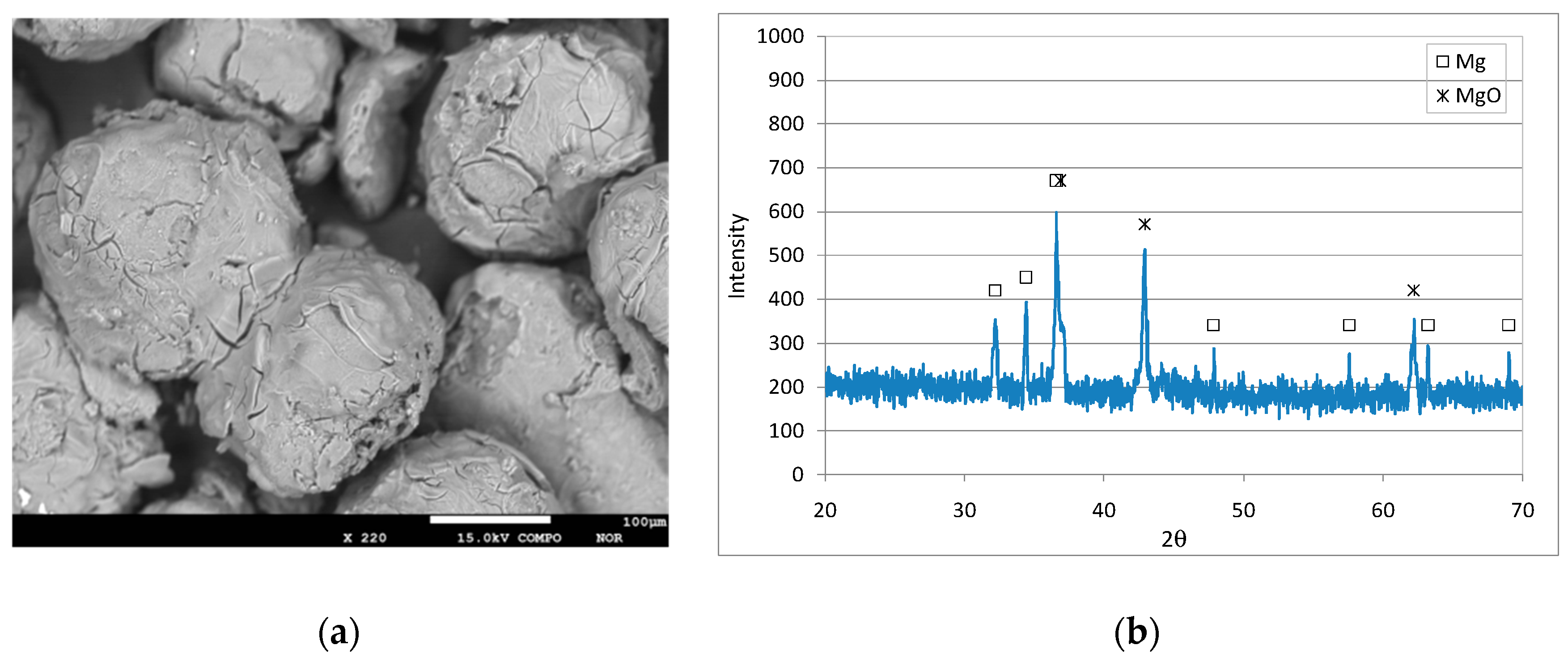
3.1. Characteristics of the Reactive Material
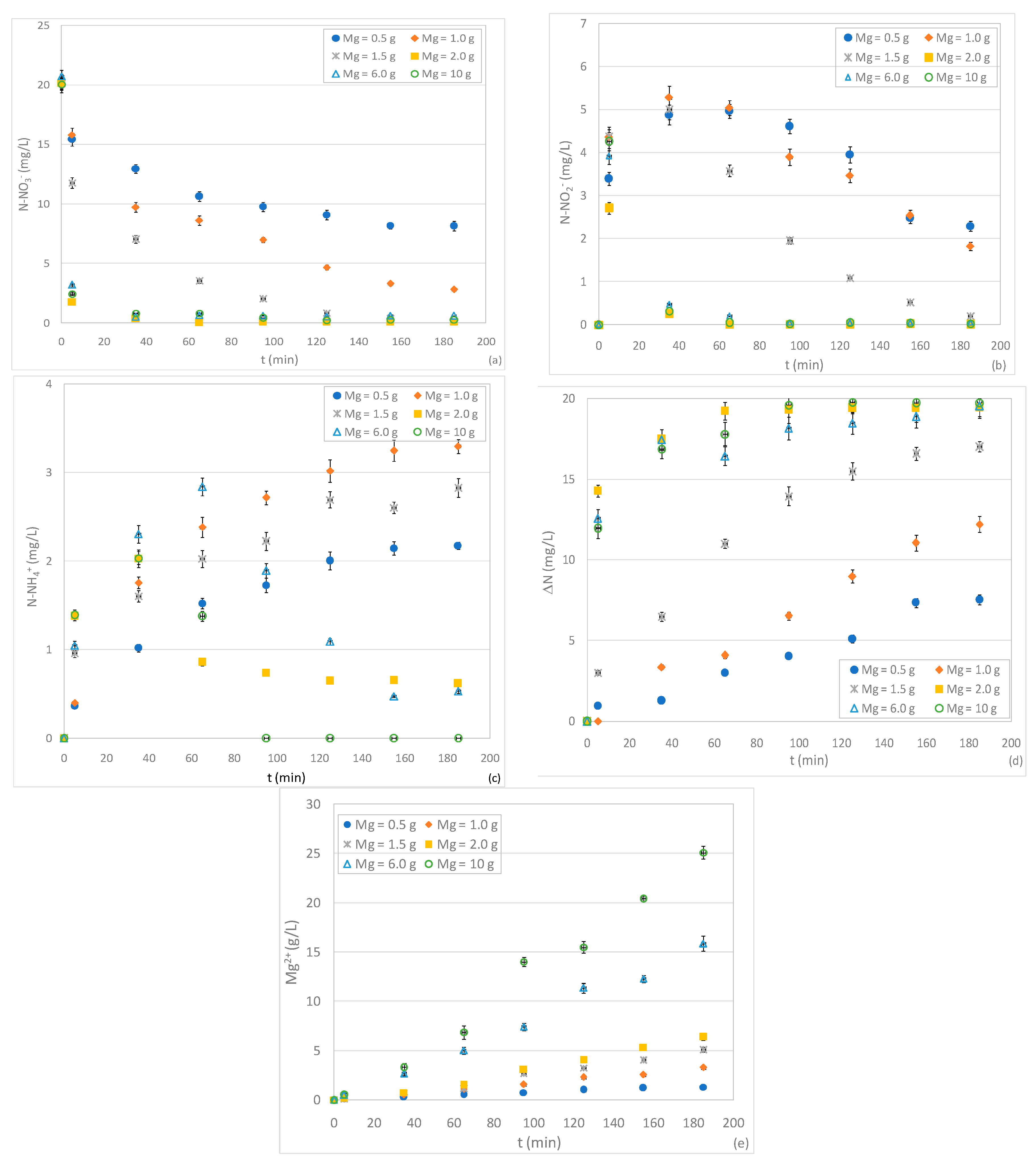
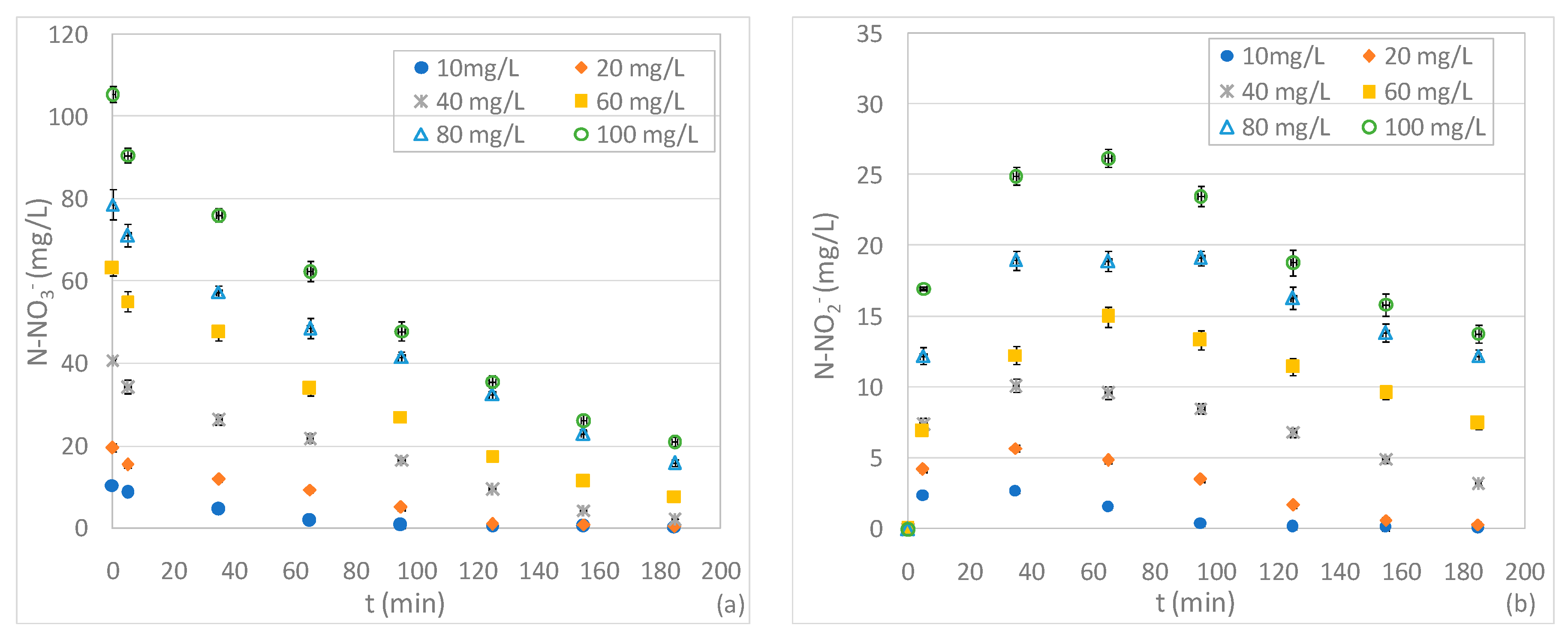
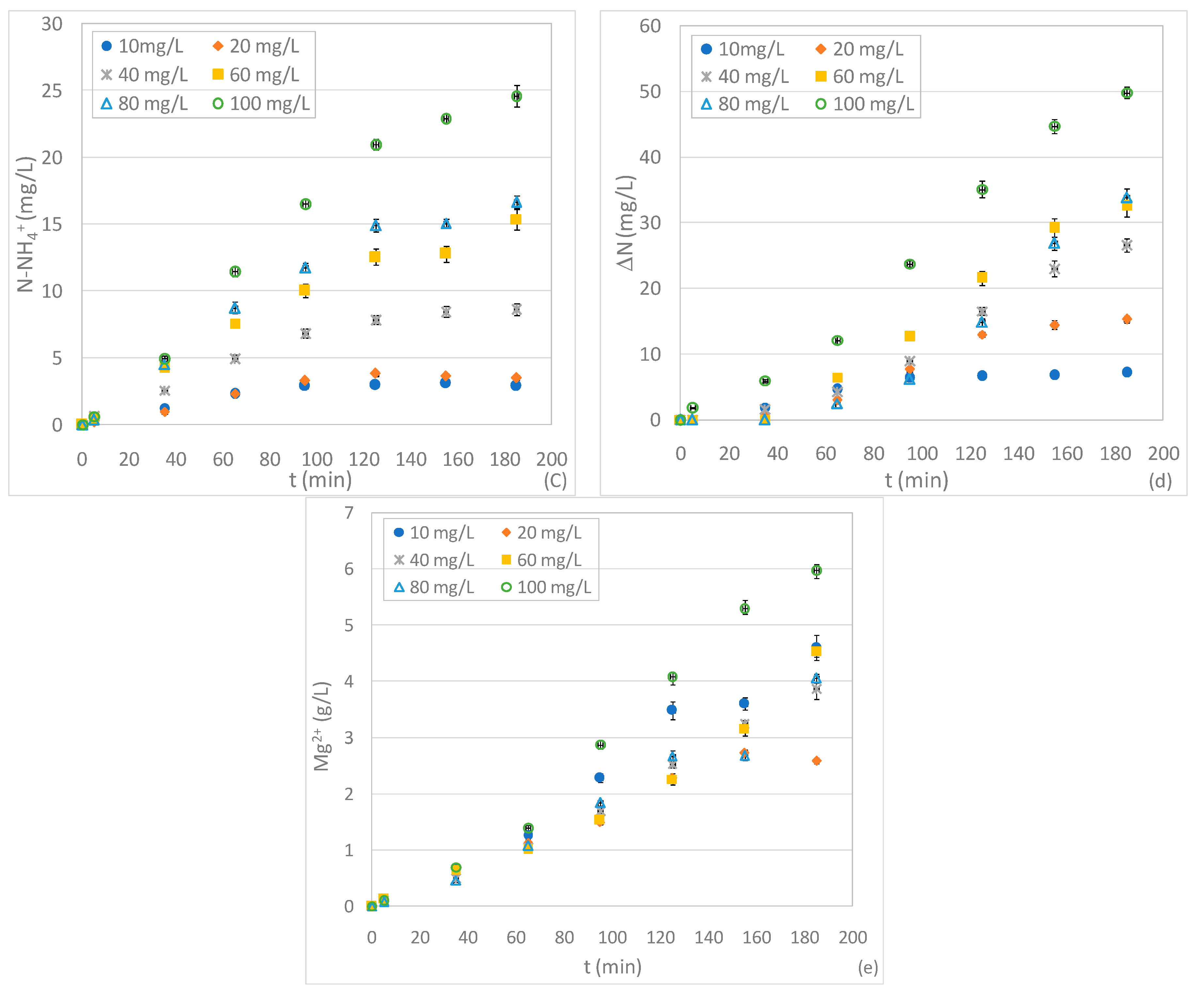
3.2. Tests Conducted with Different Quantity of ZVM
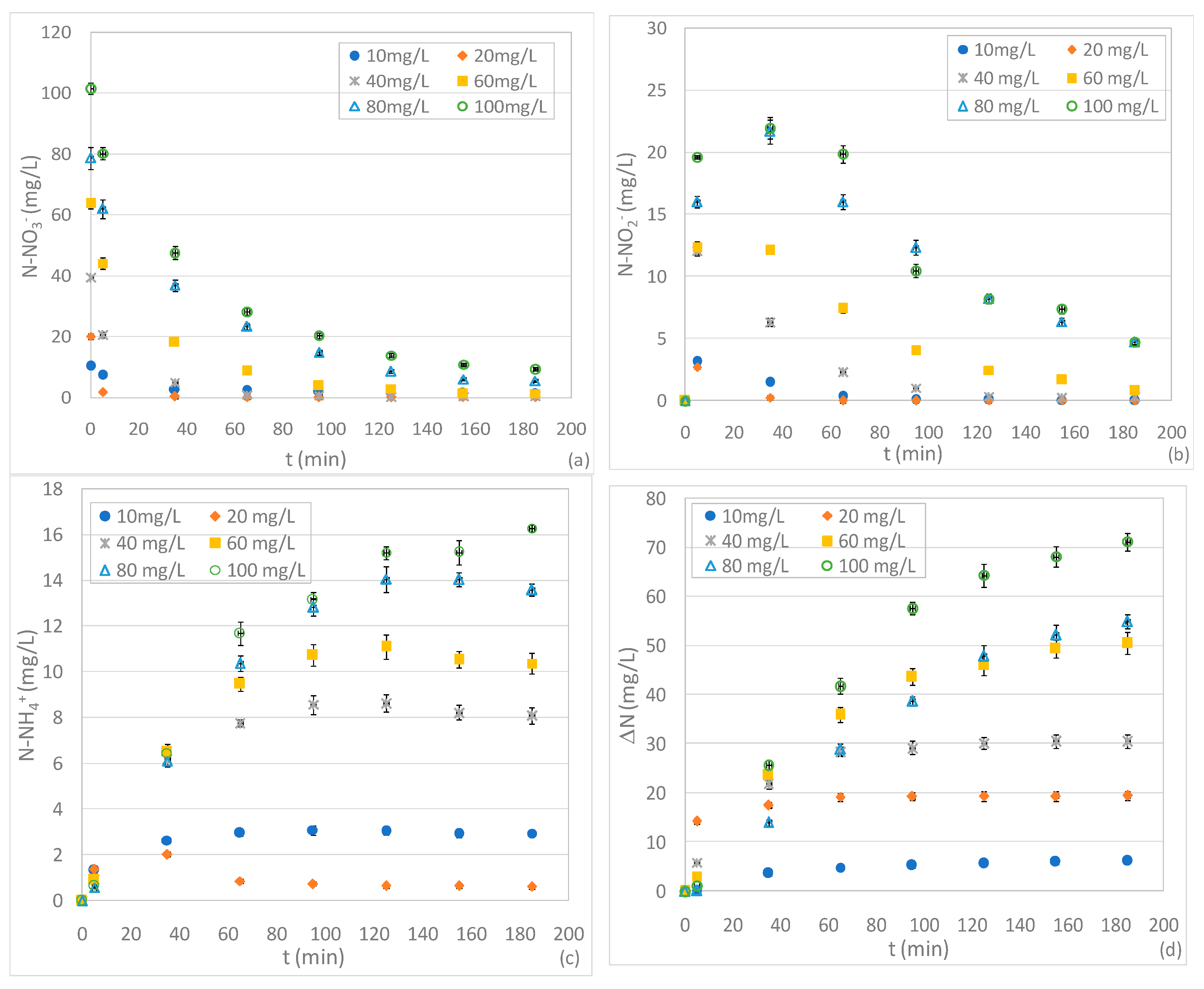
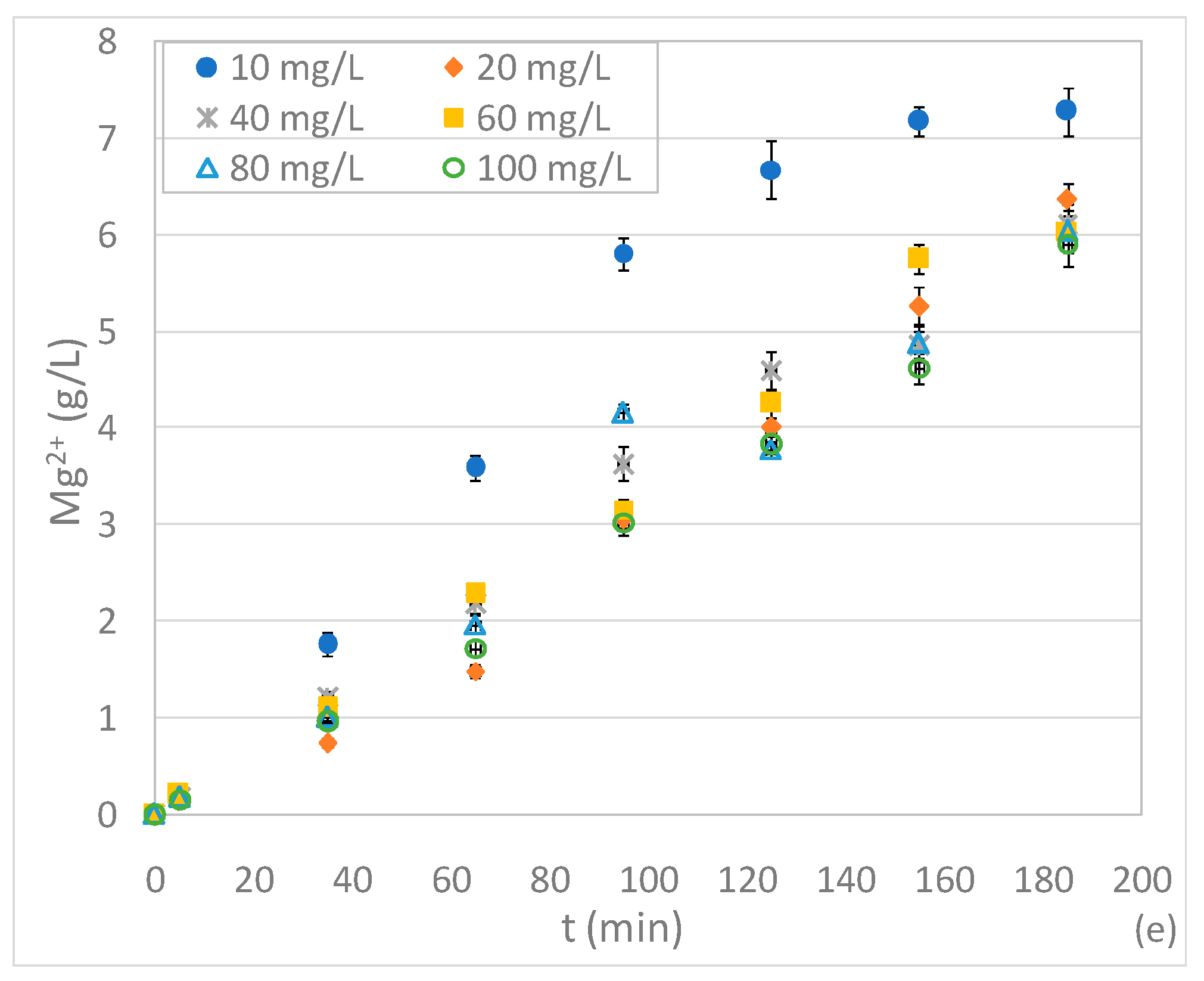
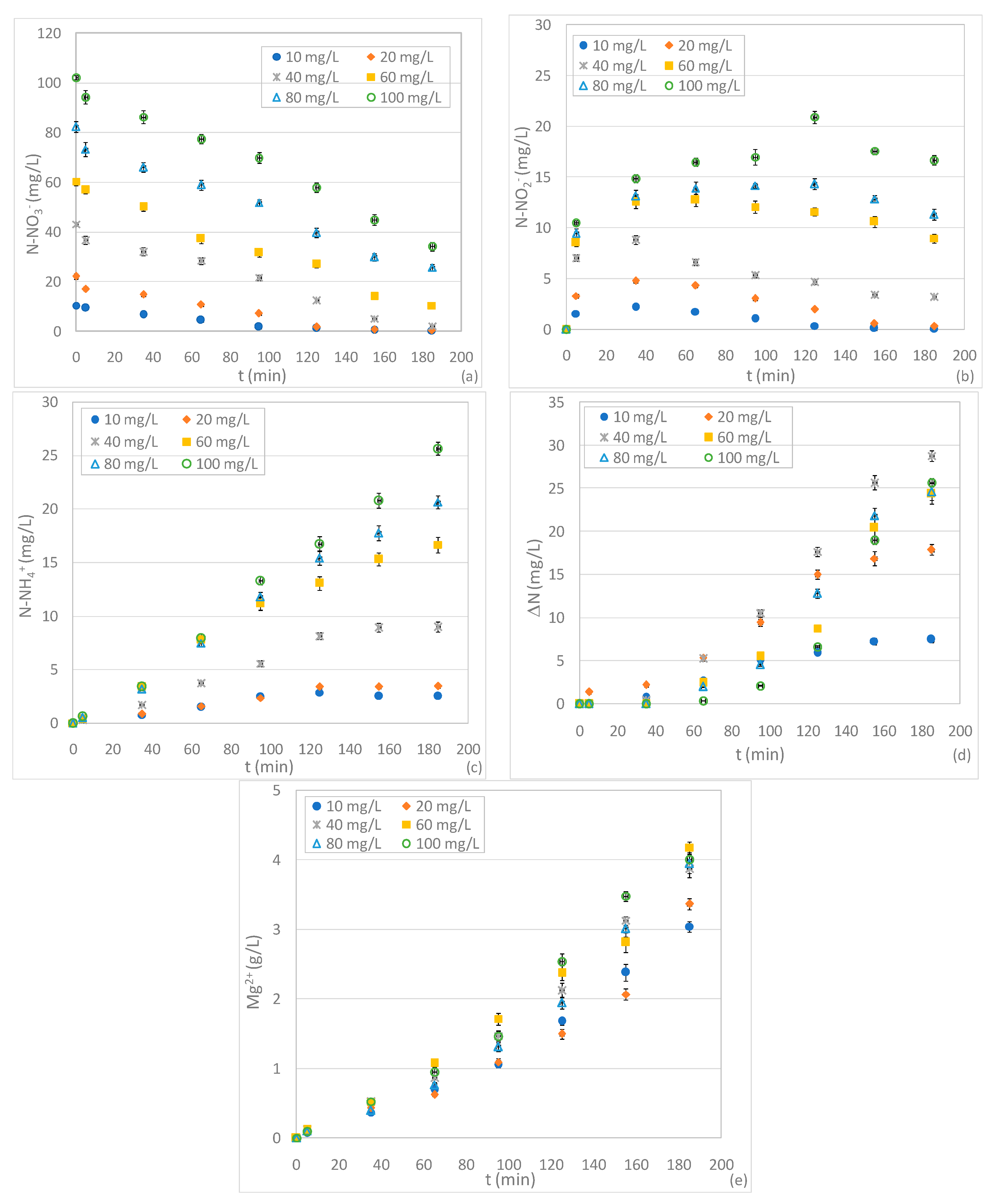
3.3. Tests Conducted at Different pHs and N-NO3− Concentrations
3.4. Kinetic Analysis and Process Modeling
3.5. Reaction Mechanisms and Characteristics of Exhausted Mg0
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Bhatnagar, A.; Sillanpää, M. A review of emerging adsorbents for nitrate removal from water. Chem. Eng. J. 2011, 168, 493–504. [Google Scholar] [CrossRef]
- Siciliano, A. Assessment of fertilizer potential of the struvite produced from the treatment of methanogenic landfill leachate using low-cost reagents. Environ. Sci. Pollut. Res. 2016, 23, 5949–5959. [Google Scholar] [CrossRef] [PubMed]
- Siciliano, A.; Stillitano, M.A.; Limonti, C.; Marchio, F. Ammonium removal from landfill leachate by means of multiple recycling of struvite residues obtained through acid decomposition. Appl. Sci. 2016, 6, 345. [Google Scholar] [CrossRef]
- Kumar, M.; Chakraborty, S. Chemical denitrification of water by zero-valent magnesium powder. J. Hazard. Mater. 2006, B135, 112–121. [Google Scholar] [CrossRef] [PubMed]
