
Next-Generation DNA Barcoding for Fish Identification Using High-Throughput Sequencing in Tai Lake, China


Abstract
:1. Introduction
2. Materials and Methods
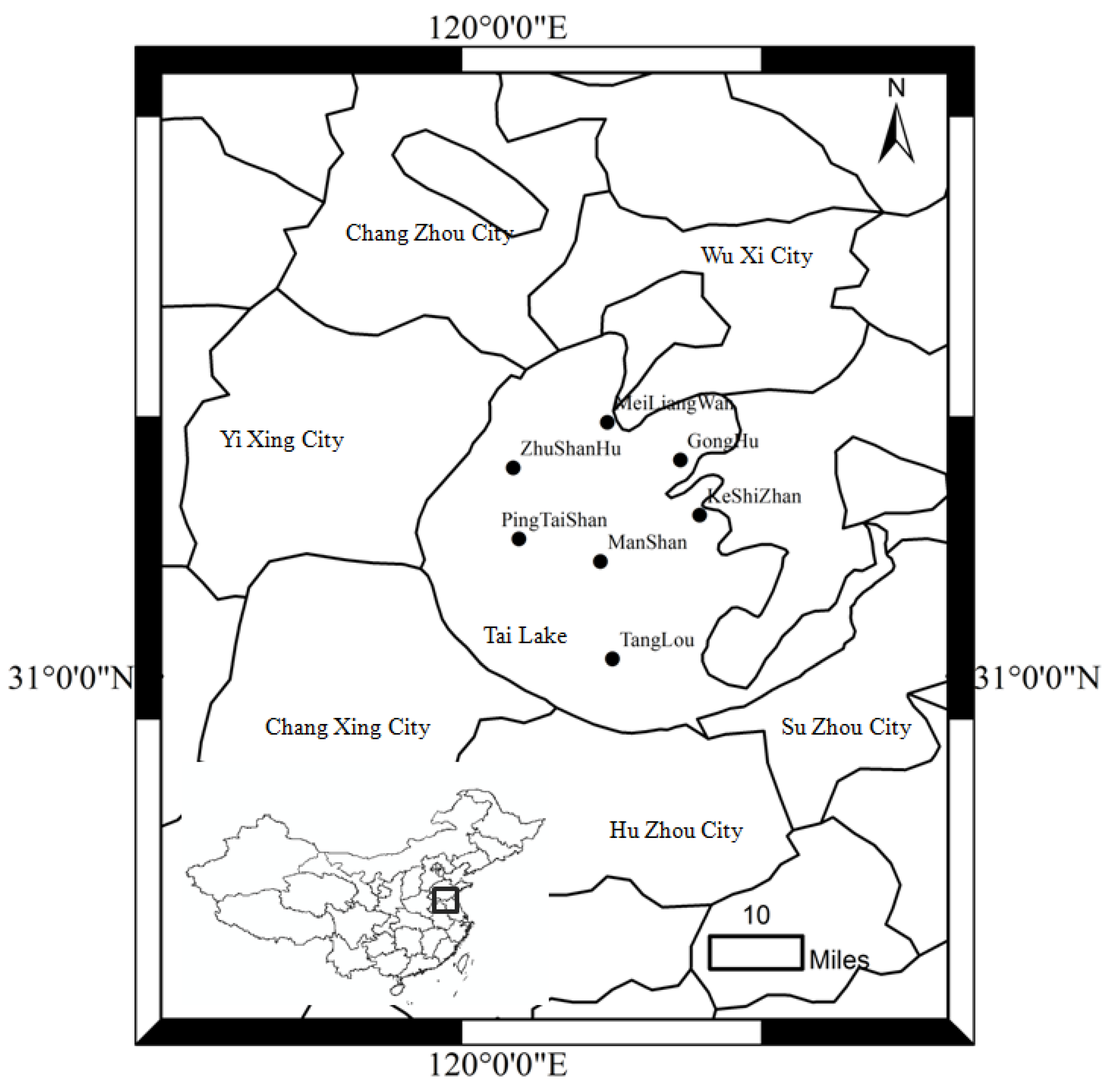
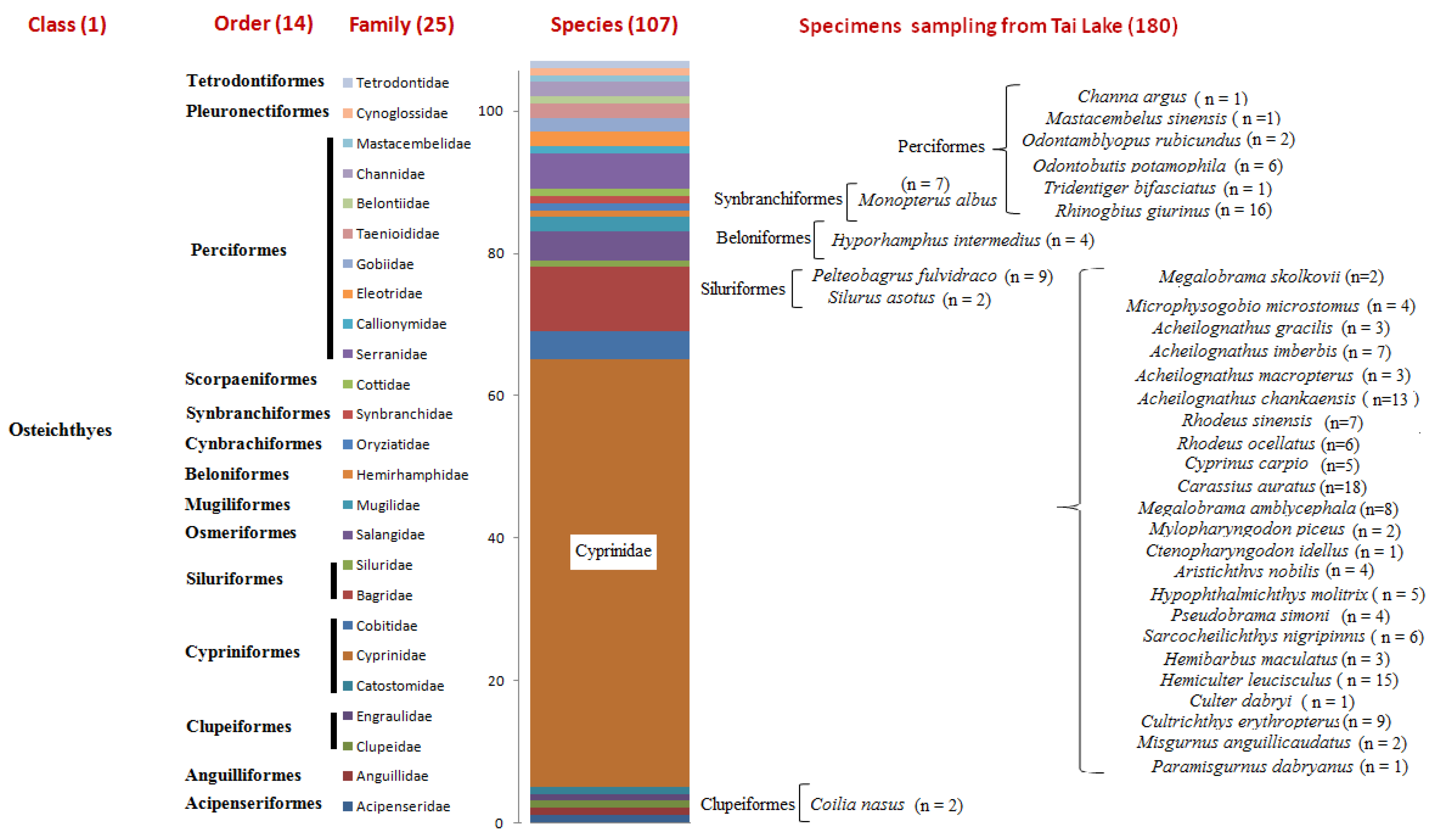
2.1. Sampling
2.2. DNA Isolation and PCR Amplification
2.3. Sequencing
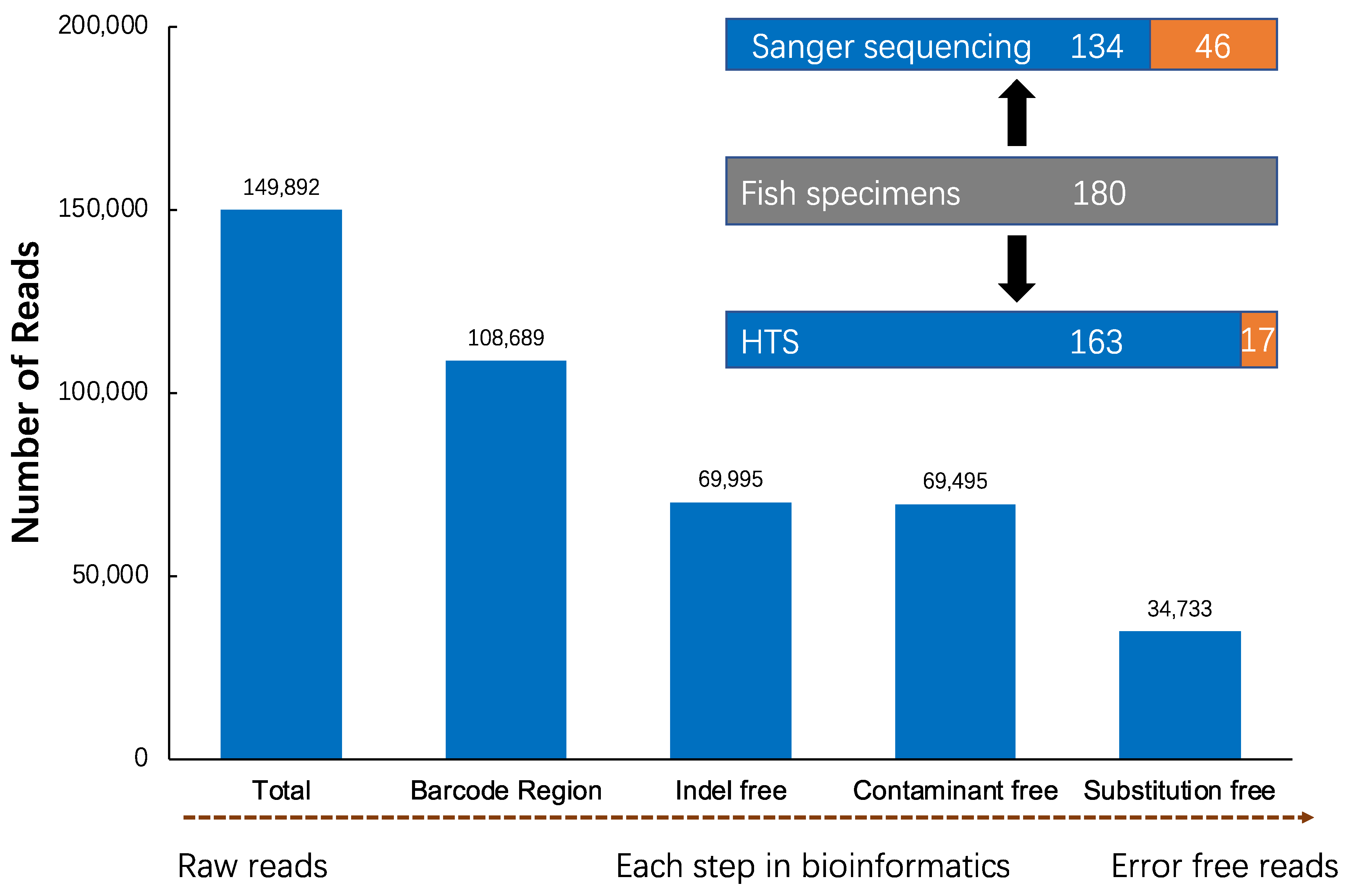
2.4. Bioinformatics
2.5. Phylogenetic Analysis
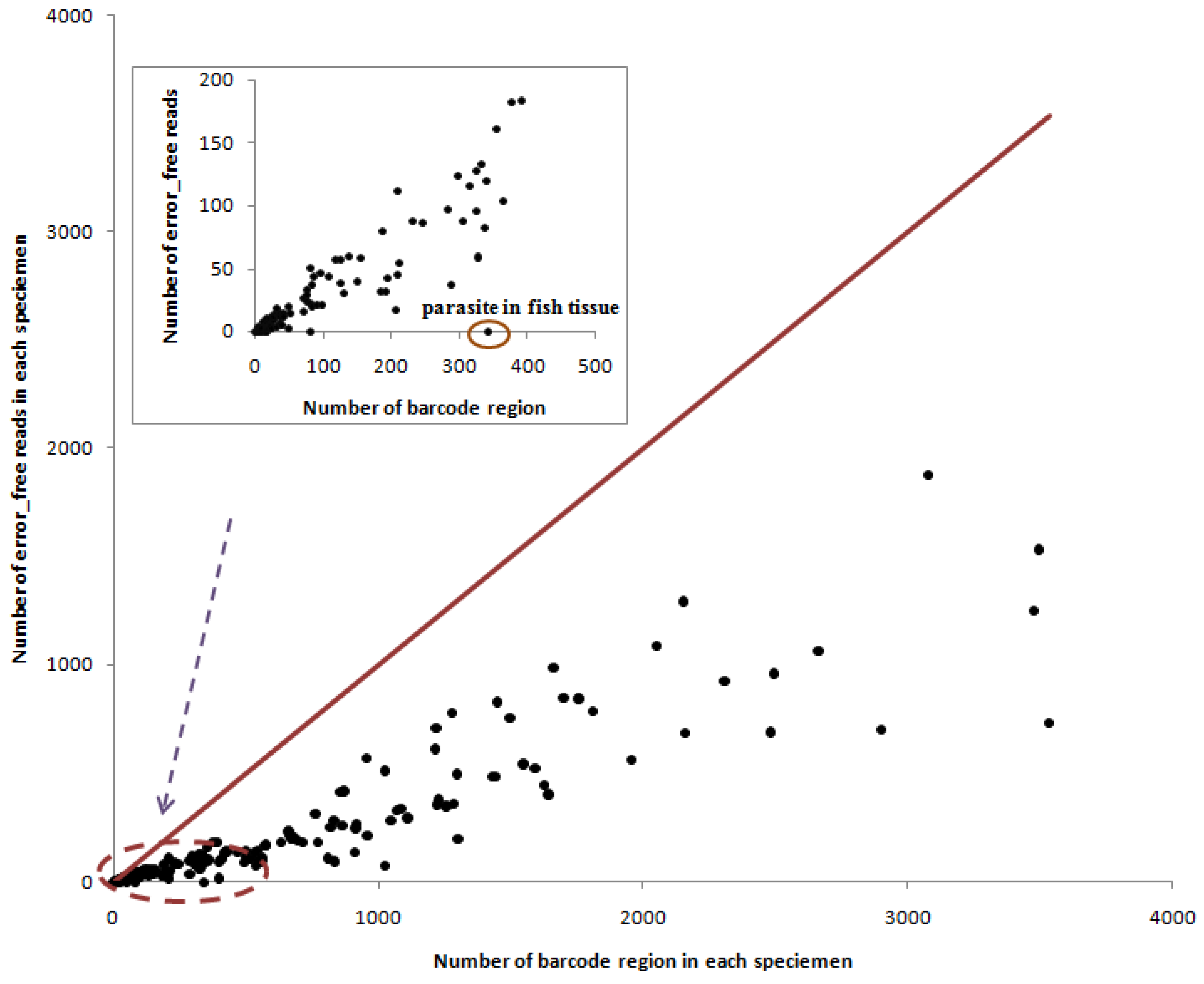
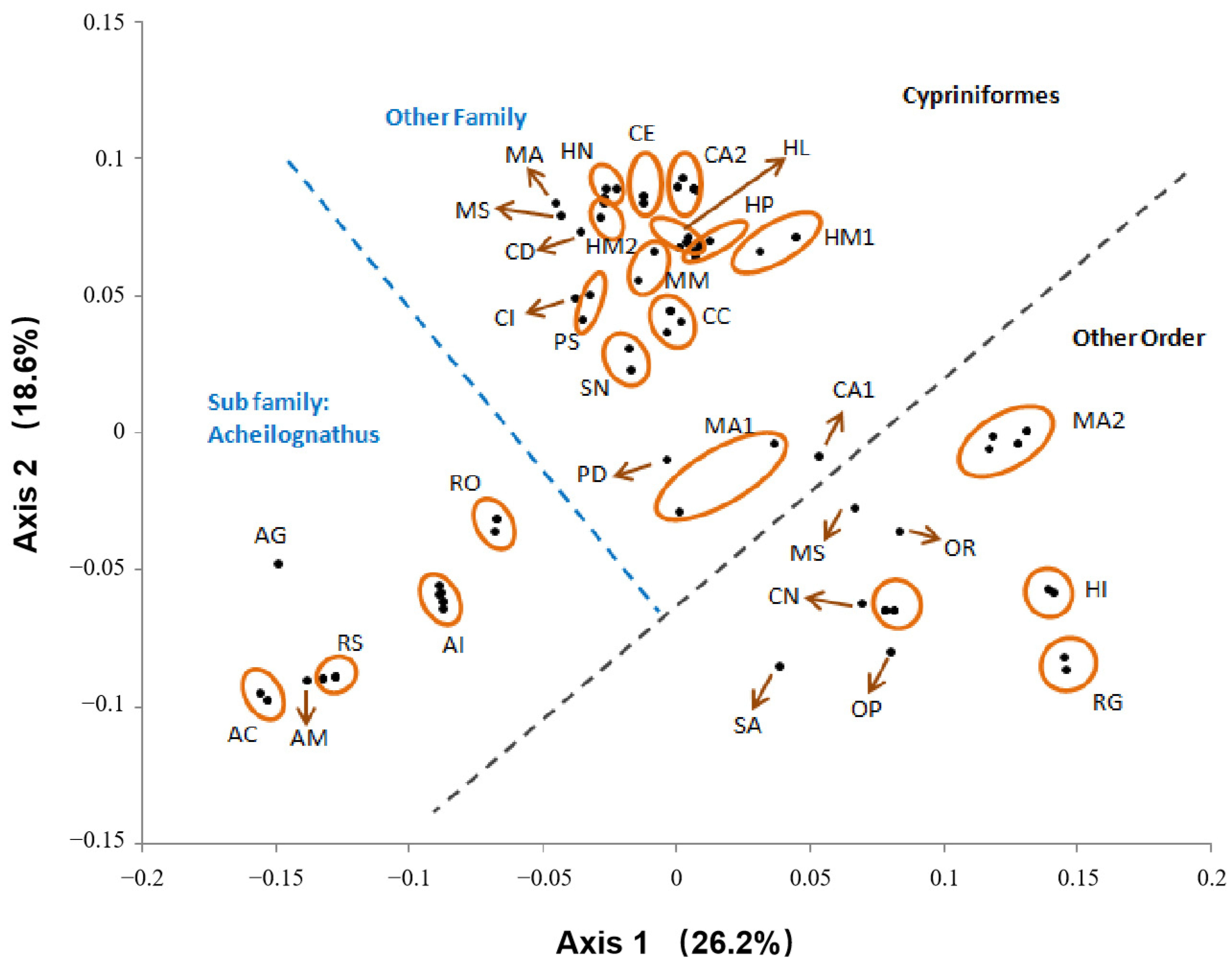
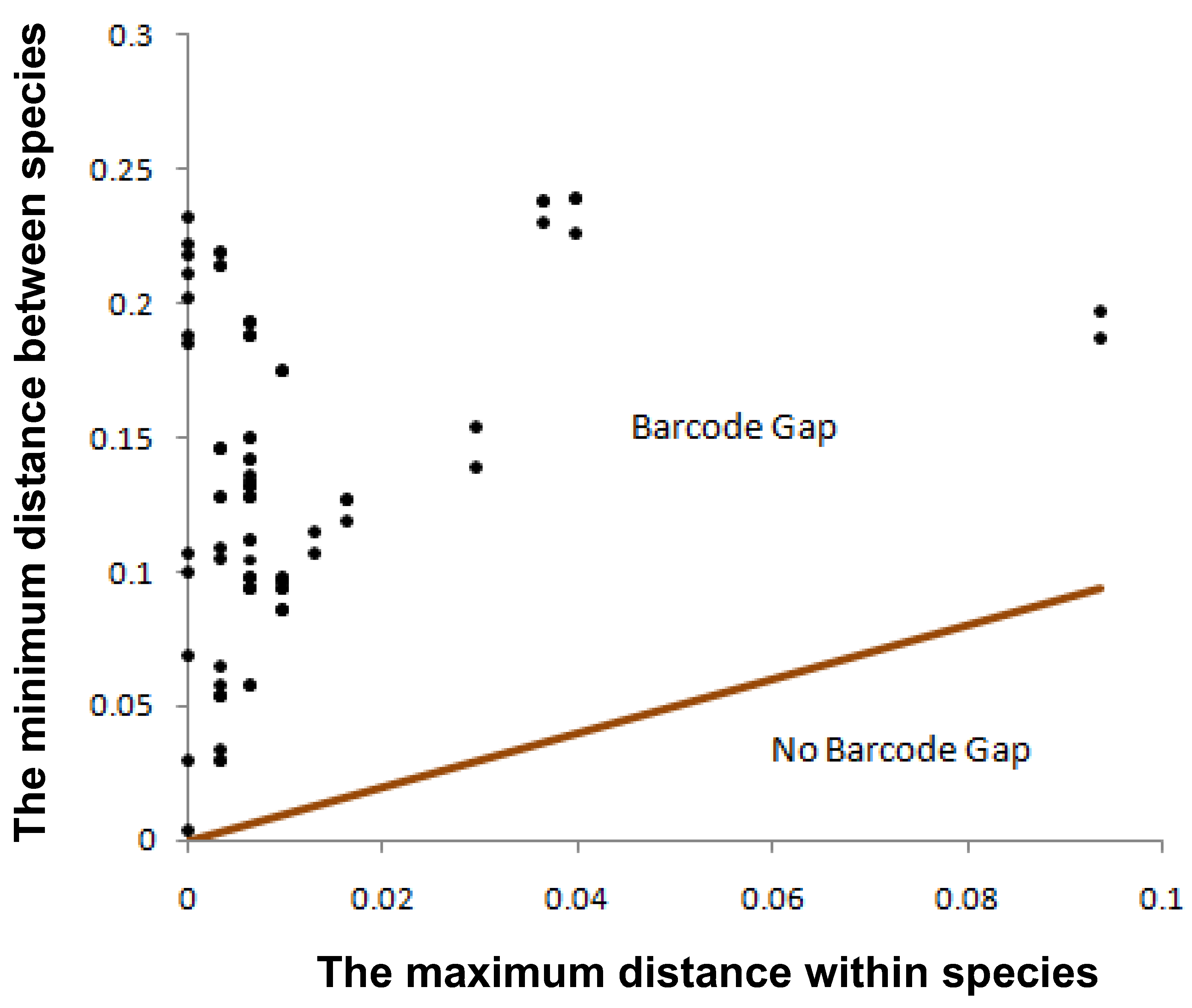
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Loreau, M.; Naeem, S.; Inchausti, P.; Bengtsson, J.; Grime, J.P.; Hector, A.; Hooper, D.U.; Huston, M.A.; Raffaelli, D.; Schmid, B.; et al. Ecology—Biodiversity and ecosystem functioning: Current knowledge and future challenges. Science 2001, 294, 804–808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paerl, H.W.; Otten, T.G. Blooms Bite the Hand That Feeds Them. Science 2013, 342, 433–434. [Google Scholar] [CrossRef] [PubMed]
- Cardinale, B.J. Biodiversity improves water quality through niche partitioning. Nature 2011, 472, 86–89. [Google Scholar] [CrossRef]
- Hajibabaei, M.; Janzen, D.H.; Burns, J.M.; Hallwachs, W.; Hebert, P.D. DNA barcodes distinguish species of tropical Lepidoptera. Proc. Natl. Acad. Sci. USA 2006, 103, 968–971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kress, W.J.; Garcia-Robledo, C.; Uriarte, M.; Erickson, D.L. DNA barcodes for ecology, evolution, and conservation. Trends Ecol. Evol. 2015, 30, 25–35. [Google Scholar] [CrossRef]
- Valentini, A.; Pompanon, F.; Taberlet, P. DNA barcoding for ecologists. Trends Ecol. Evol. 2009, 24, 110–117. [Google Scholar] [CrossRef] [PubMed]
