3.2. Semi-Pilot-Scale Evaluation
The laboratory-scale setup was scaled up by a factor of 7, while maintaining the A/V ratio used in the laboratory scale constant. The A/V ratio had a value of 10 m−1 (0.02 m2/0.002 m3 for the laboratory scale and 0.14 m2/0.014 m3 for the semi-pilot scale).
Prior to the evaluation of PFAAs removal, a mass transfer study was performed to determine the average mass-transfer coefficient (k
m) in both setups (k
m,lab for the laboratory scale and k
m,sp for the semi-pilot scale). The values of k
m were determined with Equation (
3), using the limiting-current technique—the procedure is described elsewhere [
26,
35].
where
is the limiting current (A),
n is the number of e
− exchanged,
F is Faraday’s constant (96,485 C/mol),
A is the anodic area (m
2), and
is the concentration in the bulk (mol/m
3).
Constant concentrations of potassium of 0.05 M K
4Fe(CN)
6 and 0.1 M K
3Fe(CN)
6 were used for all the experiments. The concentration of K
3Fe(CN)
6 was in excess to ensure the limiting current was at the anode. For the corresponding flow rates (2 L/min at the laboratory scale and 6 L/min at the semi-pilot scale) that provided an equivalent Re number for both setups ( 2300), k
m,lab was
m/s and k
m,sp was
m/s, giving a k
m,lab/k
m,sp ratio of 0.8. The value of k
m depends on the cell geometry and increases with a lower inter-electrode gap [
26]. Therefore, the smaller inter-electrode distance of the semi-pilot scale (2 mm, compared to 3 mm at the laboratory scale) led to an enhancement of k
m at the semi-pilot scale. An enhancement in k
SA for PFAAs degradation was also expected at the semi-pilot scale.