- Siciliano, A.; Stillitano, M.A.; De Rosa, S. Increase of the anaerobic biodegradability of olive mill wastewaters through a pre-treatment with hydrogen peroxide in alkaline conditions. Desalin. Water Treat. 2014, 55, 1735–1746. [Google Scholar] [CrossRef]
- Hwang, Y.-H.; Kim, D.-G.; Shin, H.-S. Mechanism study of nitrate reduction by nano zero valent iron. J. Hazard. Mater. 2011, 185, 1513–1521. [Google Scholar] [CrossRef] [PubMed]
- Siciliano, A.; De Rosa, S. Experimental formulation of a kinetic model describing the nitrification process in biological aerated filters filled with plastic elements. Environ. Technol. 2015, 36, 293–301. [Google Scholar] [CrossRef]
- Siciliano, A.; De Rosa, S. An experimental model of COD abatement in MBBR based on biofilm growth dynamic and on substrates’ removal kinetics. Environ. Technol. 2016, 37, 2058–2071. [Google Scholar] [CrossRef]
- Luk, G.K.; Au-Yeung, W.C. Experimental investigation on the chemical reduction of nitrate from groundwater. Adv. Environ. Res. 2002, 6, 441–453. [Google Scholar] [CrossRef]
- Huang, C.-P.; Wang, H.-W.; Chiu, P.C. Nitrate reduction by metallic iron. Water Res. 1998, 32, 2257–2264. [Google Scholar] [CrossRef]
- Ruangchainikom, C.; Liao, C.-H.; Anotai, J.; Lee, M.-T. Effects of water characteristics on nitrate reduction by the Fe0/CO2 process. Chemosphere 2006, 63, 335–343. [Google Scholar] [CrossRef] [PubMed]
- Cheng, F.; Muftikian, R.; Fernando, Q.; Korte, N. Reduction of nitrate to ammonia by zero-valent iron. Chemosphere 1997, 35, 2689–2695. [Google Scholar] [CrossRef]
