
Hydroxyapatite Coatings on Calcite Powder for the Removal of Heavy Metals from Contaminated Water


 ,
, 
 and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Bare Calcite MPs and Chemical Reagents
2.2. Synthesis Route of HAP Coatings on Calcite MPs
2.3. Characterization of the HAP-Coated MPs Samples
2.4. Sorption Performance of the Synthesized HAP-Coated Material
2.4.1. General Procedure
2.4.2. Sorption Kinetics of Zn and Cu
2.4.3. Sorption Isotherms for Single-Metal Systems
2.5. Water Analysis
3. Results and Discussion
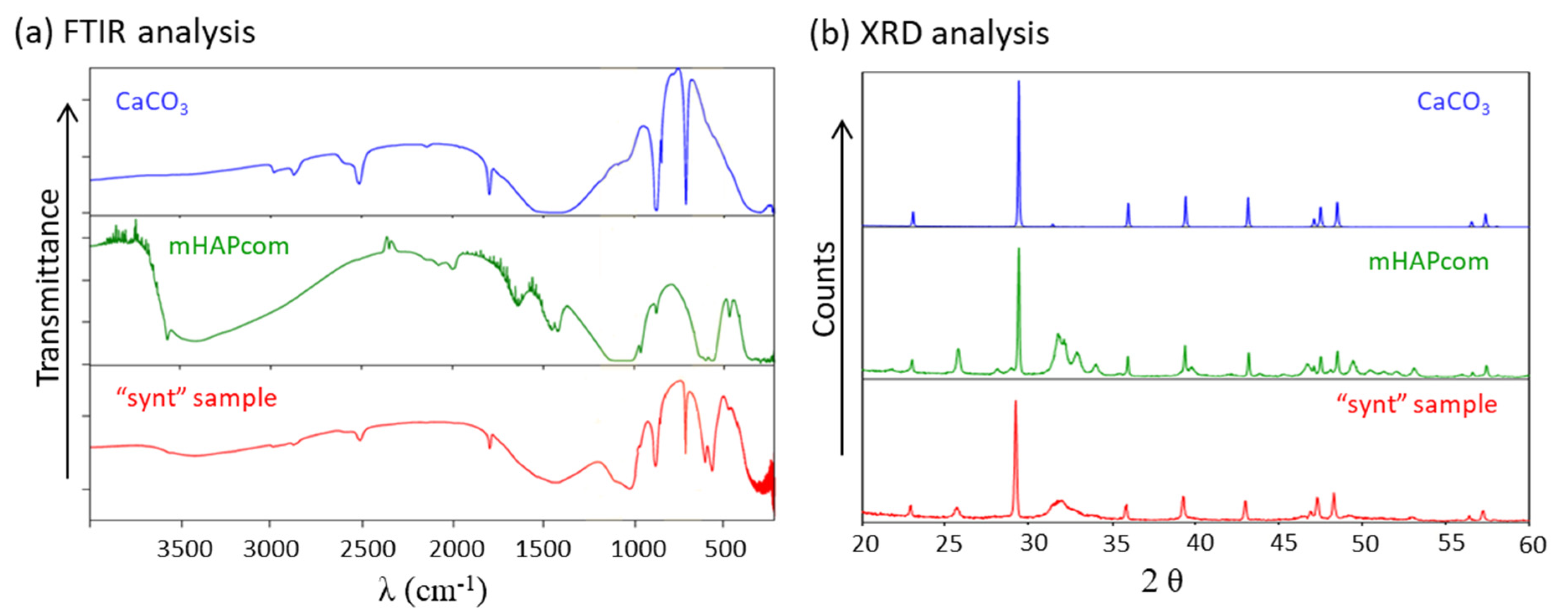
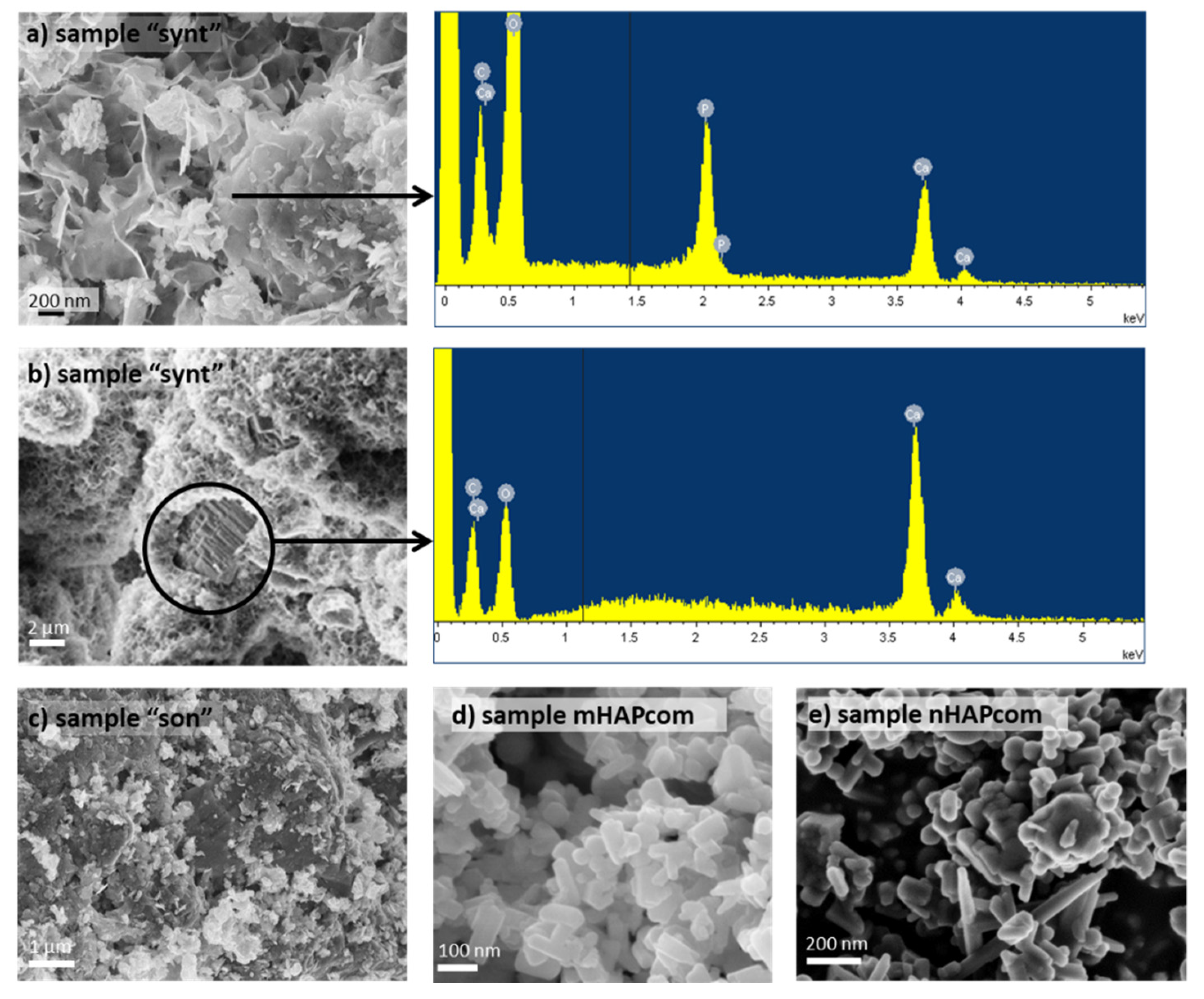
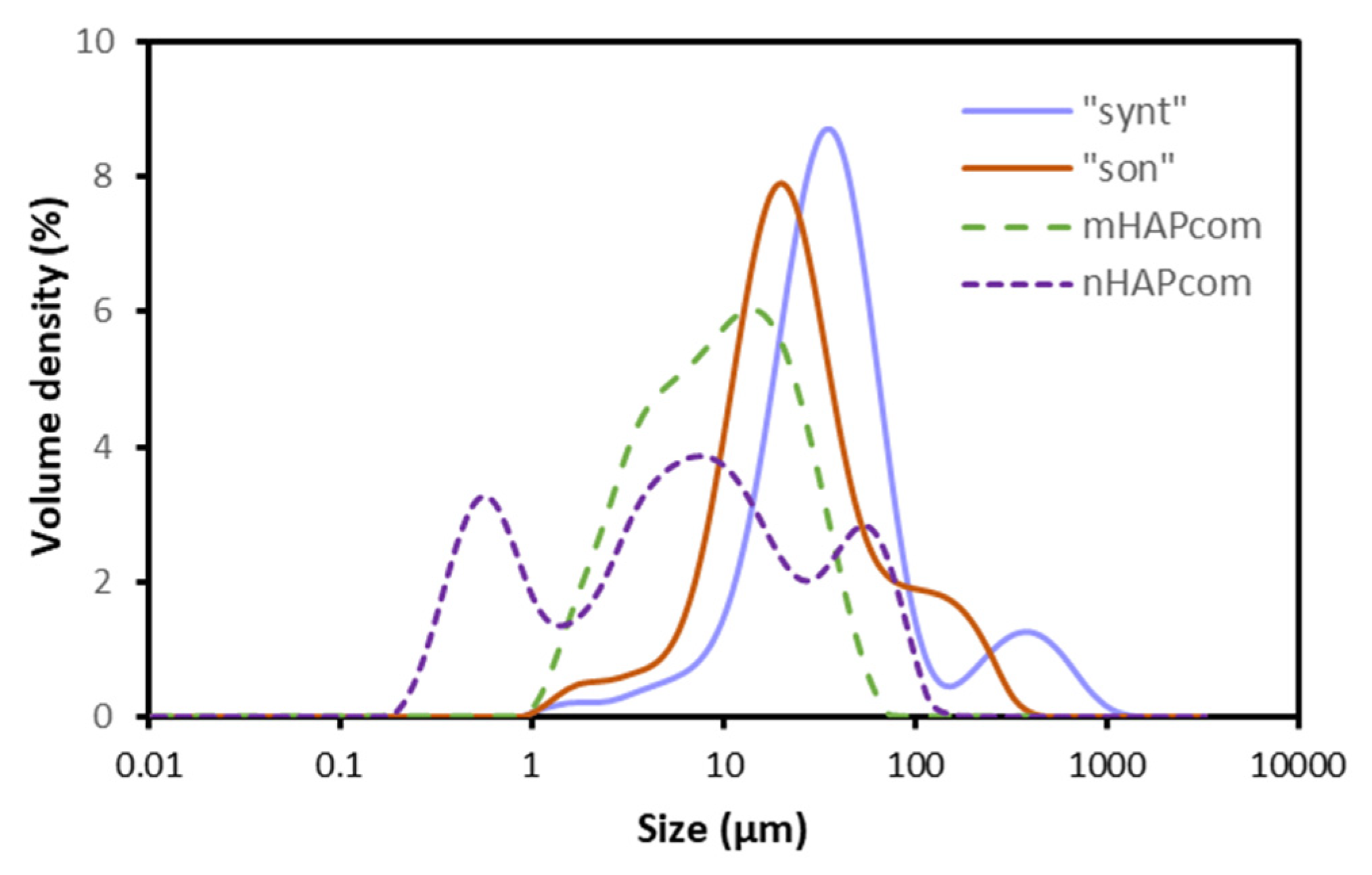
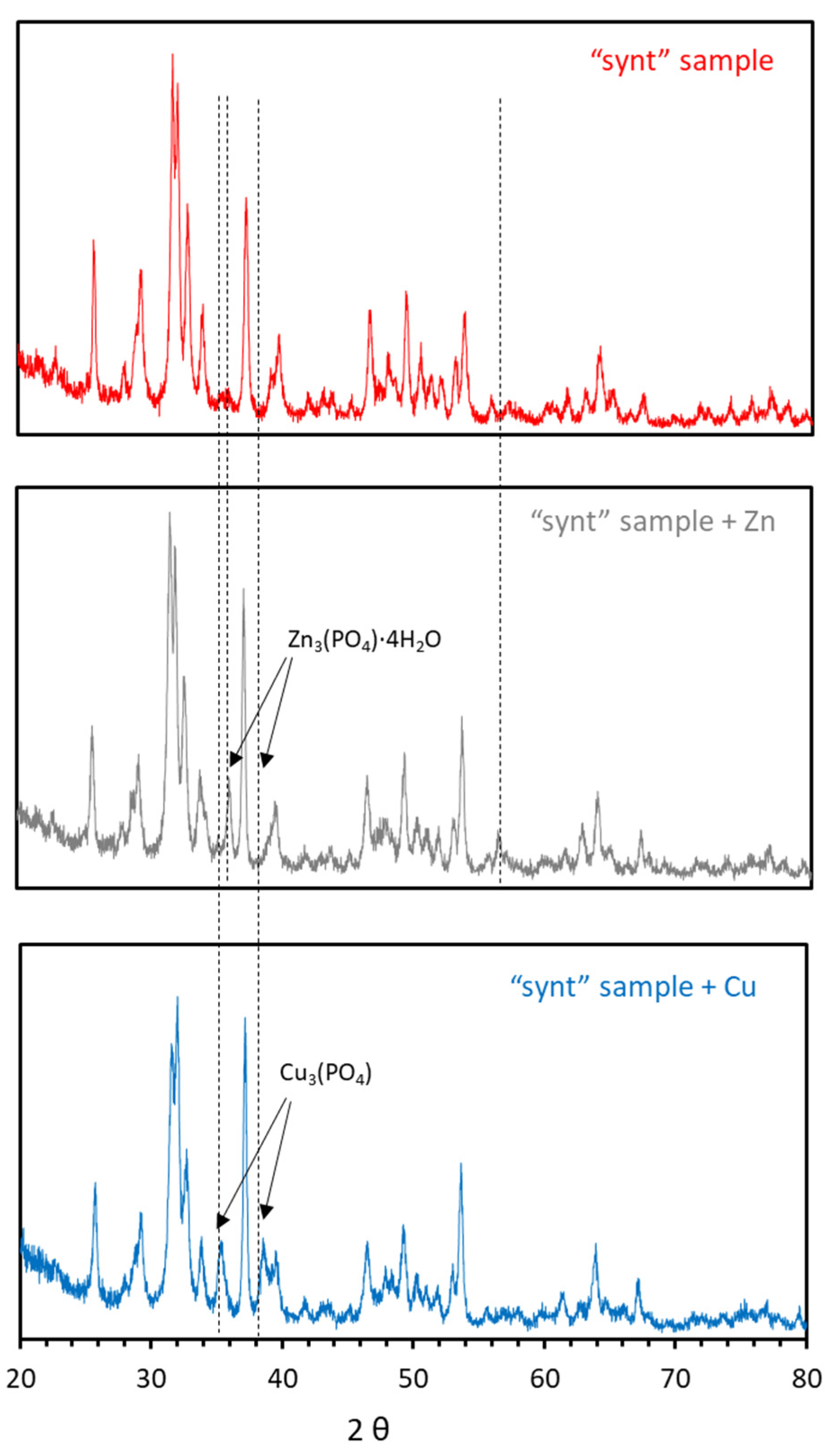
3.1. Characterization of the Synthesized Material
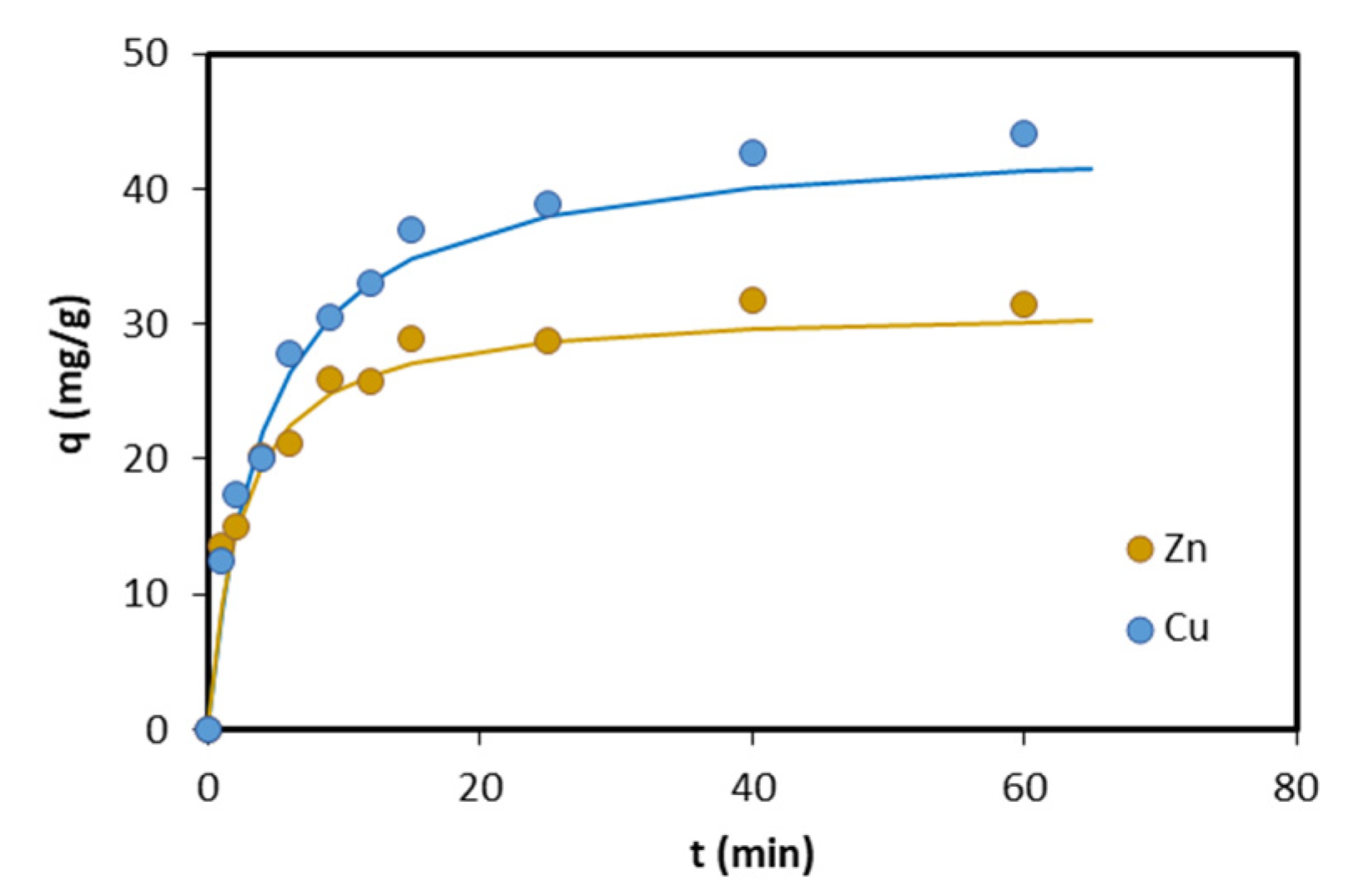
3.2. Adsorption Kinetics
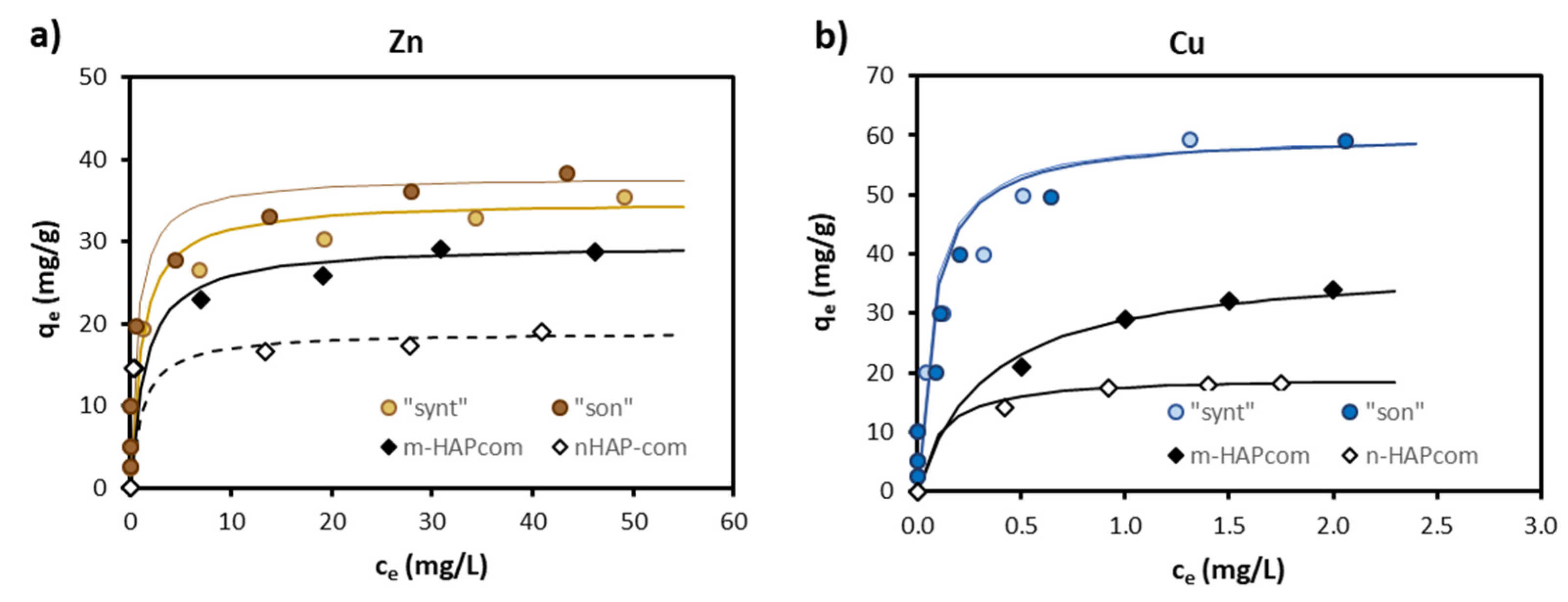
3.3. Adsorption Isotherms for Single-Metal Solutions
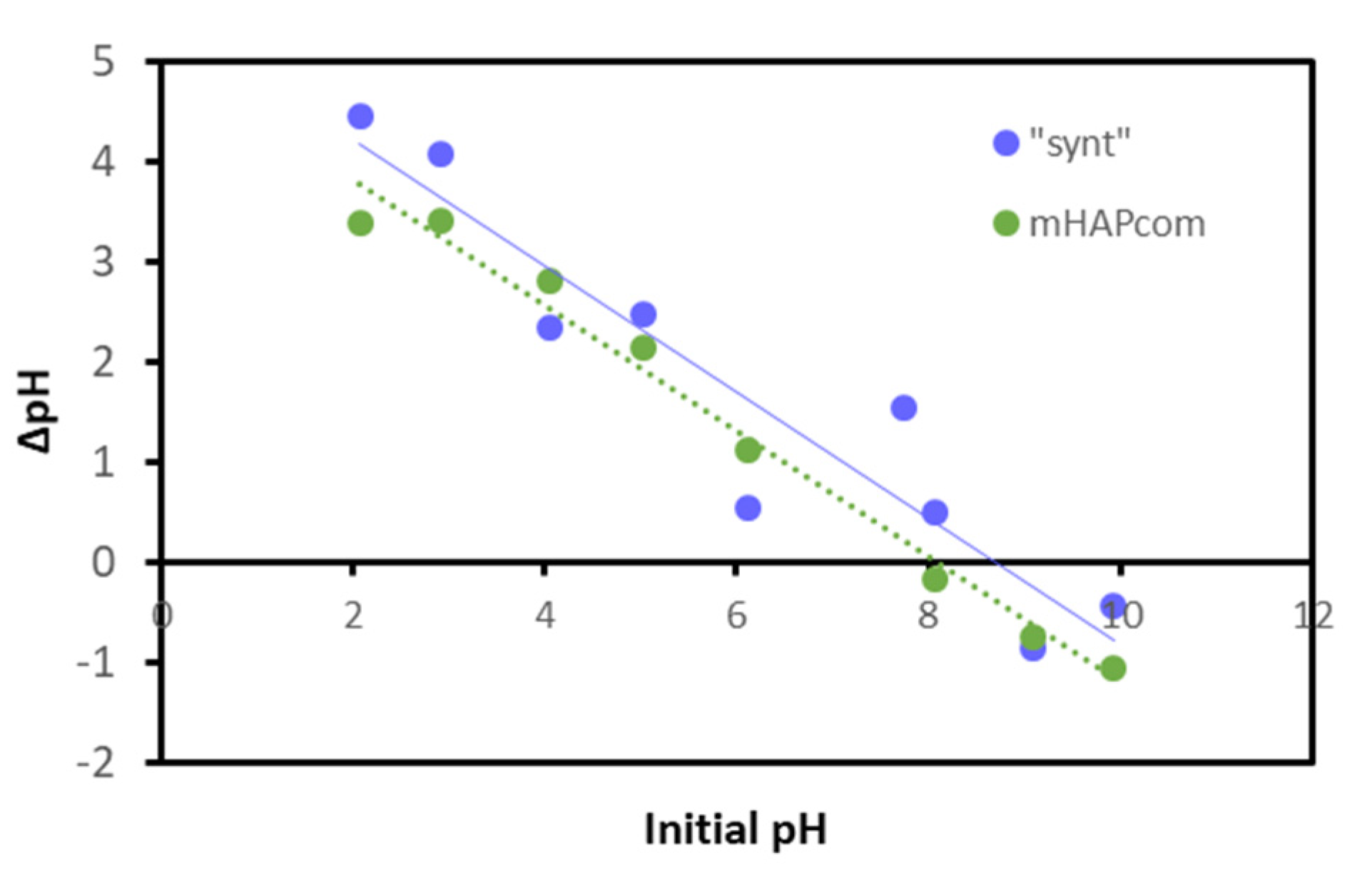
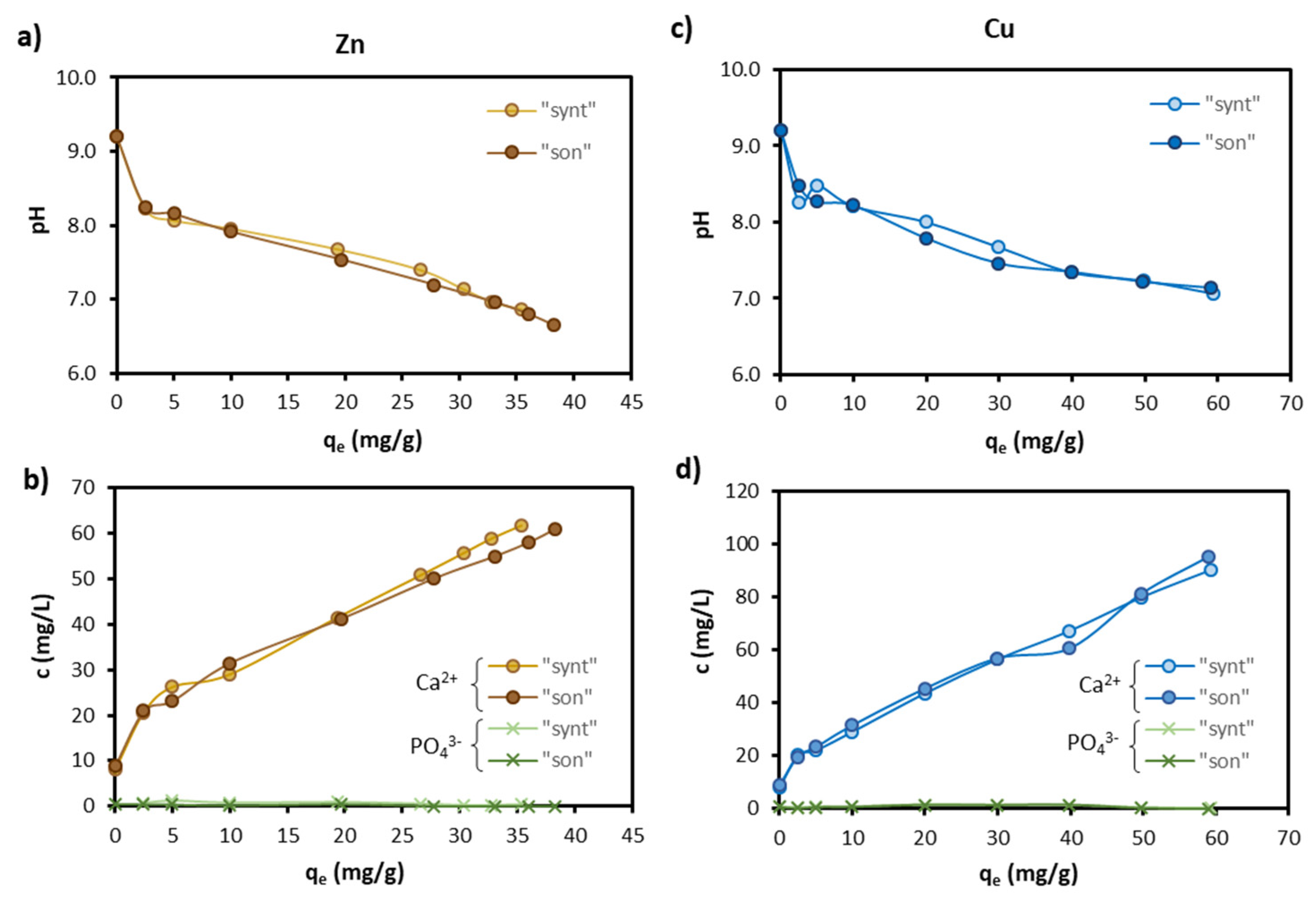
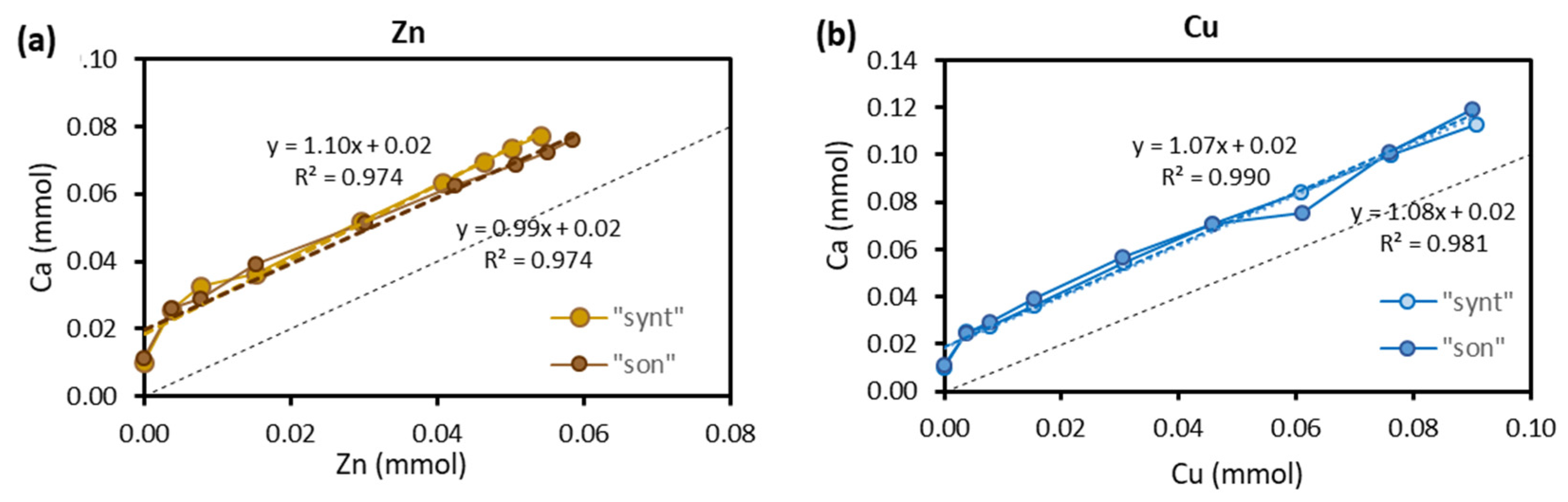
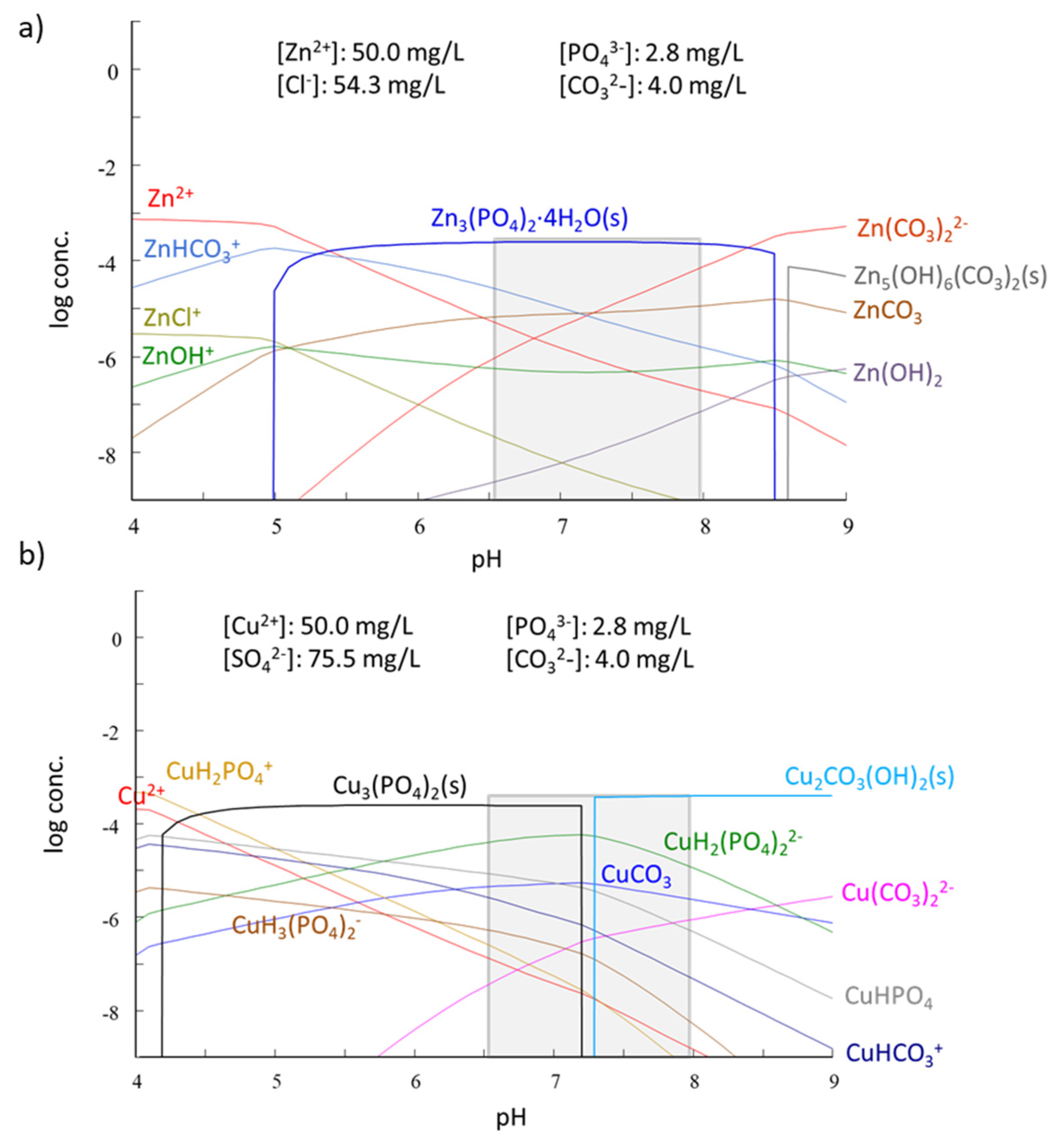
3.4. Sorption Mechanisms
2 ≡HAP-OH + M2+ ⇆ (≡HAP-O)2M + 2H+
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Burri, N.M.; Weatherl, R.; Moeck, C.; Schirmer, M. A review of threats to groundwater quality in the Anthropocene. Sci. Total Environ. 2019, 684, 136–154. [Google Scholar] [CrossRef]
- Zhou, P.; Adeel, M.; Shakoor, N.; Guo, M.; Hao, Y.; Azeem, I.; Li, M.; Liu, M.; Rui, Y. Application of nanoparticles alleviates heavy metals stress and promotes plant growth: An overview. Nanomaterials 2021, 11, 26. [Google Scholar] [CrossRef]
- Vareda, J.P.; Valente, A.J.M.; Durães, L. Assessment of heavy metal pollution from anthropogenic activities and remediation strategies: A review. J. Environ. Manag. 2019, 246, 101–118. [Google Scholar] [CrossRef]