Consecutively, the electrochemical treatment of PFAAs in a synthetic still bottoms solution was assessed at the semi-pilot scale and the results were compared with those obtained at the laboratory scale. The voltage that resulted from the galvanostatic process was lower at the semi-pilot scale (5.7 V at the semi-pilot scale vs. 5.9 V at the laboratory scale), attributed to the smaller inter-electrode distance, as previously stated.
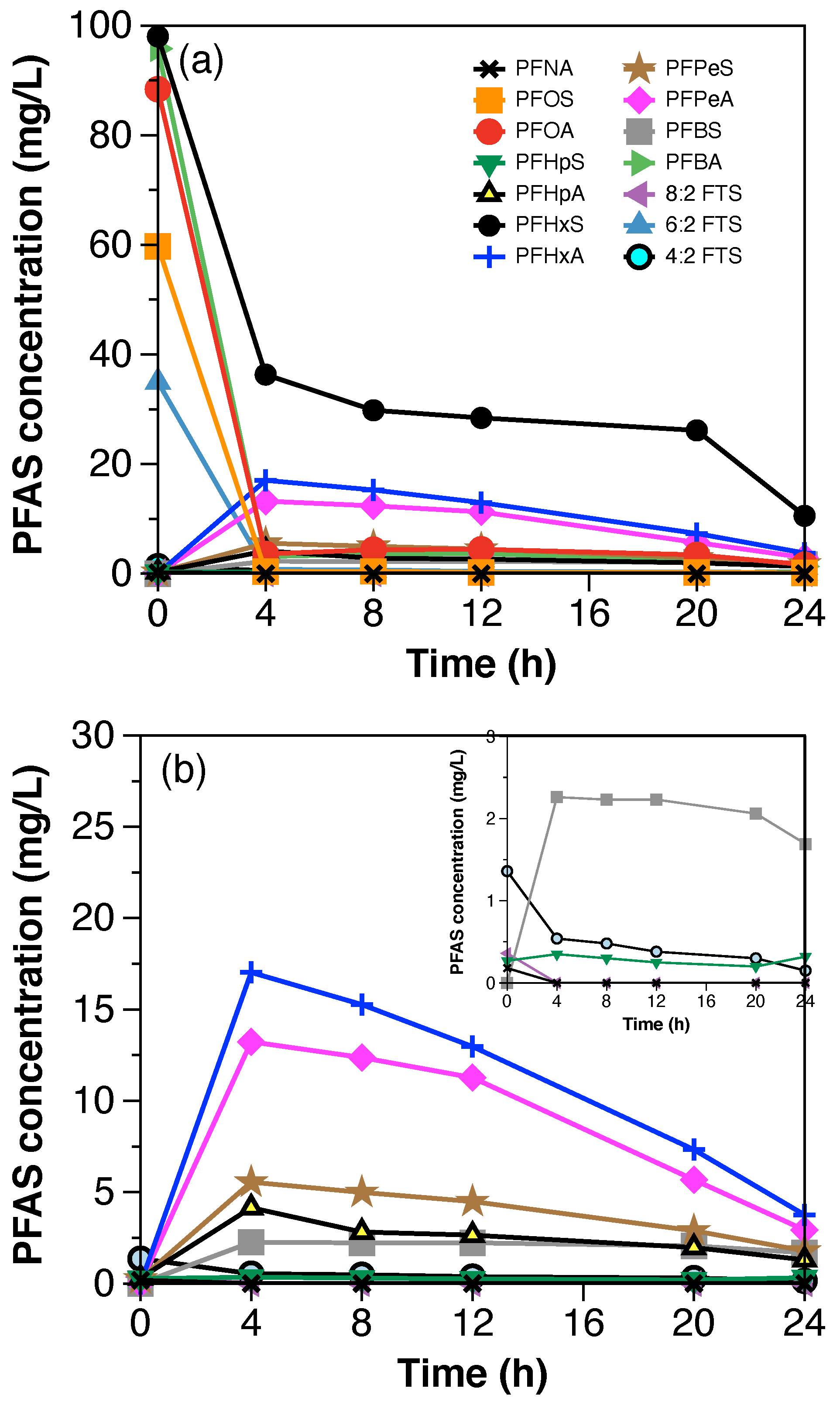
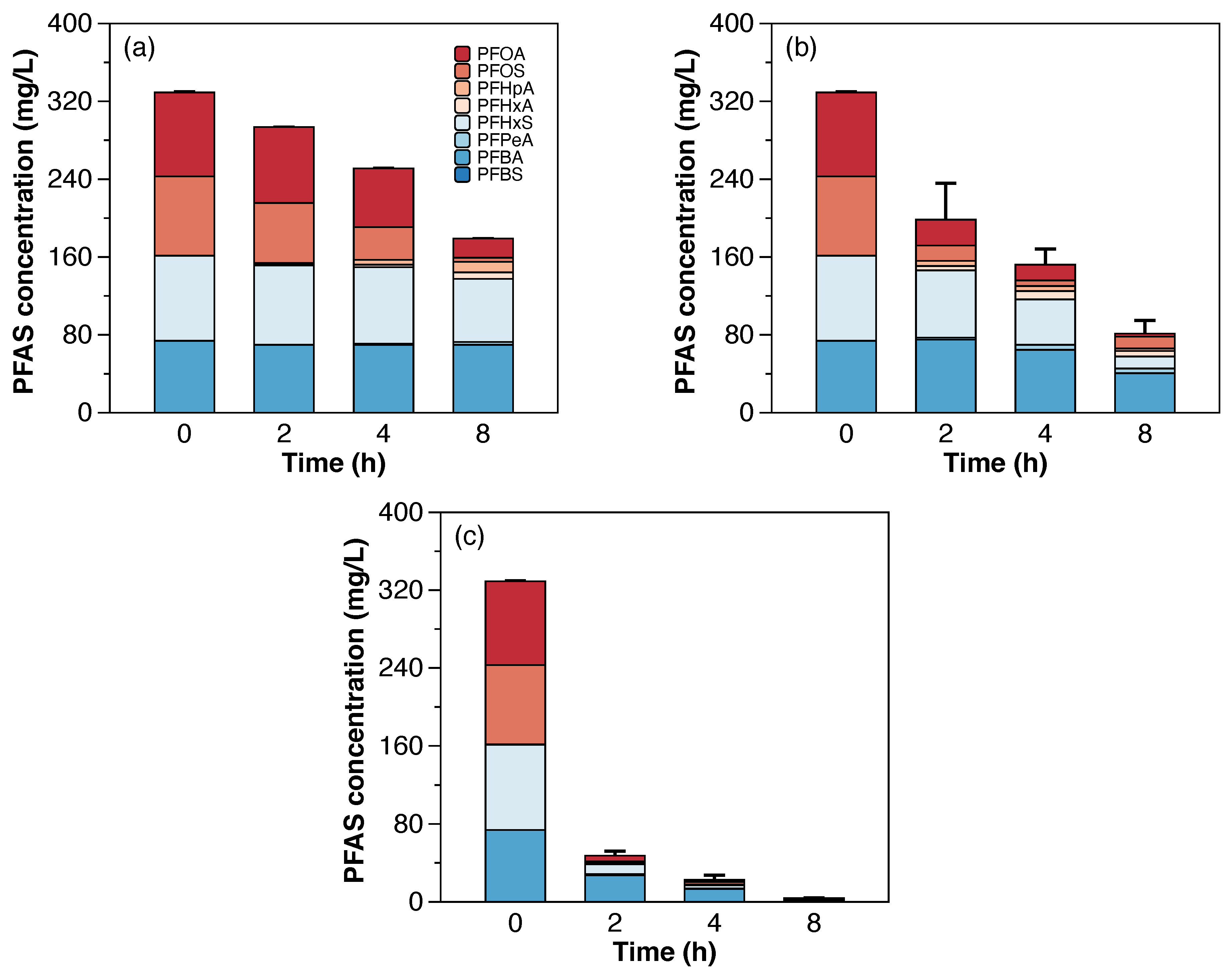
Figure 3 shows the decrease in concentration of total PFAAs from the synthetic still bottoms treated with 50 mA/cm
2 in both scales. The total PFAAs removal after 8 h of treatment was 94% in the semi-pilot-scale setup. The percentages of individual PFAAs remaining in solution after treatment with respect to their initial concentrations were 19% of PFBA, 3% of PFHxS, and <2% of PFOA and PFOS. Similar to the laboratory scale experiments, a layer of foam was observed during the electrochemical treatment of PFAAs in the semi-pilot-scale setup. Therefore, a fraction of PFAAs removal, in particular the highly hydrophobic PFAAs, was likely attributed to their partitioning into the foam. To verify this, the foam generated during the experimental time (8 h) was collected separately and sent for PFAS analysis. Results showed that the mass percentage of individual PFAAs partitioned into the foam with respect to the initial concentration of PFAAs in the solution corresponded to 61% of PFOS, 17% of PFOA, 8% of PFBA, and 2% of PFHxS. Likewise, a previous study showed that at least 80% of the PFOS-associated fluorine partitioned into the foam [
22].
