Identification of Sulfate Sources and Biogeochemical Processes in an Aquifer Affected by Peatland: Insights from Monitoring the Isotopic Composition of Groundwater Sulfate in Kampinos National Park, Poland




Abstract
:
1. Introduction
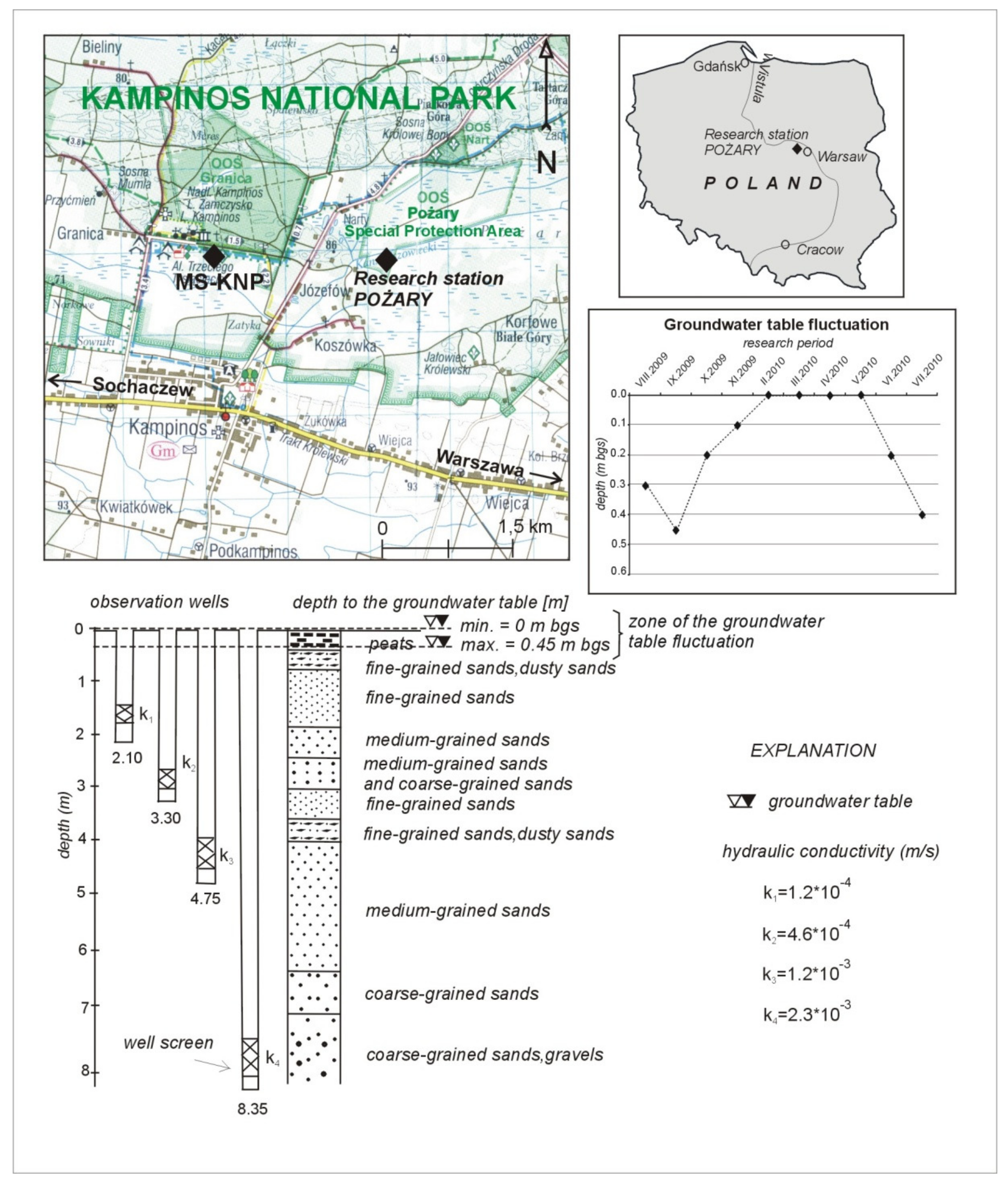
2. Study Area and Hydrogeological Settings
3. Materials and Methods
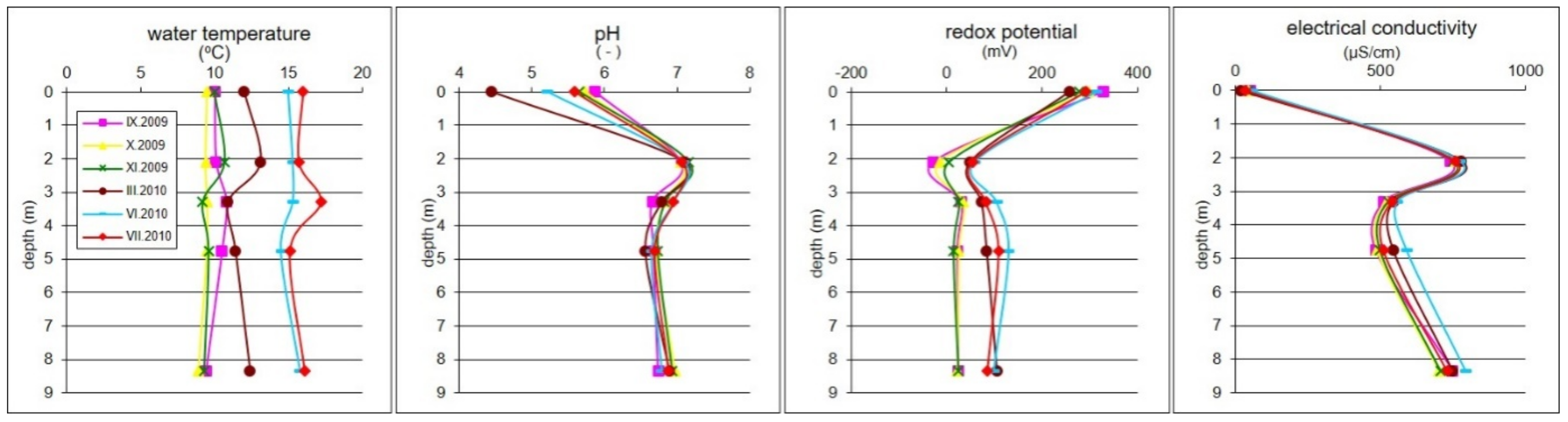
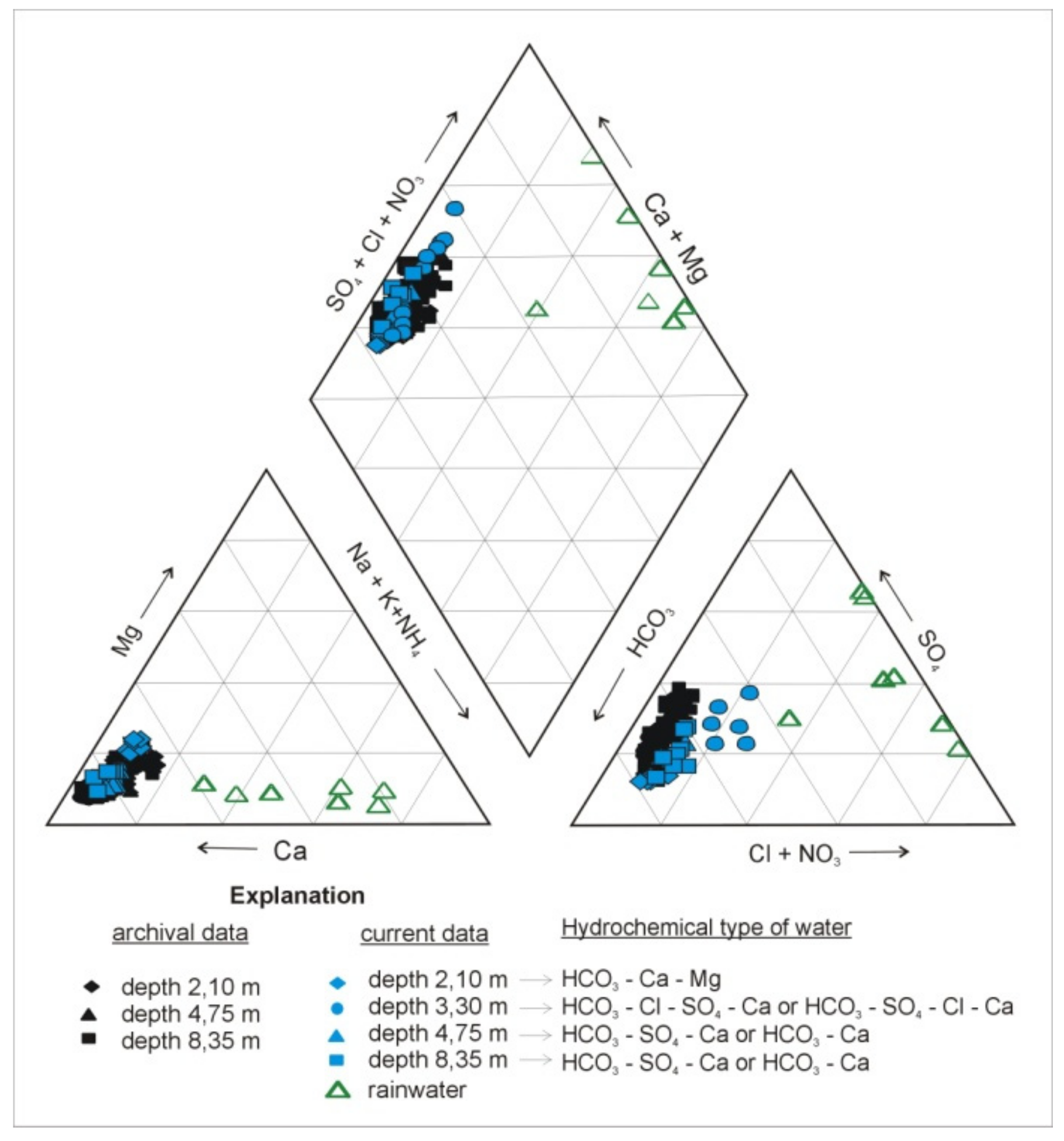
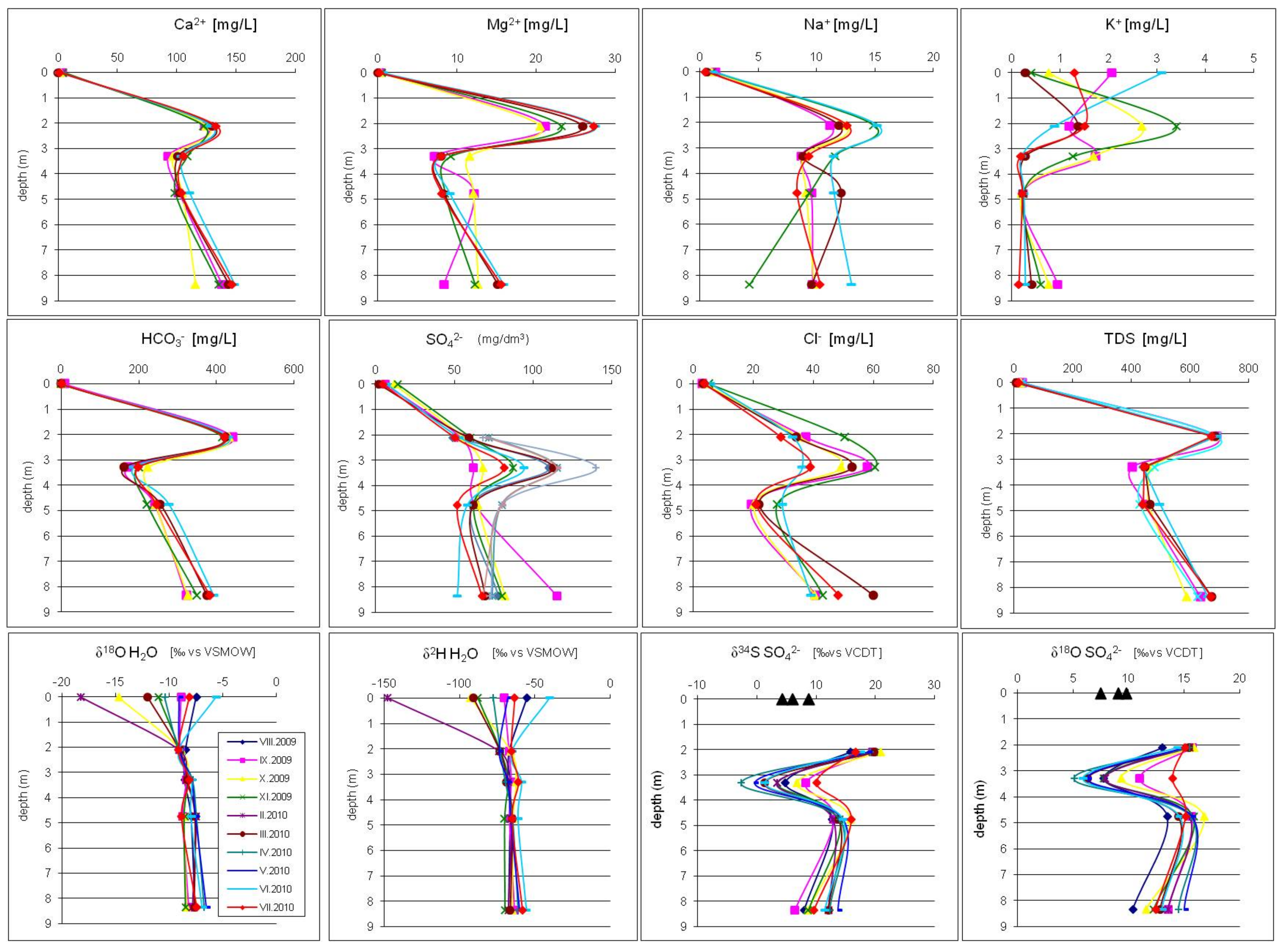