- Vidu, R.; Matei, E.; Predescu, A.M.; Alhalaili, B.; Pantilimon, C.; Tarcea, C.; Predescu, C. Removal of heavy metals from wastewaters: A challenge from current treatment methods to nanotechnology applications. Toxics 2020, 8, 101. [Google Scholar] [CrossRef]
- Carolin, C.F.; Kumar, P.S.; Saravanan, A.; Joshiba, G.J.; Naushad, M. Efficient techniques for the removal of toxic heavy metals from aquatic environment: A review. J. Environ. Chem. Eng. 2017, 5, 2782–2799. [Google Scholar] [CrossRef]
- Abdullah, N.; Yusof, N.; Lau, W.J.; Jaafar, J.; Ismail, A.F. Recent trends of heavy metal removal from water/wastewater by membrane technologies. J. Ind. Eng. Chem. 2019, 76, 17–38. [Google Scholar] [CrossRef]
- Kaushik, A.; Singh, A. Metal removal and recovery using bioelectrochemical technology: The major determinants and opportunities for synchronic wastewater treatment and energy production. J. Environ. Manag. 2020, 270, 110826. [Google Scholar] [CrossRef] [PubMed]
- Malik, L.A.; Bashir, A.; Qureashi, A.; Pandith, A.H. Detection and removal of heavy metal ions: A review. Environ. Chem. Lett. 2019, 17, 1495–1521. [Google Scholar] [CrossRef]
- Waheed, A.; Baig, N.; Ullah, N.; Falath, W. Removal of hazardous dyes, toxic metal ions and organic pollutants from wastewater by using porous hyper-cross-linked polymeric materials: A review of recent advances. J. Environ. Manag. 2021, 287, 112360. [Google Scholar] [CrossRef] [PubMed]
- Adeleye, A.S.; Conway, J.R.; Garner, K.; Huang, Y.; Su, Y.; Keller, A.A. Engineered nanomaterials for water treatment and remediation: Costs, benefits, and applicability. Chem. Eng. J. 2016, 286, 640–662. [Google Scholar] [CrossRef] [Green Version]