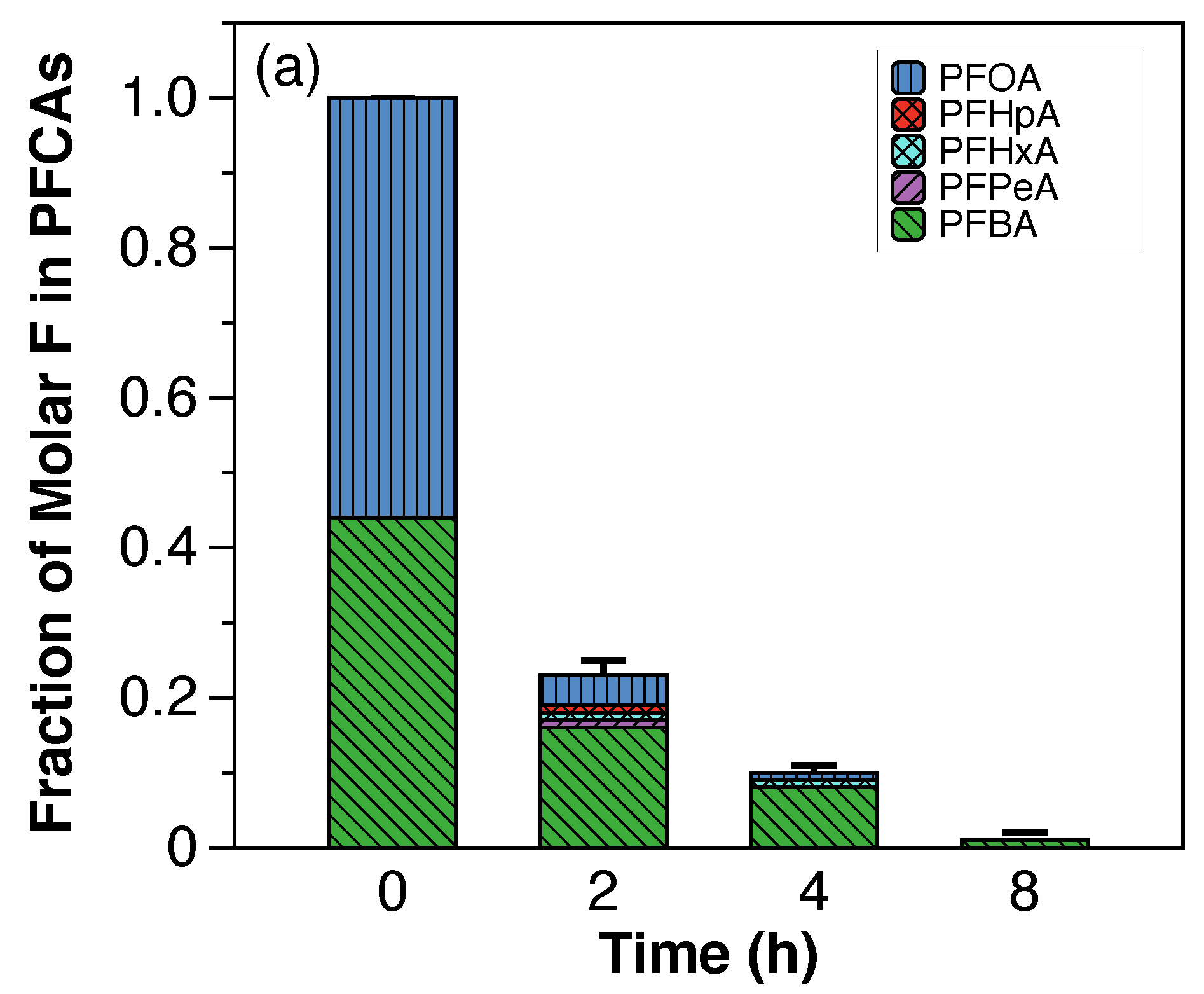
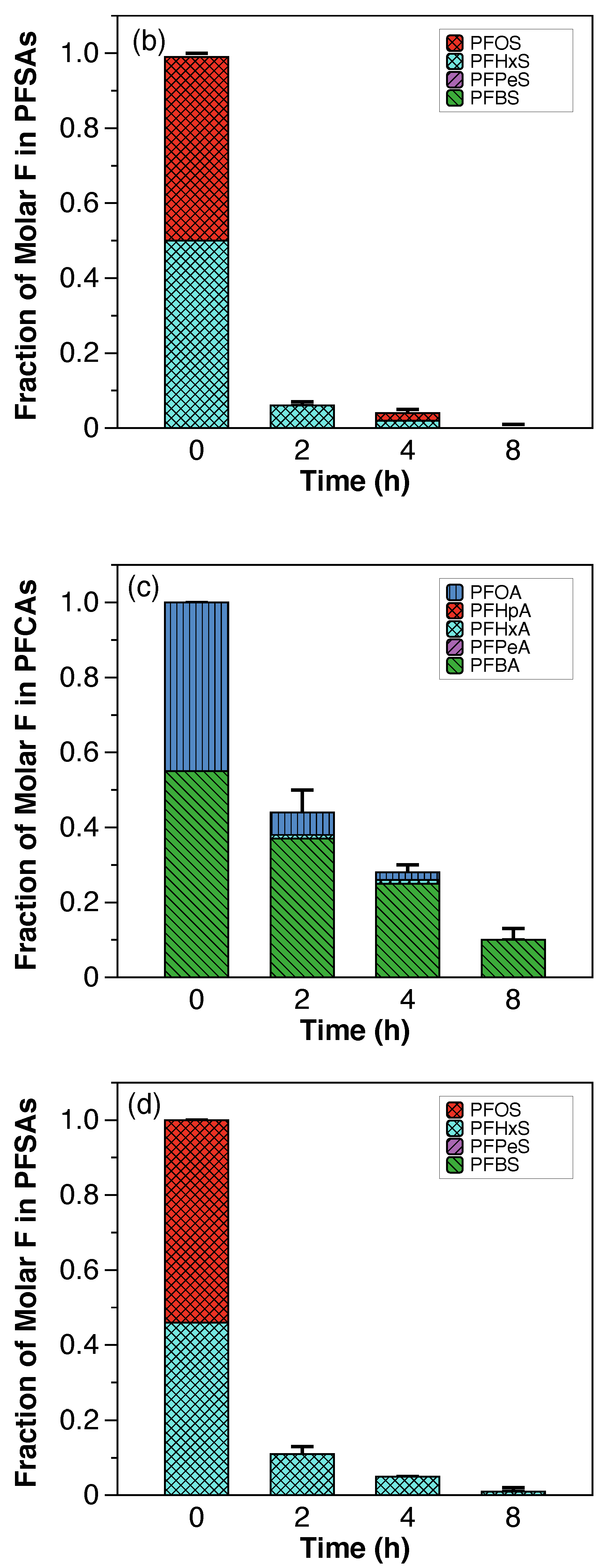
The fraction of molar F in PFCAs and PFSAs (shown in
Figure A5) was used to compare the evolution of individual PFAAs over time during the electrochemical treatment in both scales. In general, higher fractions of PFCAs, in particular PFHpA, PFHxA, and PFPeA, were generated at the laboratory scale, suggesting faster degradation kinetics at the laboratory scale and more foam partitioning at the semi-pilot scale.
The values of kSA for total PFAAs removal were m/s and m/s for the laboratory and the semi-pilot scales, respectively, giving a kSA,lab/kSA,sp ratio of 1.6. Interestingly, opposite ratios showing kSA,lab > kSA,sp and km,lab < km,sp were obtained.
The lower value of k
SA for PFAAs removal in the semi-pilot setup suggests that other factors besides fluid properties, hydrodynamics, and A/V ratio play a critical role in the treatment efficiency of PFAAs in IX still bottoms. These factors include gas evolution and current density distribution [
36]. During the electrochemical oxidation of target compounds (e.g., PFAAs), only a fraction of the applied current density, equal to the limiting current, is used in the oxidation of the target compound [
27]. The remaining fraction of current is used in side reactions including oxygen and hydrogen evolution [
37]. The previous reactions generate substantial quantities of gas (
) that are proportional to the applied current, according to Faraday’s first law of electrolysis (Equation (
4)):
where
R is the universal gas constant (8.314 J/mol
−1K
−1),