- Meyer, C.P.; Paulay, G. DNA barcoding: Error rates based on comprehensive sampling. PLoS Biol. 2005, 3, 2229–2238. [Google Scholar] [CrossRef] [Green Version]
- Moritz, C.; Cicero, C. DNA barcoding: Promise and pitfalls. PLoS Biol. 2004, 2, 1529–1531. [Google Scholar] [CrossRef] [Green Version]
- Ratnasingham, S.; Hebert, P.D. bold: The Barcode of Life Data System. Mol. Ecol. Notes 2007, 7, 355–364. [Google Scholar] [CrossRef] [Green Version]
- Ward, R.D.; Hanner, R.; Hebert, P.D. The campaign to DNA barcode all fishes, FISH-BOL. J. Fish Biol. 2009, 74, 329–356. [Google Scholar] [CrossRef]
- Ward, R.D.; Zemlak, T.S.; Innes, B.H.; Last, P.R.; Hebert, P.D. DNA barcoding Australia’s fish species. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2005, 360, 1847–1857. [Google Scholar] [CrossRef] [PubMed]
- Casiraghi, M.; Labra, M.; Ferri, E.; Galimberti, A.; De Mattia, F. DNA barcoding: A six-question tour to improve users’ awareness about the method. Brief. Bioinform. 2010, 11, 440–453. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Zhang, X.; Zhang, W.; Sun, J.; Xie, Y.; Zhang, Y.; Jr, G.A.B.; Yu, H. Indigenous species barcode database improves the identification of zooplankton. PLoS ONE 2017, 12, e0185697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shokralla, S.; Porter, T.M.; Gibson, J.F.; Dobosz, R.; Janzen, D.H.; Hallwachs, W.; Golding, G.B.; Hajibabaei, M. Massively parallel multiplex DNA sequencing for specimen identification using an Illumina MiSeq platform. Sci. Rep. 2015, 5, 9687. [Google Scholar] [CrossRef] [Green Version]
- Shokralla, S.; Gibson, J.F.; Nikbakht, H.; Janzen, D.H.; Hallwachs, W.; Hajibabaei, M. Next-generation DNA barcoding: Using next-generation sequencing to enhance and accelerate DNA barcode capture from single specimens. Mol. Ecol. Resour. 2014, 14, 892–901. [Google Scholar] [CrossRef] [Green Version]