- Henderson, A.D.; Demond, A.H. Long-term performance of zero-valent iron permeable reactive barriers: A critical review. Environ. Eng. Sci. 2007, 24, 401–423. [Google Scholar] [CrossRef] [Green Version]
- Wen, D.; Fu, R.; Li, Q. Removal of inorganic contaminants in soil by electrokinetic remediation technologies: A review. J. Hazard. Mater. 2021, 401, 123345. [Google Scholar] [CrossRef] [PubMed]
- Xin, J.; Tang, F.; Yan, J.; La, C.; Zheng, X.; Liu, W. Investigating the efficiency of microscale zero valent iron-based in situ reactive zone (mZVI-IRZ) for TCE removal in fresh and saline groundwater. Sci. Total Environ. 2018, 626, 638–649. [Google Scholar] [CrossRef] [PubMed]
- Gibert, O.; Assal, A.; Devlin, H.; Elliot, T.; Kalin, R.M. Performance of a field-scale biological permeable reactive barrier for in-situ remediation of nitrate-contaminated groundwater. Sci. Total Environ. 2019, 659, 211–220. [Google Scholar] [CrossRef] [Green Version]
- Patil, S.S.; Shedbalkar, U.U.; Truskewycz, A.; Chopade, B.A.; Ball, A.S. Nanoparticles for environmental clean-up: A review of potential risks and emerging solutions. Environ. Technol. Innov. 2016, 5, 10–21. [Google Scholar] [CrossRef]
- Luo, J.; Yu, D.; Hristovski, K.D.; Fu, K.; Shen, Y.; Westerhoff, P.; Crittenden, J.C. Critical review of advances in engineering nanomaterial adsorbents for metal removal and recovery from water: Mechanism identification and engineering design. Environ. Sci. Technol. 2021, 55, 4287–4304. [Google Scholar] [CrossRef]
- Luo, J.; Fu, K.; Yu, D.; Hristovski, K.D.; Westerhoff, P.; Crittenden, J.C. Review of advances in engineering nanomaterial adsorbents for metal removal and recovery from water: Synthesis and microstructure impacts. ACS EST Eng. 2021, 1, 623–661. [Google Scholar] [CrossRef]