- Rodríguez-Maroto, J.M.; García-Herruzo, F.; García-Rubio, A.; Gómez-Lahoz, C.; Vereda-Alonso, C. Kinetics of the chemical reduction of nitrate by zero-valent iron. Chemosphere 2009, 74, 804–809. [Google Scholar] [CrossRef] [PubMed]
- Choe, S.; Liljestrand, H.M.; Khim, J. Nitrate reduction by zero-valent iron under different pH regimes. Appl. Geochem. 2004, 19, 335–342. [Google Scholar] [CrossRef]
- Huang, Y.H.; Zhang, T.C. Effects of low pH on nitrate reduction by iron powder. Water Res. 2004, 38, 2631–2642. [Google Scholar] [CrossRef] [PubMed]
- Luo, H.; Jin, S.; Fallgren, P.H.; Colberg, P.J.S.; Johnson, P.A. Prevention of iron passivation and enhancement of nitrate reduction by electron supplementation. Chem. Eng. J. 2010, 160, 185–189. [Google Scholar] [CrossRef]
- Huang, Y.H.; Zhang, T.C. Effects of dissolved oxygen on formation of corrosion products and concomitant oxygen and nitrate reduction in zero-valent iron systems with or without aqueous Fe2+. Water Res. 2005, 39, 1751–1760. [Google Scholar] [CrossRef]
- Noubactep, C.; Caré, S. On nanoscale metallic iron for groundwater remediation. J. Hazard. Mater. 2010, 182, 923–927. [Google Scholar] [CrossRef] [Green Version]
- Yang, G.C.C.; Lee, H.L. Chemical reduction of nitrate by nanosized iron:Kinetics and Pathways. Water Res. 2005, 39, 884–894. [Google Scholar] [CrossRef]