- Rothberg, J.M.; Hinz, W.; Rearick, T.M.; Schultz, J.; Mileski, W.; Davey, M.; Leamon, J.H.; Johnson, K.; Milgrew, M.J.; Edwards, M.; et al. An integrated semiconductor device enabling non-optical genome sequencing. Nature 2011, 475, 348–352. [Google Scholar] [CrossRef] [Green Version]
- Quail, M.A.; Smith, M.; Coupland, P.; Otto, T.D.; Harris, S.R.; Connor, T.R.; Bertoni, A.; Swerdlow, H.P.; Gu, Y. A tale of three next generation sequencing platforms: Comparison of Ion Torrent, Pacific Biosciences and Illumina MiSeq sequencers. BMC Genom. 2012, 13, 341. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Erickson, R.A.; Tang, S.; Zhang, Y.; Niu, Z.C.; Liu, H.L.; Yu, H.X. Structure and spatial patterns of macrobenthic community in Tai Lake, a large shallow lake, China. Ecol. Indic. 2016, 61, 179–187. [Google Scholar] [CrossRef]
- Ri, Y.Z.; Cheng, D. The Fisheries in Taihu; Shanghai Press of Scinece and Technology: Shanghai, China, 2005; p. 20. [Google Scholar]
- Mao, Z.; Gu, X.; Zeng, Q.; Zhou, L.; Sun, M. Status and changes of fishery resources (2009–2010) in Lake Taihu and their responses to water eutrophication. J. Lake Sci. 2011, 23, 6. [Google Scholar]
- Pages, H.A.P.; Gentleman, R.; DebRoy, S. Biostrings: String objects representing biological sequences, and matching algorithms. In R Package Version 2.30.1; R Foundation: Vienna, Austria, 2014. [Google Scholar]
- Wright, E. Database Enabled Code for Ideal Probe Hybridization Employing R. In R Version: 1.10.1; R Foundation: Vienna, Austria, 2013. [Google Scholar]
- Seqinr. Biological Sequences Retrieval and Analysis. In R Package Version 3.1.3; R Foundation: Vienna, Austria, 2014. [Google Scholar]
- Morgan, M.; Lawrence, M.; Anders, S. FASTQ input and manipulation. In R Package Version 1.22.0; R Foundation: Vienna, Austria, 2014. [Google Scholar]