I is the current applied (A),
T is the average working temperature (303 K),
t is the treatment time (s),
F is the Faraday’s constant (96,485 C/mol),
P is the atmospheric pressure (
Pa), and
n is the number of e
− exchanged (2 for H
2, and 4 for O
2). A higher electrode area requires the application of a higher current to maintain a constant current density between both reactor scales, leading to the generation of a higher volume of gas in the semi-pilot-scale setup. For the corresponding currents of each setup (10.7 A at the laboratory scale and 70 A at the semi-pilot scale), the total volume of gas generated corresponds to 7.5 L/h and 49.3 L/h, approximately 7-fold more gas generation at the semi-pilot scale. Although a local increase in the mass transfer is expected if gas bubbles are generated [
27], the inherent surface-active properties of PFAAs induce their movement towards the air-water interface of the bubbles [
38], that travel to the interface of the solution (foam generation), where PFAAs are partitioned. In addition, local gas hold-up in the vicinity of the electrodes could have interfered with direct anodic oxidation of PFAAs in the liquid phase [
37]. Therefore, the probability of PFAAs reaching the anode surface decreases, slowing down the oxidation process. Thus, a lower k
SA is obtained.
Finally, possible differences in current density distributions along the electrodes in each setup could have affected the mass transfer of the process [
36,
39]. To maintain current similarity, it is recommended to increase the number of smaller modules, rather than increase the electrode size [
36].