4. Results
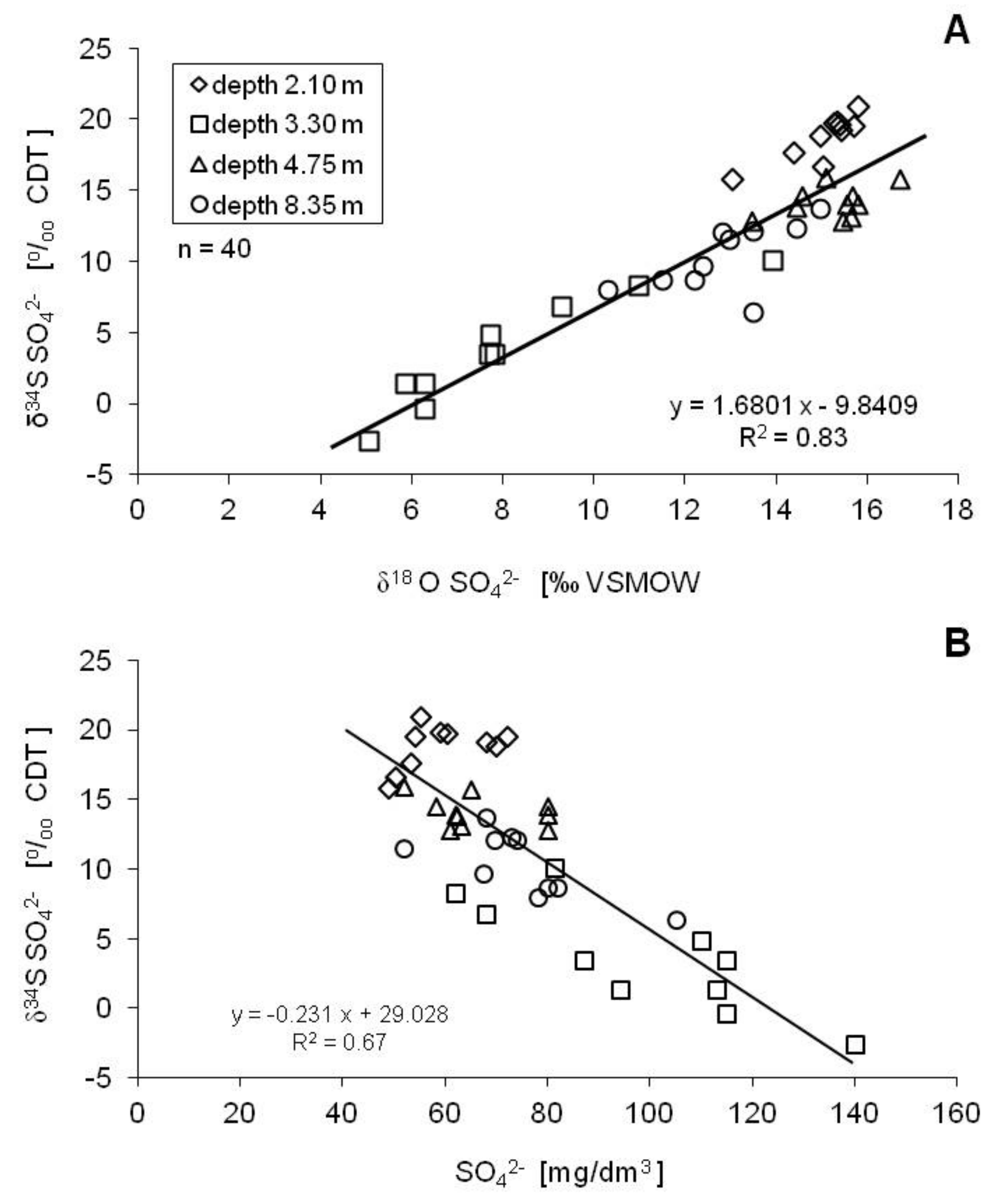
5. Discussion
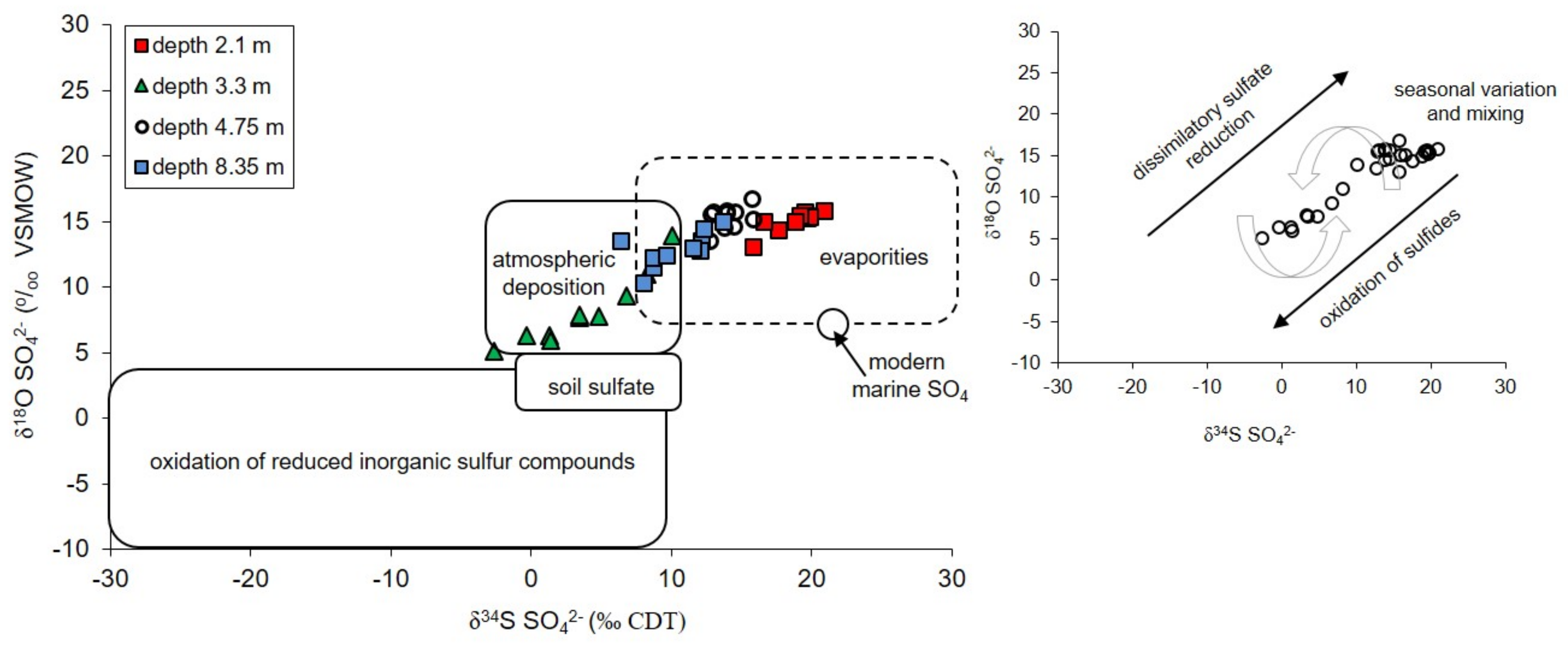
5.1. Sulfate Sources in Groundwater Studied
5.1.1. Atmospheric Sulfates
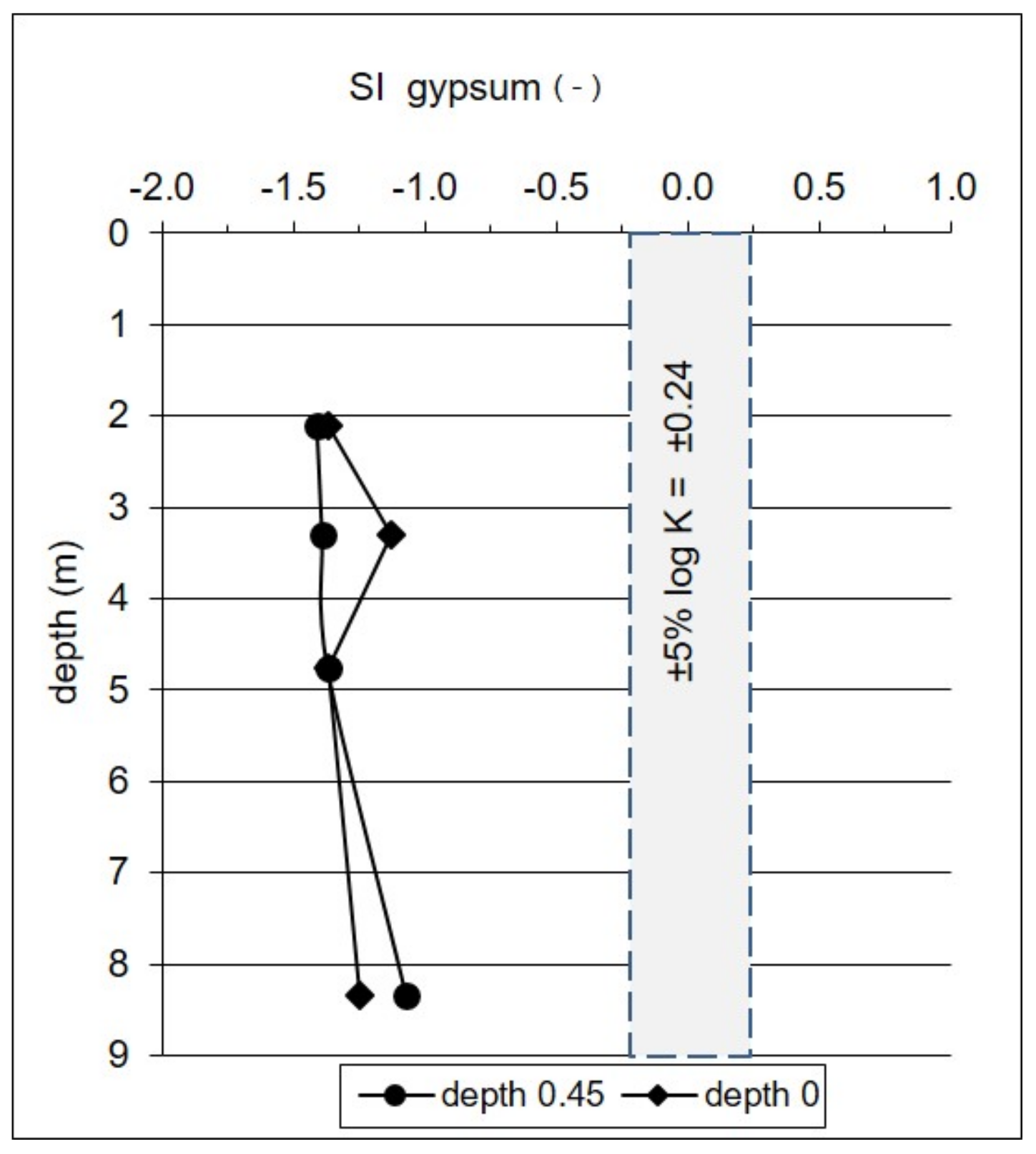
5.1.2. Dissolution of Gypsum
5.1.3. Mineralization of Carbon-Bonded Sulfur (C-S) Compounds
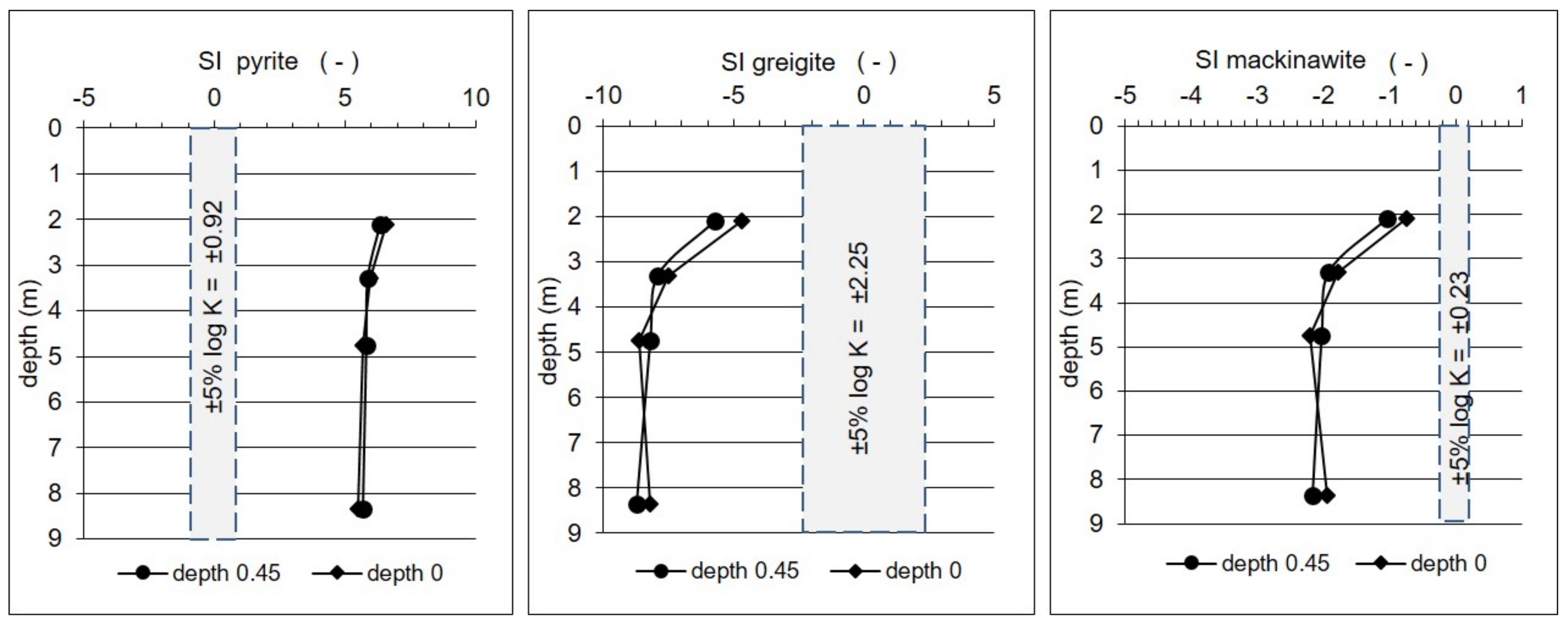
5.2. Precipitation/Oxidation of Reduced Inorganic Sulfur (RIS)