- Siciliano, A. Use of nanoscale zero-valent iron (NZVI) particles for chemical denitrification under different operating conditions. Metals 2015, 5, 1507–1519. [Google Scholar] [CrossRef]
- Kassaee, M.Z.; Motamedi, E.; Mikhak, A.; Rahnemaie, R. Nitrate removal from water using iron nanoparticles produced by arc discharge vs. Reduction. Chem. Eng. J. 2011, 166, 490–495. [Google Scholar] [CrossRef]
- Liou, Y.H.; Lo, S.-L.; Lin, C.-J.; Kuan, W.H.; Weng, S.C. Chemical reduction of an unbuffered nitrate solution using catalyzed and uncatalyzed nanoscale iron particles. J. Hazard. Mater. 2005, B127, 102–110. [Google Scholar] [CrossRef] [PubMed]
- Tsai, Y.-J.; Chou, F.-C.; Cheng, T.-C. Coupled acidification and ultrasound with iron enhances nitrate reduction. J. Hazard. Mater. 2009, 163, 743–747. [Google Scholar] [CrossRef] [PubMed]
- Siciliano, A. Removal of Cr(VI) from water using a new reactive material: Magnesium Oxide supported nanoscale zero-valent iron. Materials 2016, 9, 666. [Google Scholar] [CrossRef] [PubMed]
- Siciliano, A.; Limonti, C. Nanoscopic zero-valent iron supported on MgO for lead removal from waters. Water 2018, 10, 404. [Google Scholar] [CrossRef]
- Liou, Y.H.; Lo, S.H.; Lin, C.J.; Hu, C.Y.; Kuan, W.H.; Weng, S.C. Methods for accelerating nitrate reduction using zerovalent iron atnear-neutral pH: Effects ofH2-reducing pretreatment andcopper deposition. Environ. Sci. Technol. 2005, 39, 9643–9648. [Google Scholar] [CrossRef]