3.3. Perchlorate Formation during Electrochemical Treatment
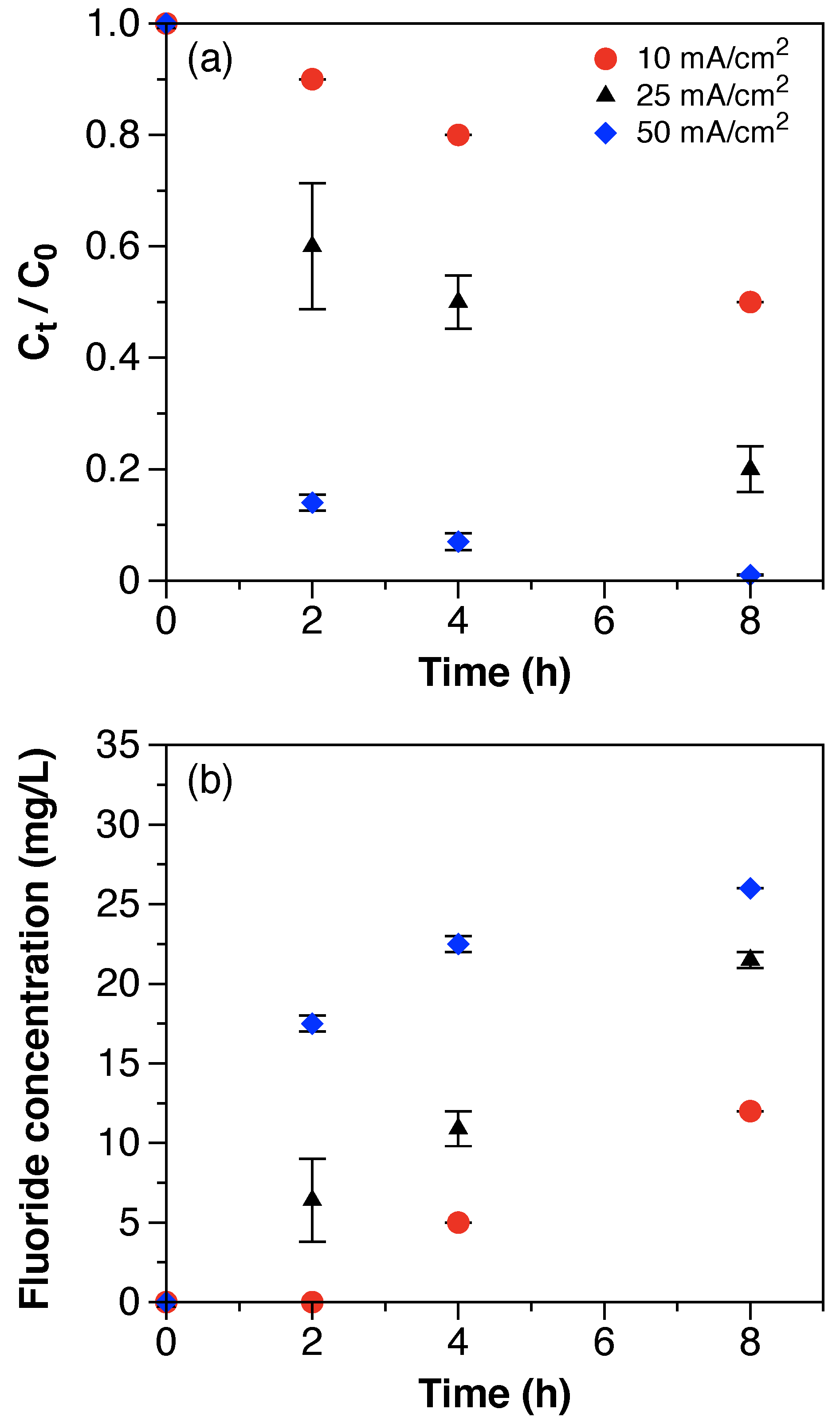
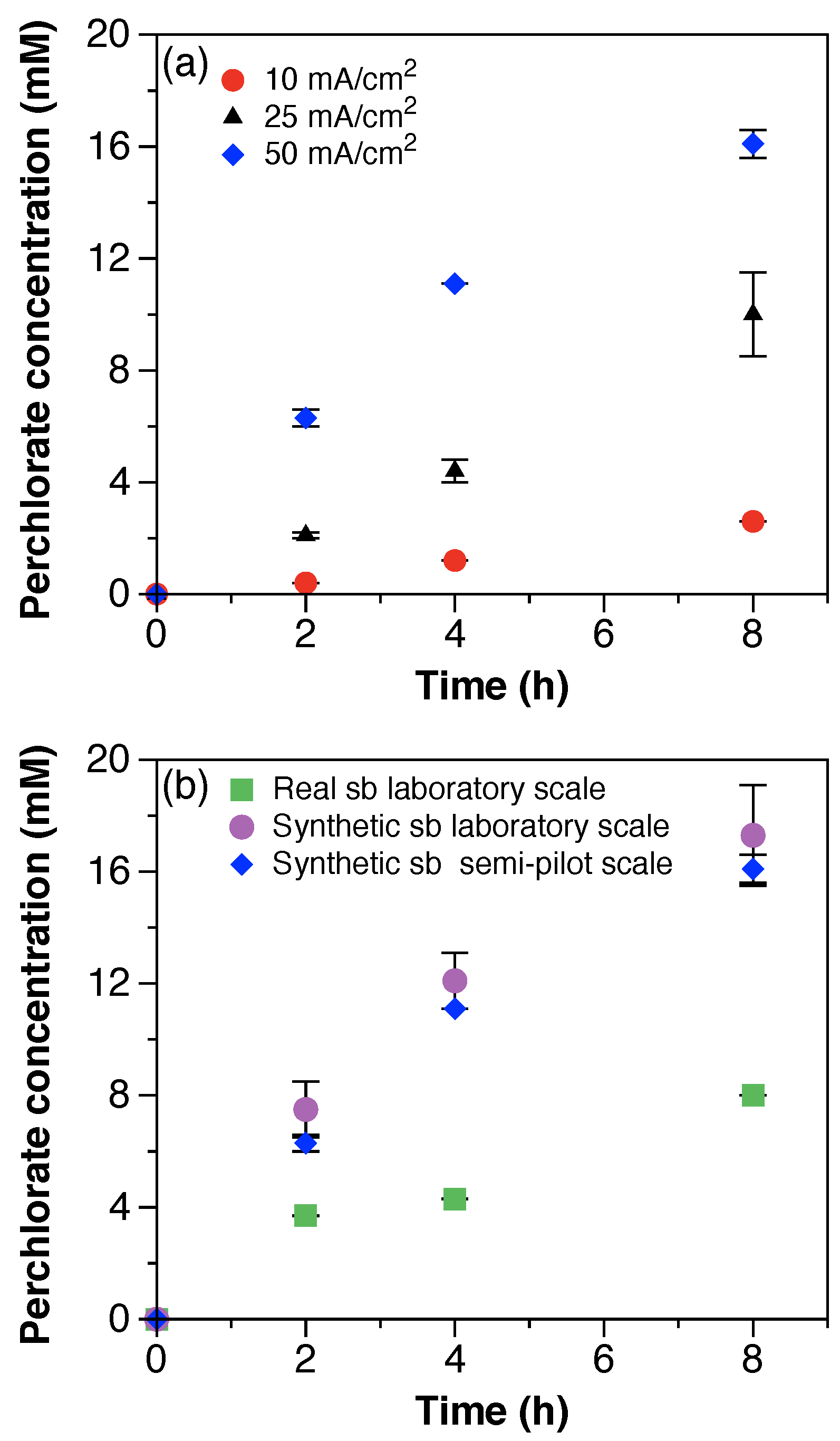
ClO
4− generation was quantified for all experiments and its evolution over time is shown in
Figure 4. For the electrochemical treatment of synthetic solutions with multiple current densities, the zero-order generation rate of ClO
4− increased with the current density (
Figure 4a) and reached concentrations of 2.6, 10.0, and 16.1 mM after 8 h of treatment with 10, 25, and 50 mA/cm
2, respectively. ClO
4− concentrations at the end of the treatment time (8 h) accounted for 0.2, 0.8, and 1.4% of the initial Cl
− concentration (1250 mM). The generation of ClO
4− was relatively low compared to the initial concentration of Cl
− available for oxidation. Although the concentration of chlorate (ClO
3−) was not quantified in this work, a recent study performed with still bottom solutions reported equimolar concentrations of ClO
3− and ClO
4− generated after 40 h of electrochemical treatment [
24]. Even assuming equivalent concentrations of ClO
3− generated, the percentage of chlorinated byproducts remains low when compared to the initial concentrations of Cl
−. These results suggest that additional species present in the solution may be competing for direct anodic oxidation or scavenging Cl
− oxidation. Wang et al. showed that the presence of methanol (100–1000 mM) in still bottom solutions scavenges chlorine radical Cl
. generation and significantly reduces the formation of chlorinated byproducts [
24]. The synthetic still bottoms solution of this work included a concentration of methanol of 312 mM, which likely contributed to the reduction of ClO
4− generation. Moreover, although having similar initial concentrations of Cl
−, the generation rate of ClO
4− during the electrochemical treatment of the real still bottoms was 2-fold slower than with the synthetic solution (
Figure 4b). The latter suggests that Cl