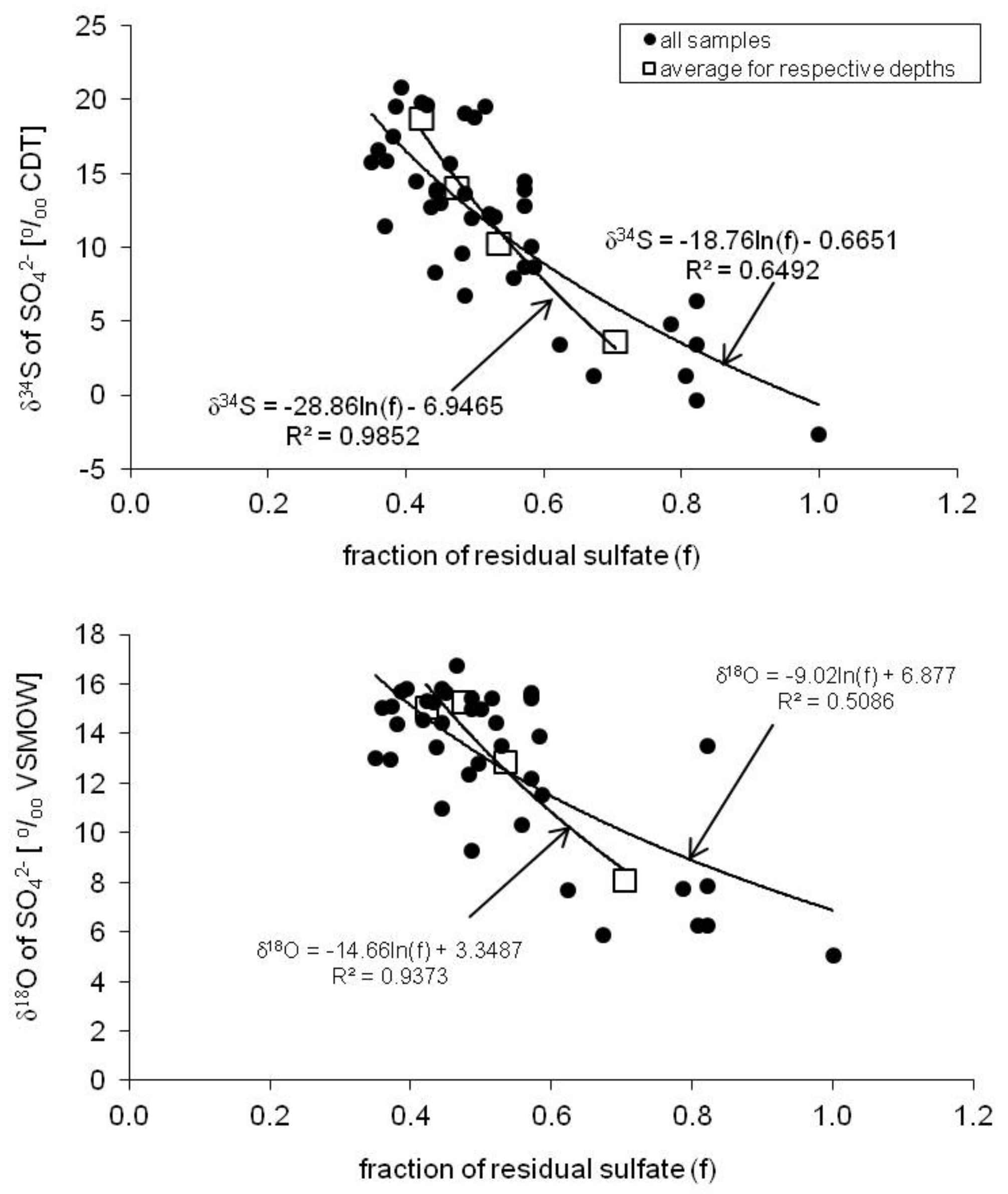
5.3. Bacterial Dissimilatory Sulfate Reduction
6. Conclusions
- (a)
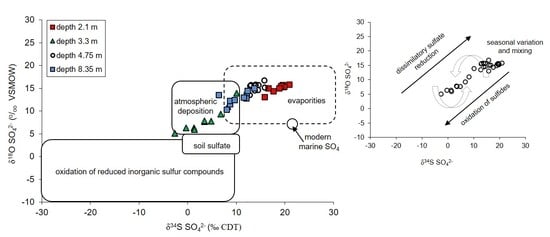
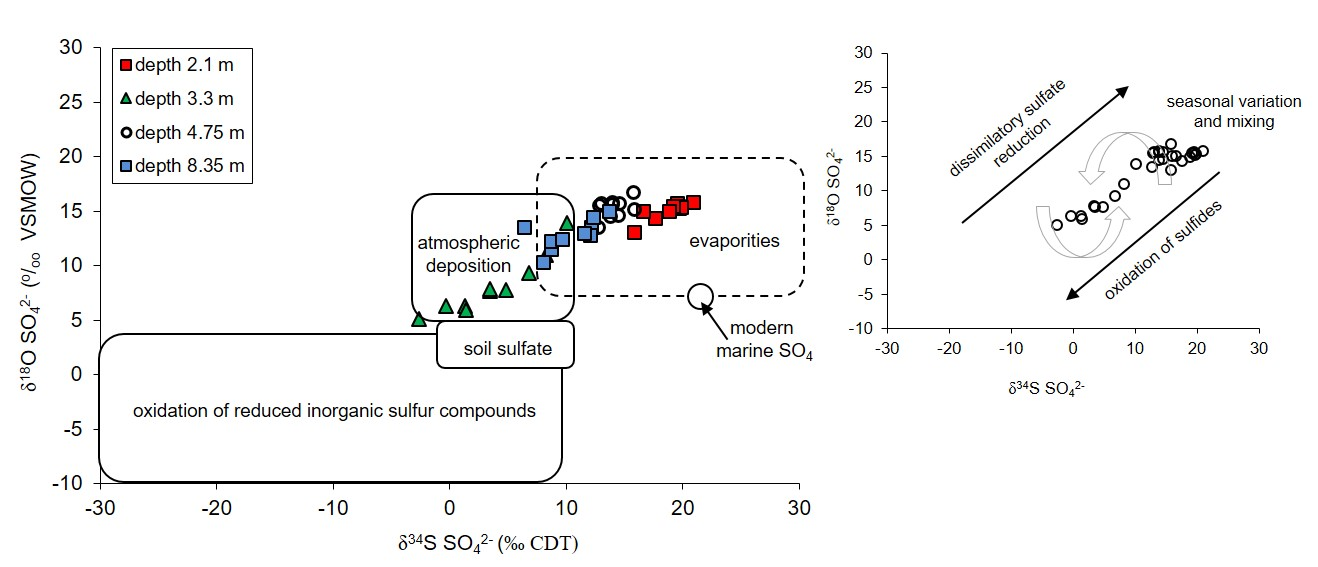
- The atmospheric deposition—sulfates which are introduce to the aquifer with the recharge water such as rain and snowmelt. Their initial values of δ34SSO4 in the area of the Pożary research station documented for rain and snow varied from +4.3 to +8.8‰ with concentrations ranging from 1.7 to 14.2 mg/dm3. Such isotopic composition is typical for sulfate of anthropogenic origin in atmospheric precipitation in industrialized regions of the northern hemisphere with δ34S values ranging from −3 to +9‰. The atmospheric sulfates reach the aquifer by direct infiltration of recharging water though the peat layer and vadose zone and by lateral groundwater inflow from an adjacent areas, where the aquifer is not cover by peat.