- Phuong, T.M.; Do, C.B.; Edgar, R.C.; Batzoglou, S. Multiple alignment of protein sequences with repeats and rearrangements. Nucleic Acids Res. 2006, 34, 5932–5942. [Google Scholar] [CrossRef] [Green Version]
- Kimura, M. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J. Mol. Evol. 1980, 16, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Oksanen, J.; Blanchet, F.G.; Kindt, R.; Legendre, P.; Minchin, P.R.; O’Hara, R.B.; Simpson, G.L.; Solymos, P.; Stevens, M.H.H.; Wagner, H. Community Ecology Package. In R Package Version 2.3.5; R Foundation: Vienna, Austria, 2016. [Google Scholar]
- Zhang, Z.; Schwartz, S.; Wagner, L.; Miller, W. A greedy algorithm for aligning DNA sequences. J. Comput. Biol. 2000, 7, 203–214. [Google Scholar] [CrossRef] [PubMed]
- Song, H.; Buhay, J.E.; Whiting, M.F.; Crandall, K.A. Many species in one: DNA barcoding overestimates the number of species when nuclear mitochondrial pseudogenes are coamplified. Proc. Natl. Acad. Sci. USA 2008, 105, 13486–13491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Brown, S.; Collins, R.; Boyer, S.; Lefort, M.-C.; Malumbres-Olarte, J.; Vink, C.; Cruickshank, R. Species Identity and Evolution in R. In R Package Version 1.3.0; R Foundation: Vienna, Austria, 2013. [Google Scholar]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [Green Version]
- Puillandre, N.; Lambert, A.; Brouillet, S.; Achaz, G. ABGD, Automatic Barcode Gap Discovery for primary species delimitation. Mol. Ecol. 2012, 21, 1864–1877. [Google Scholar] [CrossRef]
- Port, J.A.; O’Donnell, J.L.; Romero-Maraccini, O.C.; Leary, P.R.; Litvin, S.Y.; Nickols, K.J.; Yamahara, K.M.; Kelly, R.P. Assessing vertebrate biodiversity in a kelp forest ecosystem using environmental DNA. Mol. Ecol. 2016, 25, 527–541. [Google Scholar] [CrossRef] [Green Version]