- Mossa Hosseini, S.; Ataie-Ashtiani, B.; Kholghi, M. Nitrate reduction by nano-Fe/Cu particles in packed column. Desalination 2011, 276, 214–221. [Google Scholar] [CrossRef]
- Chang Ahn, S.; Oh, S.-Y.; Cha, D.K. Enhanced reduction of nitrate by zero-valent iron at elevated temperatures. J. Hazard. Mater. 2008, 156, 17–22. [Google Scholar]
- Lee, G.; Park, J. Reaction of zero-valent magnesium with water: Potential applications in environmental remediation. Geochim.Cosmochim. Acta 2013, 102, 162–174. [Google Scholar] [CrossRef]
- Lee, G.; Park, J.; Harvey, O.R. Reduction of Chromium (VI) mediated by zero-valent magnesium under neutral pH conditions. Water Res. 2013, 47, 1136–1146. [Google Scholar] [CrossRef]
- Ileri, B.; Ayyildiz, O.; Apaydin, O. Ultrasound-assisted activation of zero-valent magnesium for nitrate denitrification: Identification of reaction by-products and pathways. J. Hazard. Mater. 2015, 292, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Mirabi, M.; Ghaderi, E.; Sadabad, H.R. Nitrate reduction using hybrid system consisting of zero valent magnesium powder/activated carbon (Mg0/AC) from water. Process Saf. Environ. Prot. 2017, 111, 627–634. [Google Scholar] [CrossRef]
- APHA. Standard Methods for the Examination of Water and Wastewater, 20th eds.; American Public Health Association: Washington, DC, USA, 1998. [Google Scholar]