- (b)
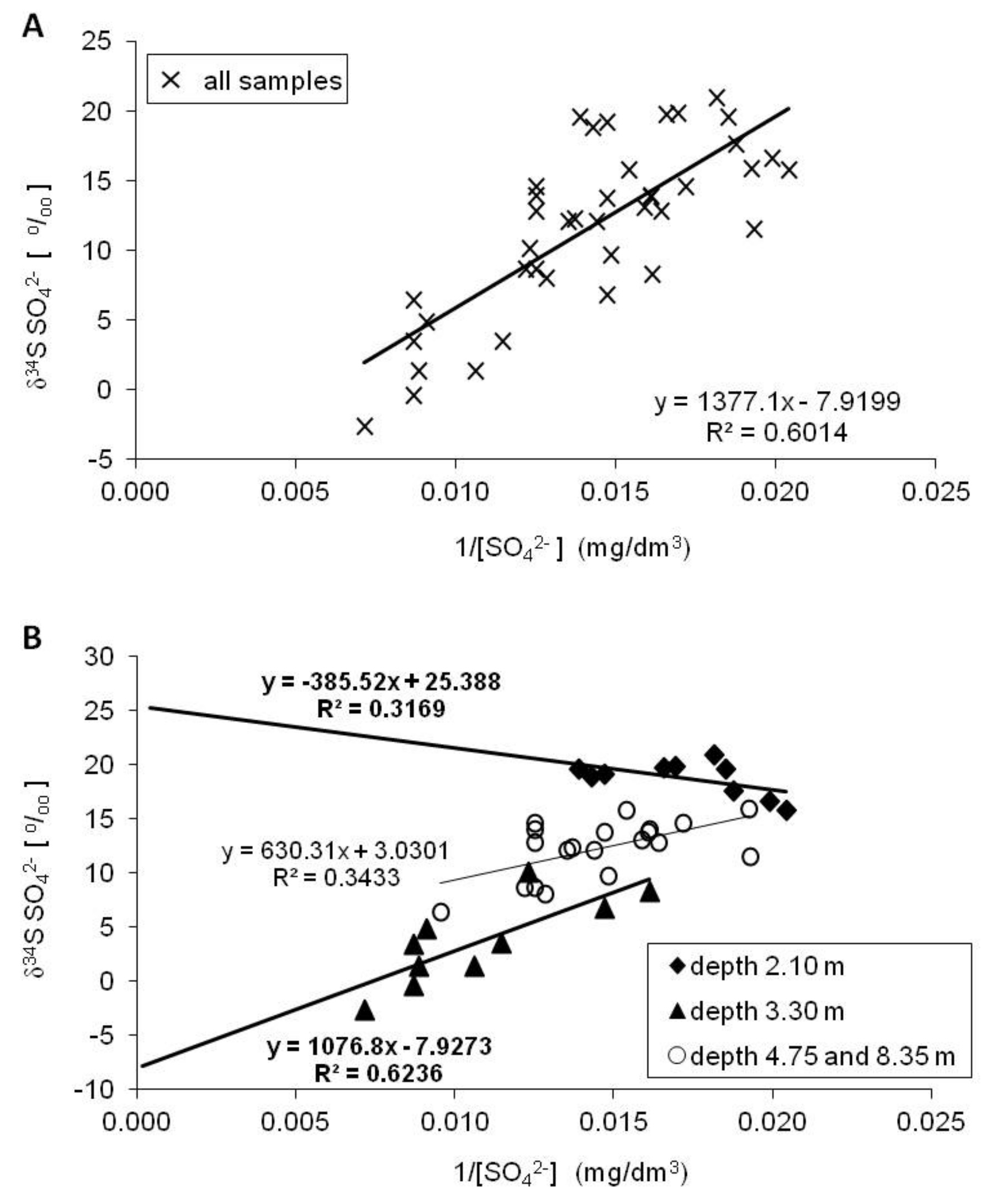
- The evaporitic sulfate minerals (lithogenic source)—sulfates enriched in heavy 18O and 34S isotopes with δ34SSO4 ranging from +15.8 to +20.9‰ and δ18OSO4 from +13.0 to +15.8‰. Such isotopic composition is typical for sulfates originating most likely from the dissolution of evaporitic gypsum which may be formed in the peat layer or in the aquifer’s vadose zone during seasons with low groundwater level. Sulfates of such isotopic composition were observed exclusively in the most upper part of the aquifer profile (monitoring depth of 2.1 m) which is the closest to the overlying peat bed. It is very likely that the source of sulfates is connected with sulfate flax from the peat layer. Calculated initial value of the dominant source of dissolved sulfate at the depth of 2.1 m yielded a δ34SI value of +25.4‰, which also strongly suggests the influence of sulfates originating from bacterial sulfate reduction.
- (c)
- The reduced inorganic sulfur compound (RIS; lithogenic source)—sulfates depleted in heavy 18O and 34S isotopes with δ34SSO4 ranging from −2.6 to +10.1‰ and δ18OSO4—from +5.10 to +13.9‰. Sulfates of such isotopic composition were observed in the aquifer exclusively in monitoring depth of 3.3 m. Depletion in heavy isotopes is connected with increasing SO42− concentration: Usually increased from about 1.5–2 times (depending on the season) in relation to the uppermost part of the aquifer profile. The source of these sulfates is connected with oxidation/re-oxidation of reduced inorganic sulfur compounds in the aquifer i.e., first of all with the dissolution/precipitation of pyrite, the dissolution of greigite and mackinawite. Calculated initial value of the dominant source of dissolved sulfate at the depth of 3.3 m yielded a δ34SI value of −7.9‰ which corroborates that the oxidation of RIS is responsible for the increase in SO42− concentration in this part of the aquifer profile.
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Johnston, S.G.; Burton, E.D.; Aaso, T.; Tuckerman, G. Sulfur, iron and carbon cycling following hydrological restoration of acidic freshwater wetlands. Chem. Geol. 2014, 371, 9–26. [Google Scholar] [CrossRef]
- Langmuir, D. Aqueous Environmental Geochemistry; Prentice Hall: Upper Saddle River, NJ, USA, 1997; 600p. [Google Scholar]
- Appelo, C.A.J.; Postma, D. Geochemistry, Groundwater and Pollution; A.A. Balkema: Rotterdam, The Netherlands, 1996; 536p. [Google Scholar]
- Chapman, S.J. Sulfur forms in open and afforested areas of two Scottish peatlands. Water Air Soil Pollut. 2002, 128, 23–39. [Google Scholar] [CrossRef]
- Novak, M.; Vile, M.A.; Bottrell, S.H.; Stepanova, M.; Jackova, I.; Buzek, F.; Prechova, E.; Newton, R.J. Isotope systematic of sulfate-oxygen and sulfate-sulfur in six European peatlands. Biogeochemistry 2005, 76, 187–213. [Google Scholar] [CrossRef]
- López-Buendia, A.M.; Whateley, M.K.G.; Bastida, J.; Urquiola, M.M. Origins of mineral matter in peat marsh and peat bog deposits, Spain. Int. J. Coal Geol. 2007, 71, 246–262. [Google Scholar] [CrossRef]
- Alewell, C.; Paul, S.; Lischeid, G.; Storck, F.R. Co-regulation of redox processes in freshwater wetlansd as a function of organic matter availability? Sci. Total Environ. 2008, 404, 335–342. [Google Scholar] [CrossRef] [PubMed]
- Schwientek, M.; Einsiedl, F.; Stichler, W.; Stögbauer, A.; Strauss, H.; Maloszewski, P. Evidence for denitrification regulated by pyrite oxidation in heterogeneous porous groundwater system. Chem. Geol. 2008, 255, 60–67. [Google Scholar] [CrossRef]
- Wu, S.; Kuschk, P.; Wiessner, A.; Müller, J.; Sasd, R.A.B.; Dong, R. Sulphur transformations in constructed wetlands for wastewater treatment: A review. Ecol. Eng. 2013, 52, 278–289. [Google Scholar] [CrossRef]
- Wiessner, A.; Kappelmeyer, U.; Kuschk, P.; Kastner, M. Sulphate reduction and the removal of carbon and ammonia in a laboratory-scale constructed wetland. Water Res. 2005, 39, 4643–4650. [Google Scholar] [CrossRef]
- Schiff, S.L.; Spoelstra, J.; Semkin, R.G.; Jeffries, D.S. Drought induced pulses of from a Canadian shield wetland: use of δ34S and δ18O in to determine sources of sulfur. Appl. Geochem. 2005, 20, 691–700. [Google Scholar] [CrossRef]
- Clark, I.; Fritz, P. Environmental Isotopes in Hydrogeology; CRC Press LLC: Boca Raton, FL, USA, 1997; 328p. [Google Scholar]
- Mandernack, K.W.; Lynch, L.; Krouse, H.R.; Morgan, M.D. Sulfur cycling in wetland peat of the New Jersey Pinelands and its effect on stream water chemistry. Gechimica Cosmochiica Acta 2000, 64, 3949–3964. [Google Scholar] [CrossRef]
- Bottrell, S.H.; Hartfield, D.; Bartlett, R.; Spence, M.J.; Bartle, K.D.; Mortimer, R.J.G. Concentrations, sulfur isotopic compositions and origin of organosulfur compounds in pore waters of a highly polluted raised peatland. Org. Geochem. 2010, 41, 55–62. [Google Scholar] [CrossRef]
- Porowska, D.; Leśniak, P.M. Identification of processes controlling groundwater chemistry below peatland—Pożary, Kampinos Park Narodowy. Geol. Surv. 2008, 56, 982–990. (In Polish) [Google Scholar]
- Słowański, W.; Piechulska-Słowańska, B.; Gogołek, W. Geological Map of Poland in the Sacle 1:200 000. Warszawa-Zachód Sheet; PIG: Warszawa, Poland, 1994. [Google Scholar]
- Fic, M.; Wierzbicki, A. Organizacja sieci monitoringu wód podziemnych na terenie rezerwatu ”Pożary” w Kampinoskim Parku Narodowym. Geol. Surv. 1994, 42, 1004–1008. (In Polish) [Google Scholar]
- Ingram, H.A.P. Hydrology. In Mires, Swamps, Bog, Fen and Moore; Gore, A.J.P., Ed.; General Studies; Elsevier: Amsterdam, The Netherlands, 1983; Volume 4A, pp. 67–158. [Google Scholar]
- Witczak, S.; Kania, J.; Kmiecik, E. Katalog Wybranych Fizycznych i Chemicznych Wskaźników Zanieczyszczeń Wód Podziemnych i Metod ich Oznaczania; IOŚ: Warszawa, Poland, 2013; 648p. [Google Scholar]
- Nielsen, D.M.; Nielsen, G.L. The Essential Handbook of Ground-Water Sampling; CRC Press: Boca Raton, FL, USA, 2007; 309p. [Google Scholar]
- Weight, W.D.; Sonderegger, J.L. Manual of Applied Field Hydrogeology; McGraw-Hill: New York, NY, USA, 2000; 608p. [Google Scholar]
- Hermanowicz, W.; Dojlido, J.; Dożańska, W.; Koziorowski, B.; Zerbe, J. Fizyczno-Chemiczne Badanie Wody i ścieków; Wydawnictwo Arkady: Warszawa, Poland, 1999; 558p. (In Polish) [Google Scholar]
- Epstein, S.; Mayeda, T. Variation of 18O content of waters from natural sources. Geochim. Cosmochim. Acta 1953, 4, 213–224. [Google Scholar] [CrossRef]
- Roether, W. Water-CO2 set-up for routine 18Oxygene assay of natural waters. Int. J. Appl. Radiat. Isot. 1970, 21, 379–387. [Google Scholar] [CrossRef]
- Coleman, M.L.; Shepherd, T.J.; Durham, J.J.; Rouse, J.E.; Moore, G.R. Reduction of water with zinc for hydrogen isotope analysis. Anal. Chem. 1982, 54, 993–995. [Google Scholar] [CrossRef]
- Kendall, C.; Coplen, T.B. Multisample conversion of water to hydrogen by zinc for stable isotope determination. Anal. Chem. 1985, 57, 1437–1440. [Google Scholar] [CrossRef]
- Schimmelmann, A.; DeNiro, M.J. Preparation of organic and water hydrogen for stable isotope analysis: Effects due to reaction vessels and zinc reagent. Anal. Chem. 1993, 65, 789–792. [Google Scholar] [CrossRef]
- Porowski, A.; Kowski, P. Determination of δ2H and δ18O in saline oil-associated waters: the question of simple vacuum distillation of water samples prior to isotopic analyses. Isot. Environ. Health Stud. 2008, 44, 227–238. [Google Scholar] [CrossRef]
- Carmody, R.W.; Plummer, L.N.; Busenberg, E.; Coplen, T.B. Methods for Collection of Dissolved Sulfate and Sulfide and Analysis of Their Sulfur Isotopic Composition; U.S. Geological Survey Open-File Report; USGS Publication Warehouse: Reston, VA, USA, 1998; pp. 97–234.