- Thanh, D.N.; Novák, P.; Vejpravova, J.; Vu, H.N.; Lederer, J.; Munshi, T. Removal of copper and nickel from water using nanocomposite of magnetic hydroxyapatite nanorods. J. Magn. Magn. Mater. 2018, 456, 451–460. [Google Scholar] [CrossRef]
- Cai, C.; Zhao, M.; Yu, Z.; Rong, H.; Zhang, C. Utilization of nanomaterials for in-situ remediation of heavy metal(loid) contaminated sediments: A review. Sci. Total Environ. 2019, 662, 205–217. [Google Scholar] [CrossRef]
- Ou, M.-Y.; Ting, Y.; Ch’ng, B.-L.; Chen, C.; Cheng, Y.-H.; Chang, T.-C.; Hsi, H.-C. Using mixed active capping to remediate multiple potential toxic metal contaminated sediment for reducing environmental risk. Water 2020, 12, 1886–1900. [Google Scholar] [CrossRef]
- Sadat-Shojai, M.; Khorasani, M.T.; Dinpanah-Khoshdargi, E.; Jamshidi, A. Synthesis methods for nanosized hydroxyapatite with diverse structures. Acta Biomater. 2013, 9, 7591–7621. [Google Scholar] [CrossRef]
- Fihri, A.; Len, C.; Varma, R.S.; Solhy, A. Hydroxyapatite: A review of syntheses, structure and applications in heterogeneous catalysis. Coordin. Chem. Rev. 2017, 347, 48–76. [Google Scholar] [CrossRef]
- Liu, C.; Huang, Y.; Shen, W.; Cui, J. Kinetics of hydroxyapatite precipitation at pH 10 to 11. Biomaterials 2001, 22, 301–3016. [Google Scholar] [CrossRef]
- Wang, P.; Li, C.; Gong, H.; Jiang, X.; Wang, H.; Li, K. Effects of synthesis conditions on the morphology of hydroxyapatite nanoparticles produced by wet chemical process. Powder Technol. 2010, 203, 315–321. [Google Scholar] [CrossRef]
- Smičiklas, I.; Onjia, A.; Raičević, S.; Janaćković, Ð.; Mitrić, M. Factors influencing the removal of divalent cations by hydroxyapatite. J. Hazard. Mater. 2008, 152, 876–884. [Google Scholar] [CrossRef] [PubMed]