- Peel, J.W.; Reddy, K.J.; Sullivan, B.P.; Bowen, J.M. Electrocatalytic reduction of nitrate in water. Water Res. 2003, 37, 2512–2519. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Operating Conditions Tested | Kinetic Constants | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| pH | Mg0 g | NNi mg/L | Mg/NNi g/mg | KN min−1 | NN,R mg/L | KNI min−1 | KI min−1 | KNA min−1 | KIA min−1 | KA min−1 | fNA - | KNG min−1 | KIG min−1 | fNG - |
| 3 | 0.5 | 20 | 0.083 | 0.03213 | 8.665 | 0.02661 | 0.01035 | 0.01022 | 0.00305 | 0.00040 | 0.1329 | 0.00347 | 0.00326 | 0.99 |
| 1 | 0.167 | 0.02243 | 3.457 | 0.02011 | 0.01945 | 0.01168 | 0.00308 | 0.00052 | 0.1815 | 0.00405 | 0.00610 | 0.99 | ||
| 1.5 | 0.250 | 0.04367 | 1.151 | 0.02913 | 0.02955 | 0.01538 | 0.00305 | 0.00101 | 0.1453 | 0.00972 | 0.0095 | 0.99 | ||
| 2 | 0.333 | 0.5310 | 0.060 | 0.142 | 0.08144 | 0.0651 | 0.09474 | 0.00358 | 0.03609 | 0.1866 | 0.0100 | 0.97 | ||
| 6 | 1.00 | 0.4474 | 0.602 | 0.1277 | 0.07423 | 0.00787 | 0.065 | 0.0071 | 0.00109 | 0.1220 | 0.0095 | 0.92 | ||
| 10 | 1.67 | 0.4736 | 0.301 | 0.1555 | 0.09147 | 0.0079410 | 0.06711 | 0.02591 | 0.00292 | 0.0870 | 0.0087 | 0.99 | ||
| Operating Conditions Tested | Kinetic Constants | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| pH | Mg0 | NNi | Mg/NNi | KN | NN,R | KNI | KI | KNA | KIA | KA | fNA | KNG | KIG | fNG |
| g | mg/L | g/mg | min−1 | mg/L | min−1 | min−1 | min−1 | min−1 | min−1 | - | min−1 | min−1 | - | |
| 3 | 2 | 10 | 0.667 | 0.07384 | 1.686 | 0.0842 | 0.0794 | 0.056 | 0.00160 | 0.00183 | 0.3618 | 0.02471 | 0.00882 | 0.65 |