- Mizutani, Y. An improvement in the carbon-reduction method for the oxygen isotopic analysis of sulphates. Geochem. J. 1971, 5, 69–77. [Google Scholar] [CrossRef]
- Halas, S.; Szaran, J.; Czarnacki, M.; Tanweer, A. Refinements in BaSO4 to CO2 preparation and δ18O-calibration of the sulphate standards NBS-127, IAEA SO-5 and IAEA SO-6. Geostand. Geoanal. Res. 2007, 31, 61–68. [Google Scholar] [CrossRef]
- Halas, S.; Szaran, J. Use of Cu2O-NaPO3 mixtures for SO2 extraction from BaSO4 for sulphur isotope analysis. Isot. Environ. Health Stud. 2004, 40, 229–231. [Google Scholar] [CrossRef] [PubMed]
- Porowska, D. Content of dissolved oxygen and carbon dioxide in groundwaters within the selected hydrogeocheical environment. Monogr. Kom. Gospod. Wodnej Pan 2004, 24, 137. [Google Scholar]
- Trudell, M.L.; Gillham, R.W.; Cherry, J.A. An in-situ study of the occurrence and rate of denitrification in a shallow unconfined sand aquifer. J. Hydrol. 1986, 86, 251–268. [Google Scholar] [CrossRef]
- Smith, R.L.; Duff, J.H. Denitrification in a sand and gravel aquifer. Appl. Environ. Microbiol. 1988, 54, 1071–1078. [Google Scholar] [PubMed]
- Böttcher, J.; Strebel, O.; Voerkelius, S.; Schmidt, H.L. Using isotope fractionation of nitrate nitrogen and nitrate oxygen for evaluation of denitrification in a sandy aquifer. J. Hydrol. 1990, 114, 413–424. [Google Scholar]
- Smith, R.L.; Howes, B.L.; Duff, L.H. Denitrification in nitrate-contaminated groundwater: occurrence in steep vertical geochemical gradients. Geochim. Cosmochim. Acta 1991, 55, 1815–1825. [Google Scholar] [CrossRef]
- Krogulec, E.; Jóźwiak, K. Mineral nitrogen in shallow waters of the Kampinos National Park. In Proceedings of the XII Sympozjum Współczesne Problemy Hydrogeologii, Kraków-Krynica, Poland, 21–23 June 2007; pp. 105–113. [Google Scholar]
- Krouse, H.R. Sulfur isotopes in our environment. In Handbook of Environmental Isotope Geochemistry. Volume 1: The Terrestrial Environment; Fritz, P., Fontes, J.C., Eds.; Elsevier: Amsterdam, The Netherlands, 1980; pp. 435–472. [Google Scholar]
- Krouse, H.R.; Mayer, B. Sulfur and oxygen isotopes in sulfate. In Environmental Tracers in Subsurface Hydrology; Cook, P., Herczeg, A.L., Eds.; Kluwer Academic Publishers: Dordrecht, The Netherlands, 2000; pp. 195–231. [Google Scholar]
- Knöller, K.; Fauville, A.; Mayer, B.; Strauch, G.; Friese, K.; Veizer, J. Sulfur cycling in an acid mining lake and its vicinity in Lusatia, Germany. Chem. Geol. 2004, 204, 303–323. [Google Scholar]
- Cook, P.G.; Herczeg, A.L. Environmental Tracers in Subsurface Hydrology; Kluwer: Boston, MA, USA, 2000; 529p. [Google Scholar]
- Porowski, A. Isotop hydrogeology. In Handbook of Engineering Hydrology. Volume 1. Fundamentals and Applications; Eslamian, S., Ed.; CRC Press: Boca Raton, FL, USA, 2014; pp. 345–378. [Google Scholar]
- Trembaczowski, A. Sulfur and oxygen isotopes behavior in sulfates of atmospheric groundwater system, observation and model. Nord. Hydrol. 1991, 22, 49–66. [Google Scholar] [CrossRef]
- Bottrell, S.H.; Coulson, J.; Spence, M.; Roworth, P.; Novak, M.; Forbes, L. Impacts of pollutant loading, climate variability and site management on the surface water quality of a lowland raised bog, Thorne Moors, E. England, UK. Appl. Geochem. 2004, 19, 413–422. [Google Scholar] [CrossRef]
- Skrzypek, G.; Akagi, T.; Drzewicki, W.; Jędrysek, M.O. Stable isotope studies of moss sulfur and sulfate from bog surface waters. Geochem. J. 2008, 42, 481–492. [Google Scholar] [CrossRef] [Green Version]
- Rozanski, K.; Araguas-Araguas, L.; Gonfiantini, R. Isotopic patterns in modern global precipitation. In Climate Change in Continental Isotopic Records. Geophysical Monograph; Stewart, P.K., Lohmann, K.C., McKenzie, J., Savin, S., Eds.; American Geophysical Union: Washington, DC, USA, 1993; pp. 1–36. [Google Scholar]
- Kroopnick, P.; Craig, H. Atmospheric oxygen: isotopic composition and solubility fractionation. Science 1972, 175, 54–55. [Google Scholar] [CrossRef] [PubMed]
- Everdingen, R.O.; Krouse, H.R. Isotope composition of sulphates generated by bacterial and abiological oxidation. Nature 1985, 315, 395–396. [Google Scholar] [CrossRef]
- Mayer, B.; Fritz, P.; Prietzel, J.; Krouse, H.R. The use of stable sulfur and oxygen isotope ratios for interpreting the mobility of sulfate in aerobic forest soils. Appl. Geochem. 1995, 10, 161–173. [Google Scholar] [CrossRef]
- Taylor, B.E.; Wheeler, M.C.; Nordstrom, D.K. Stable isotope geochemistry of acid mine drainage: experimental oxidation of pyrite. Geochim. Cosmochim. Acta 1984, 48, 2669–2678. [Google Scholar] [CrossRef]
- Porowska, D. Content of dissolved oxygen and carbon dioxide in rainwaters and groundwaters within the forest reserve of the Kampinos National Park and the urban area of Warsaw, Poland. Geol. Q. 2003, 47, 187–194. [Google Scholar]
- Rydelek, P. Origin and composition of mineral particles of selected peat deposits in Lubartowska Upland. Woda Śr. Obsz. Wiej. 2011, 11, 135–149. [Google Scholar]
- Mayer, B. Assessing sources and transformations of sulphate and nitrate in the hydrosphere using isotope techniques. In Isotopes in the Water Cycle; Past, Present and Future of a Developing Science; Aggarawal, P.K., Gat, J., Froehlich, K.F.O., Eds.; Springer: Dordrecht, The Netherlands, 2007; pp. 67–90. [Google Scholar]
- Zuber, A. Metody znacznikowe w badaniach hydrogeologicznych, poradnik metodyczny; Oficyna Wydawnicza Politechniki Wrocławskiej: Wrocław, Poland, 2007; 402p. (In Polish) [Google Scholar]
- Brown, K.A. Sulphur distribution and metabolism in waterlogged peat. Soil Biol. Biochem. 1985, 17, 39–45. [Google Scholar] [CrossRef]
- Rickard, D.; Luther, G.W., III. Chemistry of Iron Sulfides. Chem. Rev. 2007, 107, 514–562. [Google Scholar] [CrossRef]
- Rickard, D. Sulfidic Sediments and Sedimentary Rocks; Elsevier Science: Amsterdam, The Netherlands, 2012; Volume 65, 816p. [Google Scholar]