. scavenging may be affected by additional constituents of the solution, besides methanol. However, this assumption requires further studies.
ClO
4− generation was quantified for all experiments and its evolution over time is shown in
Figure 4. For the electrochemical treatment of synthetic solutions with multiple current densities, the zero-order generation rate of ClO
4− increased with the current density (
Figure 4a) and reached concentrations of 2.6, 10.0, and 16.1 mM after 8 h of treatment with 10, 25, and 50 mA/cm
2, respectively. ClO
4− concentrations at the end of the treatment time (8 h) accounted for 0.2, 0.8, and 1.4% of the initial Cl
− concentration (1250 mM). The generation of ClO
4− was relatively low compared to the initial concentration of Cl
− available for oxidation. Although the concentration of chlorate (ClO
3−) was not quantified in this work, a recent study performed with still bottom solutions reported equimolar concentrations of ClO
3− and ClO
4− generated after 40 h of electrochemical treatment [
24]. Even assuming equivalent concentrations of ClO
3− generated, the percentage of chlorinated byproducts remains low when compared to the initial concentrations of Cl
−. These results suggest that additional species present in the solution may be competing for direct anodic oxidation or scavenging Cl
− oxidation. Wang et al. showed that the presence of methanol (100–1000 mM) in still bottom solutions scavenges chlorine radical Cl
. generation and significantly reduces the formation of chlorinated byproducts [
24]. The synthetic still bottoms solution of this work included a concentration of methanol of 312 mM, which likely contributed to the reduction of ClO
4− generation. Moreover, although having similar initial concentrations of Cl
−, the generation rate of ClO
4− during the electrochemical treatment of the real still bottoms was 2-fold slower than with the synthetic solution (
Figure 4b). The latter suggests that Cl
. scavenging may be affected by additional constituents of the solution, besides methanol. However, this assumption requires further studies.