- Marchegiani, F.; Cibej, E.; Vergni, P.; Tosi, G.; Fermani, S.; Falini, G. Hydroxyapatite synthesis from biogenic calcite single crystals into phosphate solutions at ambient conditions. J. Cryst. Growth 2009, 311, 4219–4225. [Google Scholar] [CrossRef]
- Ni, M.; Ratner, B.D. Nacre surface transformation to hydroxyapatite in a phosphate buffer solution. Biomaterials 2003, 24, 4323–4331. [Google Scholar] [CrossRef]
- Guo, Y.P.; Zhou, Y. Conversion of nacre powders to apatite in phosphate buffer solutions at low temperatures. Mater. Chem. Phys. 2007, 106, 88–94. [Google Scholar] [CrossRef]
- Pham Minh, D.; Lyczko, N.; Sebei, H.; Nzihou, A.; Sharrock, P. Synthesis of calcium hydroxyapatite from calcium carbonate and different orthophosphate sources: A comparative study. Mater. Sci. Eng. B 2012, 177, 1080–1089. [Google Scholar] [CrossRef] [Green Version]
- Naidu, S.; Scherer, G.W. Nucleation, growth and evolution of calcium phosphate films on calcite. J. Colloid Interf. Sci. 2014, 435, 128–137. [Google Scholar] [CrossRef]
- Yang, F.; Liu, Y. Artificial hydroxyapatite film for the conservation of outdoor marble artworks. Mater. Lett. 2014, 124, 201–203. [Google Scholar] [CrossRef]
- Graziani, G.; Sassonia, E.; Franzonia, E.; Scherer, G.W. Hydroxyapatite coatings for marble protection: Optimization of calcite covering and acid resistance. Appl. Surf. Sci. 2016, 368, 241–257. [Google Scholar] [CrossRef]
- Ivanets, A.; Kitikova, N.; Shashkova, I.; Matrunchik, Y.; Kul’bitskaya, L.; Sillanpää, M. Non-acidic synthesis of phosphatized dolomite and its sorption behaviour towards Pb2+, Zn2+, Cu2+, Cd2+, Ni2+, Sr2+ and Co2+ ions in multicomponent aqueous solution. Environ. Technol. Innov. 2016, 6, 152–164. [Google Scholar] [CrossRef]
- Fiol, N.; Villaescusa, I. Determination of sorbent point zero charge: Usefulness in sorption studies. Environ. Chem. Lett. 2009, 7, 79–84. [Google Scholar] [CrossRef]