- Yu, D.W.; Ji, Y.Q.; Emerson, B.C.; Wang, X.Y.; Ye, C.X.; Yang, C.Y.; Ding, Z.L. Biodiversity soup: Metabarcoding of arthropods for rapid biodiversity assessment and biomonitoring. Methods Ecol. Evol. 2012, 3, 613–623. [Google Scholar] [CrossRef] [Green Version]
- Miller, S.E. DNA barcoding and the renaissance of taxonomy. Proc. Natl. Acad. Sci. USA 2007, 104, 4775–4776. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.; Ma, X.; Shen, Y.; Mao, Y.; He, S. The fish diversity in the upper reaches of the Salween River, Nujiang River, revealed by DNA barcoding. Sci. Rep. 2015, 5, 17437. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Hanner, R. Molecular approach to the identification of fish in the South China Sea. PLoS ONE 2012, 7, e30621. [Google Scholar] [CrossRef] [PubMed]
- Knebelsberger, T.; Landi, M.; Neumann, H.; Kloppmann, M.; Sell, A.F.; Campbell, P.D.; Laakmann, S.; Raupach, M.J.; Carvalho, G.R.; Costa, F.O. A reliable DNA barcode reference library for the identification of the North European shelf fish fauna. Mol. Ecol. Resour. 2014, 14, 1060–1071. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Provan, J.; Gao, L.M.; Li, D.Z. Sampling strategy and potential utility of indels for DNA barcoding of closely related plant species: A case study in taxus. Int. J. Mol. Sci. 2012, 13, 8740–8751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galan, M.; Pages, M.; Cosson, J.F. Next-generation sequencing for rodent barcoding: Species identification from fresh, degraded and environmental samples. PLoS ONE 2012, 7, e48374. [Google Scholar] [CrossRef] [PubMed]