Finally, under the same experimental conditions, the generation of ClO
4− at the semi-pilot scale was comparable to the results obtained at the laboratory scale (
Figure 4b). The ClO
4− concentration after 8 h of electrochemical treatment was of 17.3 mM. The results suggest that ClO
4− generation with BDD electrodes solely depends on the applied current density, regardless of factors associated to scale performance differences.
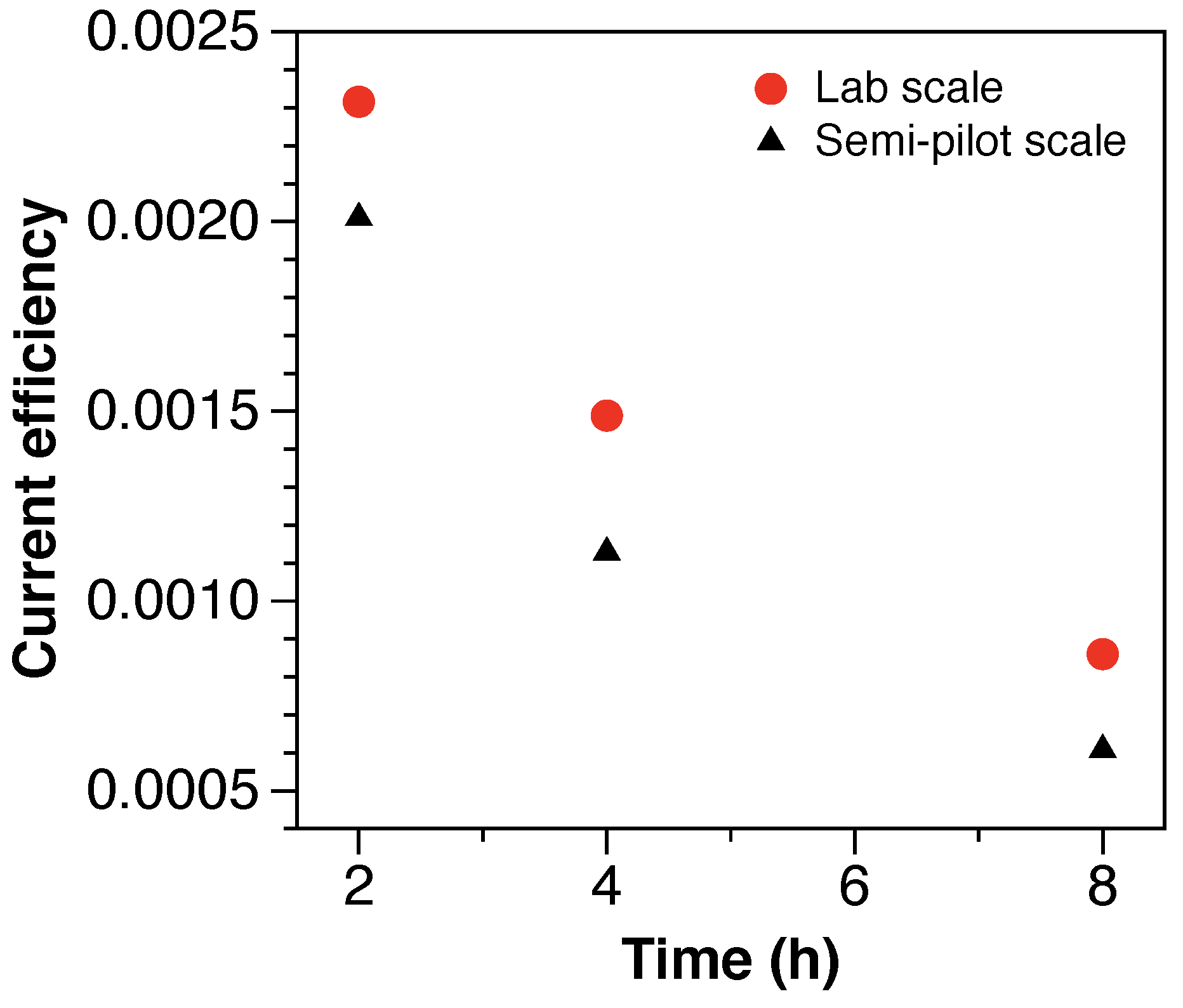
3.4. Treatment Efficiency and Energy Consumption
The coulombic efficiency (CE) was used to quantify the current efficiency for PFAAs defluorination during the electrochemical treatment and it is defined in Equation (
5) [
40,
41]:
where
F is Faraday’s constant (96,485 C/mol),
V is the volume of solution treated (L),
e is the moles of e
− needed per mole fluoride (1 electron per C-F bond [
22]),
is the fluoride concentration (mol/L),
I is the current (A), and
t is the treatment time (s).