- Sheha, R.R. Sorption behavior of Zn(II) ions on synthesized hydroxyapatites. J. Colloid Interf. Sci. 2007, 310, 18–26. [Google Scholar] [CrossRef]
- Meski, S.; Ziani, S.; Khireddine, H.; Boudboub, S.; Zaidi, S. Factorial design analysis for sorption of zinc on hydroxyapatite. J. Hazard. Mater. 2011, 186, 1007–1017. [Google Scholar] [CrossRef]
- Klinkaewnarong, J.; Utara, S. Ultrasonic-assisted conversion of limestone into needle-like hydroxyapatite nanoparticles. Ultrason. Sonochem. 2018, 46, 18–25. [Google Scholar] [CrossRef]
- Lei, S.; Shi, Y.; Qiu, Y.; Che, L.; Xue, C. Performance and mechanisms of emerging animal-derived biochars for immobilization of heavy metals. Sci. Total Environ. 2019, 646, 1281–1289. [Google Scholar] [CrossRef]
- Ramasamy, V.; Anand, P.; Suresh, G. Synthesis and characterization of polymer-mediated CaCO3 nanoparticles using limestone: A novel approach. Adv. Powder Technol. 2018, 29, 818–834. [Google Scholar] [CrossRef]
- Salimi, M.N.; Bridson, R.H.; Grover, L.M.; Leeke, G.A. Effect of processing conditions on the formation of hydroxyapatite nanoparticles. Powder Technol. 2012, 218, 109–118. [Google Scholar] [CrossRef]
- Harding, I.S.; Rashid, N.; Hing, K.A. Surface charge and the effect of excess calcium ions on the hydroxyapatite surface. Biomaterials 2005, 26, 6818–6826. [Google Scholar] [CrossRef]
- Corami, A.; Mignardi, S.; Ferrini, V. Copper and zinc decontamination from single- and binary-metal solutions using hydroxyapatite. J. Hazard. Mater. 2007, 146, 164–170. [Google Scholar] [CrossRef]
- Šljivić, M.; Smičiklas, I.; Plećaš, I.; Mitrić, M. The influence of equilibration conditions and hydroxyapatite physico-chemical properties onto retention of Cu2+ ions. Chem. Eng. J. 2009, 148, 80–88. [Google Scholar] [CrossRef]