- Pratyush, D.D.; Tiwari, S.; Kumar, A.; Singh, S.K. A new approach to touch down method using betaine as co-solvent for increased specificity and intensity of GC rich gene amplification. Gene 2012, 497, 269–272. [Google Scholar] [CrossRef]
- Bragg, L.M.; Stone, G.; Butler, M.K.; Hugenholtz, P.; Tyson, G.W. Shining a light on dark sequencing: Characterising errors in Ion Torrent PGM data. PLoS Comput. Biol. 2013, 9, e1003031. [Google Scholar] [CrossRef] [Green Version]
- Piry, S.; Guivier, E.; Realini, A.; Martin, J.F. |SE|S|AM|E| Barcode: NGS-oriented software for amplicon characterization--application to species and environmental barcoding. Mol. Ecol. Resour. 2012, 12, 1151–1157. [Google Scholar] [CrossRef]
- Meglecz, E.; Piry, S.; Desmarais, E.; Galan, M.; Gilles, A.; Guivier, E.; Pech, N.; Martin, J.F. SESAME (SEquence Sorter & AMplicon Explorer): Genotyping based on high-throughput multiplex amplicon sequencing. Bioinformatics 2011, 27, 277–278. [Google Scholar]
- Minamoto, T.; Yamanaka, H.; Takahara, T.; Honjo, M.N.; Kawabata, Z.I. Surveillance of fish species composition using environmental DNA. Limnology 2012, 13, 193–197. [Google Scholar] [CrossRef] [Green Version]
- Smith, M.A.; Bertrand, C.; Crosby, K.; Eveleigh, E.S.; Fernandez-Triana, J.; Fisher, B.L.; Gibbs, J.; Hajibabaei, M.; Hallwachs, W.; Hind, K.; et al. Wolbachia and DNA barcoding insects: Patterns, potential, and problems. PLoS ONE 2012, 7, e36514. [Google Scholar] [CrossRef] [Green Version]
- Viñas, L.; Besada, V.; Sericano, J.L. Sampling of fish, benthic species, and seabird eggs in pollution assessment A2—Pawliszyn, Janusz. In Comprehensive Sampling and Sample Preparation; Academic Press: Oxford, UK, 2012; pp. 349–372. [Google Scholar]