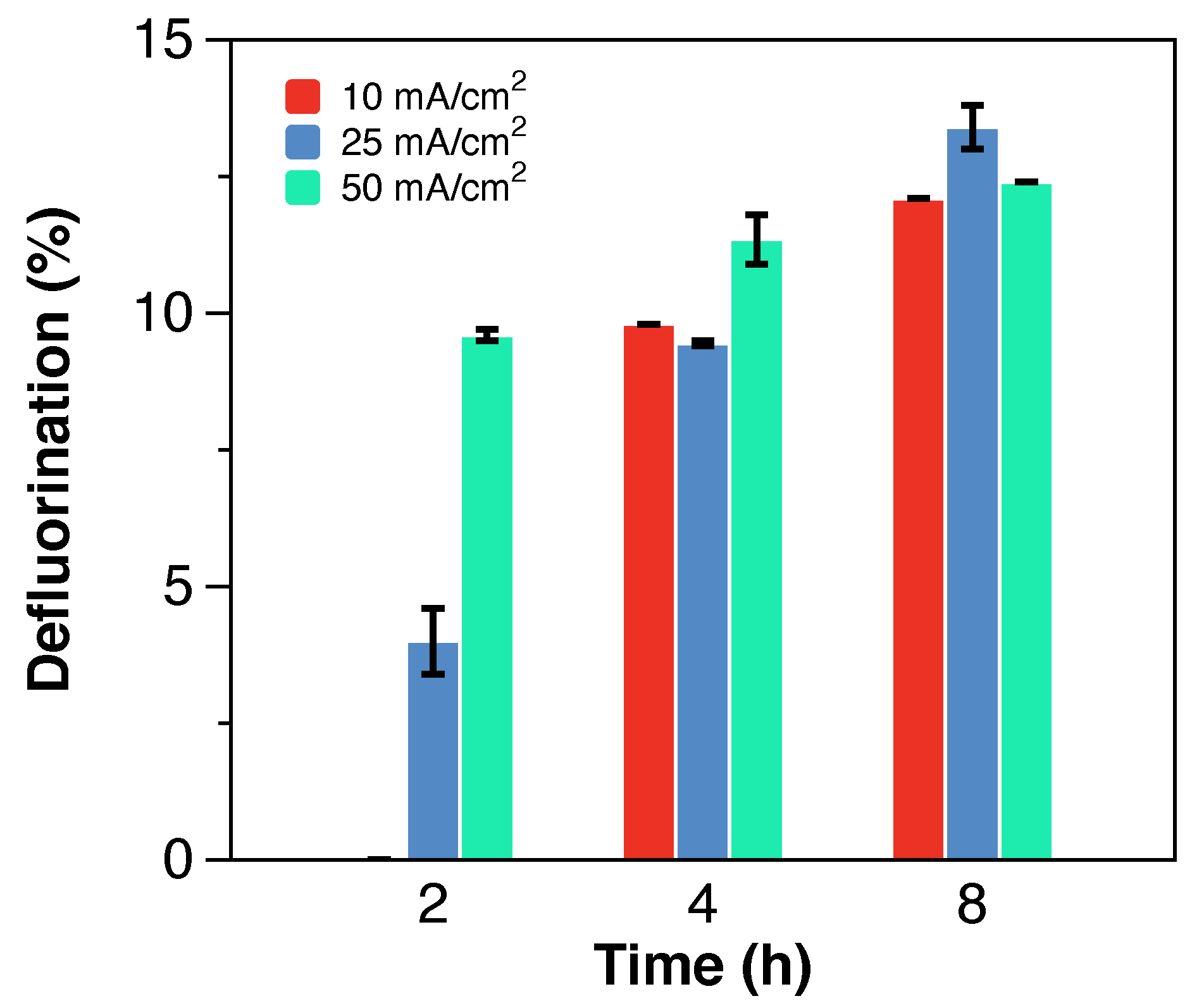
As shown in
Figure 5, the CE decreases over time from
at 2 h of treatment to
at 8 h of treatment. A comparable but lower decreasing trend was observed at the semi-pilot scale, with 15 and 40% lower CE at 2 and 8 h of electrochemical treatment, respectively. The low and decreasing CE values, characteristic of mass-transfer limited electrochemical reactions with applied potentials above the water oxidation threshold, are attributed to competitive oxidation reactions from additional components of the solution (e.g., Cl
−, additional TOC) and water electrolysis reactions [
27]. Nevertheless, the reported CE values are 5-fold greater than the values reported for the electrochemical treatment of low concentrations of PFAS in groundwater [
29], showing that the efficiency of the electrochemical treatment of PFAS increases with highly concentrated solutions, such as still bottoms from IX spent regenerant solutions.
Finally, the electric energy per order (E
EO) was determined using Equation (
6) as follows [
42]:
where
P is the power of the system (W),
V is the treatment volume (L),
t is the treatment time (h), and
and
C are the initial and final PFAAs concentration. The energy required for 90% PFAAs removal with a current density of 50 mA/cm
2 was 173 and 194 Wh/L for the laboratory and semi-pilot scales, respectively. Although the smaller inter-electrode distance in the semi-pilot-scale system provided a lower voltage, the faster degradation kinetics in the laboratory scale setup compensated the energy losses that result from a wider electrode gap, leading to a lower energy consumption required for the same order of removal. The latter highlights the importance of a fast degradation rate in the electrochemical process that allows for energy optimization.
Last, it is important to consider that the energy consumption for the electrochemical treatment of PFAS from still bottoms accounts for less than 0.01% of the total volume of water pre-treated with IX resins [
10]. Therefore, the calculated energy required for the electrochemical treatment of the total volume of pre-treated water with IX is 0.017 Wh/L at the laboratory scale and 0.019 Wh/L at the semi-pilot scale. This outcome illustrates the benefits of a combined tandem IX- electrochemical oxidation process that allows for >99.9% energy reduction for the combined IX/EO technologies when compared to electrochemical oxidation of PFAAs alone.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}