- Cohen, A.D.; Spackman, W. Methods in peat petrology and their application to reconstruction of paleoenvironments. Geol. Soc. Am. Bull. 1972, 83, 129–142. [Google Scholar] [CrossRef]
- Casagrande, D.J.; Seiffert, K.; Berschinski, C.; Sutton, N. Sulfur in peat forming systems of the Oekefenokee Swamp and Florida Everglades: origins of sulfur in coal. Geochim. Cosmochim. Acta 1977, 41, 161–167. [Google Scholar] [CrossRef]
- Philips, S.; Bustin, R.M. Sulfur in Changuinola peat deposit, Panama, as an indicator of the environments of deposition of peat and coal. J. Sediment. Res. 1996, 66, 184–196. [Google Scholar] [CrossRef]
- Berner, R.A.; Raiswell, R. Burial of organic carbon and pyrite sulfur ion sediments over Phanerozoic time: A new theory. Geochim. Cosmochim. Acta 1983, 47, 855–862. [Google Scholar] [CrossRef]
- Jóźwiak, K. Bogs iron ore in the marshy ground areas—e.g., Kampinoski National Park. Biul. Pig 2011, 445, 237–244. [Google Scholar]
- Wallace, A.; Wallace, G.A. Factors influencing oxidation of iron pyrite in soil. J. Plant Nutr. 1992, 15, 1579–1587. [Google Scholar] [CrossRef]
- Balci, N.; Shanks, W.C., III; Mayer, B.; Mandernack, K. Oxygen and sulfur isotope systematics of sulfate produced by bacterial and abiotic oxidation of pyrite. Geochim. Cosmochim. Acta 2007, 71, 3796–3811. [Google Scholar] [CrossRef]
- Canifeld, D.E. Isotope fractionation by natural populations of sulfate-reducing bacteria. Cosmochim. Acta 2001, 65, 1117–1124. [Google Scholar] [CrossRef]
- Samborska, K.; Halas, S. 34S and 18O in dissolved sulfate as tracers of hydrogeochemical evolution of the Triassic carbonate aquifer exposed to intense groundwater exploitation (Olkusz–Zawiercie region, southern Poland). Appl. Geochem. 2010, 25, 1397–1414. [Google Scholar] [CrossRef]
- Miao, Z.; Brusseau, M.L.; Carroll, K.C.; Carreón-Diazconti, C.; Johnson, B. Sulfate reduction in groundwater: characterization and applications for remediation. Environ. Geochem. Health 2012, 34, 539–550. [Google Scholar] [CrossRef]
- Kaplan, I.R.; Rittenberg, S.C. Microbiological fractionation of sulfur isotopes. J. Gen. Microbiol. 1964, 34, 195–212. [Google Scholar] [CrossRef]
- Rees, C.E. A steady-state model for sulfur isotope fractionation in bacterial reduction process. Geochim. Cosmochim. Acta 1973, 37, 1141–1162. [Google Scholar]
- Fritz, P.; Basharmal, G.M.; Drimmie, R.J.; Isen, J.; Qureshi, R.M. Oxygen isotope exchange between sulphate and water during bacterial sulfate reduction. Chem. Geol. 1989, 79, 99–105. [Google Scholar]
- Habicht, K.S.; Canfield, D.E. Sulfur isotope fractionation during bacterial sulfate reduction in organic-rich sediments. Geochim. Cosmochim. Acta 1997, 61, 5351–5361. [Google Scholar] [CrossRef]
- Mizutani, Y.; Rafter, T.A. Oxygen isotopic composition of sulphates, part 3: Oxygen isotopic fractionation in the bisulphate ion-water system. N. Z. J. Sci. 1969, 12, 54–59. [Google Scholar]
- Ahron, P.; Fu, B. Microbial sulfate reduction rates and sulfur and oxygen isotope ratios at oil and gas seeps in deepwater Gulf Mexico. Geochim. Cosmochim. Acta 2000, 64, 233–246. [Google Scholar] [CrossRef]
- Hoefs, J. Stable Isotope Geochemistry, 6th ed.; Springer: Berlin, Germany, 2009; 286p. [Google Scholar]
- Bottrell, S.H.; Smart, P.I.; Whitaker, F.; Raiswell, R. Geochemistry and isotope systematic of sulfur in the mixing zone of Bahamian blue holes. Appl. Geochem. 1991, 6, 97–103. [Google Scholar] [CrossRef]
- Bottrell, S.H.; Hayes, P.J.; Bannon, M.; Williams, G.M. Bacterial sulfate reduction and pyrite formation in a polluted sand aquifer. Geomicrobiol. J. 1995, 12, 75–90. [Google Scholar] [CrossRef]
- Bottrell, S.H.; Moncaster, S.M.; Tellam, J.H.; Lloyd, J.W.; Fisher, Q.J.; Newton, R.J. Control on bacterial sulfate reduction in a dual porosity aquifer system: The Lincoln Limestone aquifer, England. Chem. Geol. 2000, 169, 461–470. [Google Scholar] [CrossRef]
- Asmussen, G.; Strauch, G. Sulfate reduction in a lake and the groundwater of a former lignite mining area studied by stable sulfure and carbon isotopes. Water Air Soil Pollut. 1998, 108, 271–284. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Date | D | T | pH | Eh | SC | Ca2+ | Mg2+ | Na+ | K+ | HCO3− | SO42− | Cl− | NO3− | NH4+ | TDS |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| (m) | (°C) | (−) | (mV) | (uS/cm) | (mg/dm3) | ||||||||||
| IX.2009 | rain | 10 | 5.87 | 328 | 52 | 3.9 | 0.4 | 1.4 | 2.1 | 8.6 | 6.1 | 3.3 | 0.6 | 2.3 | 28.7 |
| 2.1 | 10.1 | 7.06 | −28 | 741 | 124.0 | 21.2 | 11.2 | 1.2 | 441.9 | 54.0 | 37.6 | <0.5 | na | 691.1 | |
| 3.3 | 10.8 | 6.66 | 31 | 512 | 93.0 | 7.2 | 8.7 | 1.7 | 172.0 | 62.0 | 58.3 | <0.5 | na | 402.9 | |
| 4.75 | 10.5 | 6.68 | 23 | 484 | 102.0 | 12.2 | 9.6 | 0.2 | 241.0 | 63.0 | 19.5 | <0.5 | na | 447.5 | |
| 8.35 | 9.4 | 6.74 | 25 | 750 | 138.0 | 8.4 | 9.7 | 1.0 | 322.0 | 105.0 | 40.8 | <0.5 | na | 624.9 | |
| X.2009 | rain | 9.5 | 5.74 | 298 | 44 | 3.0 | 0.4 | 1.1 | 0.8 | nd | 11.5 | 4.0 | 0.9 | 2.0 | 23.7 |
| 2.1 | 9.4 | 7.05 | −14 | 758 | 125.0 | 20.6 | 12.5 | 2.7 | 431.8 | 55.1 | 35.5 | <0.5 | na | 683.2 | |
| 3.3 | 9.5 | 6.83 | 35 | 521 | 96.7 | 11.6 | 8.9 | 1.7 | 222.8 | 68.0 | 49.5 | <0.5 | na | 459.2 | |
| 4.75 | 9.5 | 6.71 | 26 | 489 | 104.6 | 12.1 | 9.1 | 0.2 | 244.0 | 65.0 | 20.3 | <0.5 | na | 455.3 | |
| 8.35 | 8.9 | 6.97 | 23 | 702 | 116.0 | 12.7 | 9.8 | 0.8 | 326.0 | 82.0 | 40.5 | <0.5 | na | 587.8 | |
| XI.2009 | rain | 10 | 5.65 | 278 | 45 | 4.1 | 0.5 | 1.0 | 0.4 | nd | 14.2 | 5.5 | 0.79 | 1.1 | 27.59 |
| 2.1 | 10.7 | 7.16 | 5 | 752 | 122.7 | 23.2 | 14.9 | 3.4 | 416.2 | 60.4 | 50.5 | <0.5 | na | 691.3 | |
| 3.3 | 9.2 | 6.83 | 26 | 528 | 109.0 | 9.2 | 11.6 | 1.3 | 199.6 | 87.2 | 60.5 | <0.5 | na | 478.4 | |
| 4.75 | 9.6 | 6.73 | 14 | 499 | 98.9 | 8.5 | 9.4 | 0.2 | 221.5 | 62.1 | 28.0 | <0.5 | na | 428.6 | |
| 8.35 | 9.3 | 6.93 | 24 | 708 | 135.5 | 12.3 | 4.3 | 0.6 | 349.7 | 80.0 | 43.2 | <0.5 | na | 625.6 | |