| 20 | 0.333 | 0.53100 | 0.06 | 0.1420 | 0.08144 | 0.0651 | 0.09474 | 0.00358 | 0.03609 | 0.1866 | 0.0100 | 0.97 | ||
| 40 | 0.167 | 0.13040 | 1.16 | 0.0977 | 0.05408 | 0.00876 | 0.01056 | 0.00149 | 0.2570 | 0.03921 | 0.0085 | 0.78 | ||
| 60 | 0.111 | 0.04569 | 2.889 | 0.0280 | 0.0330 | 0.00688 | 0.00986 | 0.00100 | 0.1862 | 0.0154 | 0.00675 | 0.83 | ||
| 80 | 0.083 | 0.02384 | 5.338 | 0.0256 | 0.0314 | 0.00321 | 0.00657 | 0.00079 | 0.1397 | 0.00854 | 0.00722 | 0.78 | ||
| 100 | 0.067 | 0.02649 | 10.45 | 0.0210 | 0.0258 | 0.00295 | 0.00600 | 0.00054 | 0.14495 | 0.00937 | 0.00635 | 0.85 | ||
| 5 | 2 | 10 | 0.667 | 0.02545 | 0.00070 | 0.0400 | 0.0750 | 0.00815 | 0.01186 | 0.00057 | 0.278 | 0.00511 | 0.01710 | 0.81 |
| 20 | 0.333 | 0.01527 | 0.00026 | 0.0380 | 0.0701 | 0.00680 | 0.00609 | 0.00049 | 0.151 | 0.00353 | 0.01378 | 0.76 | ||
| 40 | 0.167 | 0.01150 | 0.00011 | 0.0241 | 0.0482 | 0.00627 | 0.00607 | 0.00002 | 0.126 | 0.00111 | 0.0110 | 0.81 | ||
| 60 | 0.111 | 0.00992 | 0.00011 | 0.01082 | 0.02434 | 0.00651 | 0.00627 | 0.00001 | 0.162 | 0.00088 | 0.0116 | 0.87 | ||
| 80 | 0.083 | 0.00764 | 0.00034 | 0.01007 | 0.02278 | 0.00655 | 0.00522 | 0.00005 | 0.134 | 0.00006 | 0.00960 | 0.55 | ||
| 100 | 0.067 | 0.00879 | 0.00034 | 0.01006 | 0.0166 | 0.00650 | 0.00358 | 0.00002 | 0.241 | 0.00003 | 0.01248 | 0.50 | ||
| 7 | 2 | 10 | 0.667 | 0.01317 | 0.00012 | 0.02180 | 0.0730 | 0.00186 | 0.01293 | 0.00051 | 0.24 | 0.00508 | 0.01495 | 0.83 |