- April, J.; Mayden, R.L.; Hanner, R.H.; Bernatchez, L. Genetic calibration of species diversity among North America’s freshwater fishes. Proc. Natl. Acad. Sci. USA 2011, 108, 10602–10607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hubert, N.; Hanner, R.; Holm, E.; Mandrak, N.E.; Taylor, E.; Burridge, M.; Watkinson, D.; Dumont, P.; Curry, A.; Bentzen, P.; et al. Identifying Canadian freshwater fishes through DNA barcodes. PLoS ONE 2008, 3, e2490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mabragana, E.; Diaz de Astarloa, J.M.; Hanner, R.; Zhang, J.; Gonzalez Castro, M. DNA barcoding identifies Argentine fishes from marine and brackish waters. PLoS ONE 2011, 6, e28655. [Google Scholar] [CrossRef] [Green Version]
- McCusker, M.R.; Denti, D.; Van Guelpen, L.; Kenchington, E.; Bentzen, P. Barcoding Atlantic Canada’s commonly encountered marine fishes. Mol. Ecol. Resour. 2013, 13, 177–188. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Li, Q.; Kong, L.; Yu, H. How DNA Barcodes Complement Taxonomy and Explore Species Diversity: The Case Study of a Poorly Understood Marine Fauna. PLoS ONE 2011, 6, e21326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hebert, P.D.N.; Cywinska, A.; Ball, S.L.; deWaard, J.R. Biological identifications through DNA barcodes. Proc. R. Soc. Lond. B Biol. Sci. 2003, 270, 313–321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hebert, P.D.; Ratnasingham, S.; deWaard, J.R. Barcoding animal life: Cytochrome c oxidase subunit 1 divergences among closely related species. Proc. Biol. Sci. R. Soc. 2003, 270 (Suppl. S1), S96–S99. [Google Scholar] [CrossRef] [Green Version]
- Hebert, P.D.N.; Stoeckle, M.Y.; Zemlak, T.S.; Francis, C.M. Identification of Birds through DNA Barcodes. PLoS Biol. 2004, 2, e312. [Google Scholar] [CrossRef] [Green Version]
- Steinke, D.; Zemlak, T.S.; Hebert, P.D. Barcoding nemo: DNA-based identifications for the ornamental fish trade. PLoS ONE 2009, 4, e6300. [Google Scholar] [CrossRef]