- Wang, Y.J.; Chen, J.H.; Cui, Y.X.; Wang, S.Q.; Zhou, D.M. Effects of low-molecular-weight organic acids on Cu(II) adsorption onto hydroxyapatite nanoparticles. J. Hazard. Mater. 2009, 162, 1135–1140. [Google Scholar] [CrossRef]
- Feng, Y.; Gong, J.L.; Zeng, G.M.; Niu, Q.Y.; Zhang, H.Y.; Niu, C.G.; Deng, J.H.; Yan, M. Adsorption of Cd (II) and Zn (II) from aqueous solutions using magnetic hydroxyapatite nanoparticles as adsorbents. Chem. Eng. J. 2010, 162, 487–494. [Google Scholar] [CrossRef]
- Xu, Y.; Schwartz, F.W.; Traína, S.J. Sorption of Zn2+ and Cd2+ on hydroxyapatite surfaces. Environ. Sci. Technol. 1994, 28, 1472–1480. [Google Scholar] [CrossRef]
- Zhu, R.; Yu, R.; Yao, J.; Mao, D.; Xing, C.; Wang, D. Removal of Cd2+ from aqueous solutions by hydroxyapatite. Catal. Today 2008, 139, 94–99. [Google Scholar] [CrossRef]
- Smičiklas, I.D.; Milonjić, S.K.; Pfendt, P.; Raičević, S. The point of zero charge and sorption of cadmium (II) and strontium (II) ions on synthetic hydroxyapatite. Sep. Purif. Technol. 2000, 18, 185–194. [Google Scholar] [CrossRef]
- Fulmer, M.T.; Ison, I.C.; Hankermayer, C.R.; Constantz, B.R.; Ross, J. Measurements of the solubilities and dissolution rates of several hydroxyapatites. Biomaterials 2002, 23, 751–755. [Google Scholar] [CrossRef]
- Stötzel, C.; Müller, F.A.; Reinert, F.; Niederdraenk, F.; Barralet, J.E.; Gbureck, U. Ion adsorption behaviour of hydroxyapatite with different crystallinities. Colloid Surface B 2009, 74, 91–95. [Google Scholar] [CrossRef]
- Chemical Equilibrium Software Hydra and Medusa. Inorganic Chemistry Department, Royal Institute of Technology: Stockholm, Sweden. Available online: https://www.kth.se/che/medusa (accessed on 3 February 2021).