| 20 | 0.333 | 0.01329 | 0.00034 | 0.02028 | 0.06056 | 0.00151 | 0.00750 | 0.00044 | 0.15 | 0.00346 | 0.0138 | 0.98 | ||
| 40 | 0.167 | 0.00953 | 0.00024 | 0.01340 | 0.04552 | 0.00148 | 0.00716 | 0.00001 | 0.28 | 0.00111 | 0.01424 | 0.98 | ||
| 60 | 0.111 | 0.00762 | 0.00043 | 0.00871 | 0.0206 | 0.00137 | 0.00792 | 0.00009 | 0.33 | 0.00027 | 0.0088 | 0.96 | ||
| 80 | 0.083 | 0.00622 | 0.00021 | 0.00609 | 0.0188 | 0.00124 | 0.00711 | 0.00001 | 0.39 | 0.00002 | 0.00668 | 0.98 | ||
| 100 | 0.067 | 0.00494 | 0.00081 | 0.00496 | 0.01547 | 0.00116 | 0.00674 | 0.00001 | 0.38 | 0.00001 | 0.00653 | 0.98 | ||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Siciliano, A.; Curcio, G.M.; Limonti, C. Experimental Analysis and Modeling of Nitrate Removal through Zero-Valent Magnesium Particles. Water 2019, 11, 1276. https://doi.org/10.3390/w11061276
Siciliano A, Curcio GM, Limonti C. Experimental Analysis and Modeling of Nitrate Removal through Zero-Valent Magnesium Particles. Water. 2019; 11(6):1276. https://doi.org/10.3390/w11061276
Chicago/Turabian StyleSiciliano, Alessio, Giulia Maria Curcio, and Carlo Limonti. 2019. "Experimental Analysis and Modeling of Nitrate Removal through Zero-Valent Magnesium Particles" Water 11, no. 6: 1276. https://doi.org/10.3390/w11061276
APA StyleSiciliano, A., Curcio, G. M., & Limonti, C. (2019). Experimental Analysis and Modeling of Nitrate Removal through Zero-Valent Magnesium Particles. Water, 11(6), 1276. https://doi.org/10.3390/w11061276