- Weitschek, E.; Van Velzen, R.; Felici, G.; Bertolazzi, P. BLOG 2.0: A software system for character-based species classification with DNA Barcode sequences. What it does, how to use it. Mol. Ecol. Resour. 2013, 13, 1043–1046. [Google Scholar] [CrossRef]
- Hebert, P.D.; Penton, E.H.; Burns, J.M.; Janzen, D.H.; Hallwachs, W. Ten species in one: DNA barcoding reveals cryptic species in the neotropical skipper butterfly Astraptes fulgerator. Proc. Natl. Acad. Sci. USA 2004, 101, 14812–14817. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Comparisons | Min | Mean | Max | SE | |
|---|---|---|---|---|---|
| Within Species | 593 | 0 | 0.30% | 9.35% | 0.03 |
| Within Genera | 819 | 0 | 3.82% | 21.07% | 0.23 |
| Within Families | 6495 | 0 | 17.40% | 29.31% | 0.09 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mu, Y.; Song, C.; Yang, J.; Zhang, Y.; Zhang, X. Next-Generation DNA Barcoding for Fish Identification Using High-Throughput Sequencing in Tai Lake, China. Water 2023, 15, 774. https://doi.org/10.3390/w15040774
Mu Y, Song C, Yang J, Zhang Y, Zhang X. Next-Generation DNA Barcoding for Fish Identification Using High-Throughput Sequencing in Tai Lake, China. Water. 2023; 15(4):774. https://doi.org/10.3390/w15040774
Chicago/Turabian StyleMu, Yawen, Chao Song, Jianghua Yang, Yong Zhang, and Xiaowei Zhang. 2023. "Next-Generation DNA Barcoding for Fish Identification Using High-Throughput Sequencing in Tai Lake, China" Water 15, no. 4: 774. https://doi.org/10.3390/w15040774
APA StyleMu, Y., Song, C., Yang, J., Zhang, Y., & Zhang, X. (2023). Next-Generation DNA Barcoding for Fish Identification Using High-Throughput Sequencing in Tai Lake, China. Water, 15(4), 774. https://doi.org/10.3390/w15040774