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sorbent | SBET (m2/g) |
|---|---|
| Initial calcite | 0.87 ± 0.01 |
| Synt | 58.25 ± 0.08 |
| Son | 53.68 ± 0.16 |
| mHAPcom | 60.77 ± 0.20 |
| nHAPcom | 18.93 ± 0.11 |
| Metal | Pseudo-First Order Kinetic Model | Pseudo-Second Order Kinetic Model | ||||
|---|---|---|---|---|---|---|
| qe | k1 | R2 | qe | k2 | R2 | |
| (mg/g) | (1/min) | (mg/g) | g/(mg·min) | |||
| Zn | 31.38 | 0.099 | 0.840 | 31.37 | 0.014 | 0.998 |
| Cu | 44.08 | 0.079 | 0.966 | 44.05 | 0.057 | 0.998 |
| Metal | Sorbent | qmax | KL | R2 | |
|---|---|---|---|---|---|
| (mg/g) | (mg/m2) | (L/mg) | |||
| Zn | synt | 34.97 | 0.600 | 0.91 | 0.9978 |
| son | 37.88 | 0.706 | 1.47 | 0.9984 | |
| mHAPcom | 29.67 | 0.489 | 0.68 | 0.9985 | |
| nHAPcom | 18.98 | 1.003 | 0.85 | 0.9967 | |
| Cu | synt | 60.24 | 1.034 | 15.09 | 0.9905 |
| son | 60.24 | 1.122 | 13.83 | 0.9955 | |
| mHAPcom | 38.61 | 0.635 | 2.98 | 0.9997 | |
| nHAPcom | 19.27 | 1.018 | 9.88 | 0.9977 | |
| HAP Synthesis and Characteristics | Adsorption | Ref | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Zn | Cu | ||||||||
| Precursors | pH | T | SBET (m2/g) | pH | qmax (mg/g) | qmax (mg/m2) | qmax (mg/g) | qmax (mg/m2) | |
| Ca(OH)2 + H3PO4 | n.r. | 20 | 67 | 5.0 | 37.53 | 0.560 | - | - | [25] |
| Ca(NO3)2 + (NH4)HPO4 (+Fe3O4/Fe2O3) | 11 | 90 | 142.5 | 5.0 (a) | 140.6 (b) | 0.99 | - | - | [45] |
| Ca(OH)2 + H3PO4 | n.r | 100 | 76.6 | 6.0 (a) | 102.04 | 1.332 | - | - | [35] |
| 5.5 (a) | 37.27 | 0.750 | - | - | |||||
| Ca(NO3)2 + (NH4)2HPO4 | 11 | n.r. | n.r. | 5.0 (a) | 10.75 | n.r. | - | - | [36] |
| commercial | 77 | 6.0 | 37.14 | 0.482 | - | - | [46] | ||
| commercial | 50 | 6.6 | 95.89 | 1.92 | 76.49 | 1.53 | [42] | ||
| CaCl2 + NH4H2PO4 + EtOH (+Fe3O4) | 11 | 20 | 101.2 | 5.0 (a) | - | - | 48.78 | 0.482 | [18] |
| Ca(OH)2 + H3PO4 | n.r. | n.r | 58 | 5.0 | - | - | 37.17 | 0.641 | [43] |
| Ca(NO3)2 + H3PO4 + NH4+-salt | 10 | 40 | 49.7 | 4.5 (a) | - | - | 29.23 | 0.588 | [44] |
| 5.5 (a) | - | - | 37.30 | 0.751 | |||||
| Calcite + (NH4)2PO4 (+EtOH) | 8 | 25 | 58.28 | 4.6 | 34.97 | 0.600 | 60.24 | 1.034 | This study |
| Calcite + (NH4)2PO4 (+EtOH) (+ sonication) | 53.68 | 4.6 | 37.88 | 0.706 | 60.24 | 1.122 | |||
| Commercial (mHAPcom) | 60.77 | 4.6 | 29.67 | 0.489 | 38.61 | 0.635 | |||
| Commercial (nHAPcom) | 18.93 | 4.6 | 18.98 | 1.003 | 19.27 | 0.985 | |||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gibert, O.; Valderrama, C.; Martínez, M.M.; Darbra, R.M.; Moncunill, J.O.; Martí, V. Hydroxyapatite Coatings on Calcite Powder for the Removal of Heavy Metals from Contaminated Water. Water 2021, 13, 1493. https://doi.org/10.3390/w13111493
Gibert O, Valderrama C, Martínez MM, Darbra RM, Moncunill JO, Martí V. Hydroxyapatite Coatings on Calcite Powder for the Removal of Heavy Metals from Contaminated Water. Water. 2021; 13(11):1493. https://doi.org/10.3390/w13111493
Chicago/Turabian StyleGibert, Oriol, César Valderrama, María M. Martínez, Rosa Mari Darbra, Josep Oliva Moncunill, and Vicenç Martí. 2021. "Hydroxyapatite Coatings on Calcite Powder for the Removal of Heavy Metals from Contaminated Water" Water 13, no. 11: 1493. https://doi.org/10.3390/w13111493
APA StyleGibert, O., Valderrama, C., Martínez, M. M., Darbra, R. M., Moncunill, J. O., & Martí, V. (2021). Hydroxyapatite Coatings on Calcite Powder for the Removal of Heavy Metals from Contaminated Water. Water, 13(11), 1493. https://doi.org/10.3390/w13111493