| III.2010 | rain | 12 | 4.44 | 257 | 18 | 0.5 | 0.1 | 0.6 | 0.3 | nd | 2.1 | 3.5 | 0.67 | 1.0 | 8.77 |
| 2.1 | 13.1 | 7.1 | 49 | 780 | 130.0 | 25.9 | 11.9 | 1.4 | 423.7 | 59.1 | 34.5 | <0.5 | na | 686.5 | |
| 3.3 | 10.9 | 6.78 | 73 | 545 | 101.0 | 8.0 | 8.8 | 0.3 | 161.5 | 113.2 | 52.9 | <0.5 | na | 445.7 | |
| 4.75 | 11.4 | 6.56 | 84 | 544 | 103.0 | 8.3 | 12.1 | 0.2 | 255.7 | 62.2 | 22.0 | <0.5 | na | 463.5 | |
| 8.35 | 12.4 | 6.89 | 105 | 749 | 144.0 | 15.2 | 9.6 | 0.4 | 374.9 | 69.5 | 60.1 | <0.5 | na | 673.7 | |
| VI.2010 | rain | 15 | 5.22 | 315 | 58 | 1.7 | 0.7 | 1.1 | 3.1 | 1.0 | 7.1 | 6.0 | 1.27 | 5.1 | 27.07 |
| 2.1 | 15.3 | 7.09 | 60 | 776 | 129.0 | 27.5 | 15.2 | 0.9 | 430.9 | 53.3 | 33.3 | <0.5 | na | 690.1 | |
| 3.3 | 15.3 | 6.95 | 106 | 560 | 104.0 | 8.1 | 11.5 | 0.2 | 186.0 | 94.2 | 36.4 | <0.5 | na | 440.4 | |
| 4.75 | 14.5 | 6.65 | 131 | 591 | 111.0 | 9.1 | 11.5 | 0.3 | 277.0 | 58.2 | 29.7 | <0.5 | na | 496.8 | |
| 8.35 | 15.8 | 6.79 | 100 | 795 | 149.0 | 15.9 | 13.0 | 0.3 | 392.0 | 51.8 | 39.3 | <0.5 | na | 661.3 | |
| VII.2010 | rain | 16 | 5.59 | 292 | 35 | 1.3 | 0.2 | 0.5 | 1.3 | 1.1 | 4.7 | 4.0 | 0.60 | 1.8 | 15.5 |
| 2.1 | 15.7 | 7.06 | 55 | 759 | 133.0 | 27.3 | 12.6 | 1.5 | 420.6 | 50.3 | 29.3 | <0.5 | na | 674.6 | |
| 3.3 | 17.2 | 6.95 | 83 | 542 | 106.0 | 8.0 | 9.3 | 0.2 | 198.6 | 81.4 | 39.0 | <0.5 | na | 442.5 | |
| 4.75 | 15.1 | 6.69 | 109 | 511 | 104.0 | 8.1 | 8.3 | 0.2 | 245.5 | 52.0 | 21.0 | <0.5 | na | 439.1 | |
| 8.35 | 16.1 | 6.88 | 86 | 731 | 147.0 | 15.6 | 10.3 | 0.2 | 383.0 | 67.4 | 48.4 | <0.5 | na | 671.9 | |
| Date | D | δ18O H2O | δ2H H2O | δ18O SO4 | δ34S SO4 | SO42− | Date | D | δ18O H2O | δ2H H2O | δ18O SO4 | δ34S SO4 | SO42− |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| (m) | (‰) vs. VSMOW | (‰) vs. VCDT | (mg/dm3) | (m) | (‰) vs. VSMOW | (‰) vs. VCDT | (mg/dm3) | ||||||
| VIII.2009 | rain | −7.48 | −55.4 | na | na | 2.3 | II.2010 | snow | −18.29 | −148.0 | 13.2 | 8.8 | 3.65 |
| 2.1 | −8.48 | −67.0 | 13.0 | 15.8 | 49.0 | 2.1 | −9.11 | −73.7 | 15.4 | 19.6 | 72.0 | ||
| 3.3 | −8.57 | −66.5 | 7.7 | 4.8 | 110.0 | 3.3 | −8.45 | −68.1 | 7.8 | 3.4 | 115.0 | ||
| 4.75 | −7.96 | −66.4 | 13.5 | 12.8 | 61.0 | 4.75 | −7.56 | −65.4 | 15.5 | 12.8 | 80.0 | ||
| 8.35 | −7.58 | −67.4 | 10.3 | 7.9 | 78.0 | 8.35 | −7.85 | −67.7 | 13.5 | 12.1 | 74.0 | ||
| IX.2009 | rain | −8.92 | −70.4 | Na | na | 6.10 | III.2010 | rain | −12.04 | −91.0 | na | na | 2.1 |
| 2.1 | −9 | −67.2 | 15.7 | 19.5 | 54.0 | 2.1 | −9.08 | −73.3 | 15.4 | 19.8 | 59.1 | ||
| 3.3 | −8.45 | −65.7 | 11.0 | 8.3 | 62.0 | 3.3 | −8.33 | −68.8 | 6.3 | 1.4 | 113.2 | ||
| 4.75 | −8.75 | −66.8 | 15.7 | 13.1 | 63.0 | 4.75 | −7.64 | −65.0 | 14.5 | 13.8 | 62.2 | ||
| 8.35 | −8.2 | −63.4 | 13.5 | 6.4 | 105.0 | 8.35 | −7.7 | −66.4 | 12.8 | 12.1 | 69.5 | ||
| X.2009 | rain | −14.75 | −93.0 | Na | na | 11.5 | IV.2010 | rain | −10.42 | −77.8 | na | na | na |
| 2.1 | −8.93 | −66.1 | 15.8 | 20.9 | 55.1 | 2.1 | −9.12 | −73.0 | 15.5 | 19.2 | 68.0 | ||
| 3.3 | −8.26 | −62.6 | 9.3 | 6.8 | 68.0 | 3.3 | −7.84 | −69.7 | 5.1 | −2.6 | 140.0 | ||
| 4.75 | −8.64 | −64.6 | 16.8 | 15.7 | 65.0 | 4.75 | −7.47 | −64.9 | 15.6 | 13.9 | 80.0 | ||
| 8.35 | −8.4 | −65.1 | 11.5 | 8.7 | 82.0 | 8.35 | −6.76 | −61.1 | 14.5 | 12.3 | 73.0 | ||
| XI.2009 | rain | −11.03 | −87.9 | na | na | 14.2 | V.2010 | rain | −9.1 | −68.1 | na | na | 2.1 |
| 2.1 | −9 | −73.0 | 15.3 | 19.7 | 60.4 | 2.1 | −9.03 | −73.9 | 15.0 | 18.8 | 70.0 | ||
| 3.3 | −8.29 | −68.1 | 7.7 | 3.5 | 87.2 | 3.3 | −7.95 | −67.8 | 6.3 | −0.4 | 115.0 | ||
| 4.75 | −8.6 | −70.3 | 15.8 | 13.9 | 62.1 | 4.75 | −7.58 | −66.1 | 15.7 | 14.6 | 80.0 | ||
| 8.35 | −8.47 | −70.1 | 12.2 | 8.7 | 80.0 | 8.35 | −6.58 | −60.1 | 15.0 | 13.7 | 68.0 | ||
| XII.2009 | snow | −15.6 | −128.0 | 9.1 | 4.3 | 2.3 | VI.2010 | rain | −5.64 | −40.2 | 9.8 | 6.0 | 7.1 |
| 2.1 | na | na | na | na | na | 2.1 | −9.11 | −65.3 | 14.4 | 17.6 | 53.3 | ||
| 3.3 | na | na | na | na | na | 3.3 | −7.93 | −58.9 | 5.9 | 1.4 | 94.2 | ||
| 4.75 | na | na | na | na | na | 4.75 | −7.98 | −61.2 | 14.6 | 14.5 | 58.2 | ||
| 8.35 | na | na | na | na | na | 8.35 | −6.94 | −55.6 | 12.99 | 11.5 | 51.8 | ||
| I.2010 | snow | −15.67 | −124.9 | na | na | 1.7 | VII.2010 | rain | −8.17 | −63.5 | na | na | 4.7 |
| 2.1 | na | na | na | na | na | 2.1 | −9.17 | −65.7 | 15.1 | 16.6 | 50.3 | ||
| 3.3 | na | na | na | na | na | 3.3 | −8.16 | −61.1 | 13.9 | 10.1 | 81.4 | ||
| 4.75 | na | na | na | na | na | 4.75 | −8.88 | −65.3 | 15.1 | 15.9 | 52 | ||
| 8.35 | na | na | na | na | na | 8.35 | −7.47 | −58.5 | 12.4 | 9.7 | 67.4 | ||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Porowski, A.; Porowska, D.; Halas, S. Identification of Sulfate Sources and Biogeochemical Processes in an Aquifer Affected by Peatland: Insights from Monitoring the Isotopic Composition of Groundwater Sulfate in Kampinos National Park, Poland. Water 2019, 11, 1388. https://doi.org/10.3390/w11071388
Porowski A, Porowska D, Halas S. Identification of Sulfate Sources and Biogeochemical Processes in an Aquifer Affected by Peatland: Insights from Monitoring the Isotopic Composition of Groundwater Sulfate in Kampinos National Park, Poland. Water. 2019; 11(7):1388. https://doi.org/10.3390/w11071388
Chicago/Turabian StylePorowski, Adam, Dorota Porowska, and Stanislaw Halas. 2019. "Identification of Sulfate Sources and Biogeochemical Processes in an Aquifer Affected by Peatland: Insights from Monitoring the Isotopic Composition of Groundwater Sulfate in Kampinos National Park, Poland" Water 11, no. 7: 1388. https://doi.org/10.3390/w11071388
APA StylePorowski, A., Porowska, D., & Halas, S. (2019). Identification of Sulfate Sources and Biogeochemical Processes in an Aquifer Affected by Peatland: Insights from Monitoring the Isotopic Composition of Groundwater Sulfate in Kampinos National Park, Poland. Water, 11(7), 1388. https://doi.org/10.3390/w11071388