Modification of Aquifer Pore-Water by Static Diffusion Using Nano-Zero-Valent Metals

Abstract
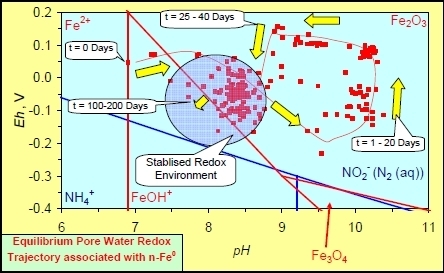
:
1. Introduction
2. Materials and Methods
2.1. Experimental Micro-SDR
- Fresh water containing n-ZVM + Ca-montmorillonite (MR9–MR11)—Figure A9, Figure A10 and Figure A11 in Appendix.
- Saline water containing n-ZVM + Ca-montmorillonite (MR12–MR16)—Figure A12, Figure A13, Figure A14, Figure A15 and Figure A16 in Appendix.
2.2. Characterization of n-ZVM

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Spherical Form | Cube Form | ||||||
|---|---|---|---|---|---|---|---|
| m | mm | m2 | g | m2 g Fe0 | m2 | g | m2 g Fe0 |
| 2.00 10−03 | 2 | 1.26 10−05 | 3.30 10−02 | 0.00038 | 6.00 10−06 | 6.30 10−02 | 0.00010 |
| 1.00 10−03 | 1 | 3.14 10−06 | 4.12 10−03 | 0.00076 | 1.50 10−06 | 7.87 10−03 | 0.00019 |
| 5.00 10−04 | 0.5 | 7.85 10−07 | 5.15 10−04 | 0.00152 | 3.75 10−07 | 9.84 10−04 | 0.00038 |
| 1.00 10−04 | 0.1 | 3.14 10−08 | 4.12 10−06 | 0.00762 | 1.50 10−08 | 7.87 10−06 | 0.00191 |
| 6.60 10−05 | 0.066 | 1.37 10−08 | 1.19 10−06 | 0.01155 | 6.53 10−09 | 2.26 10−06 | 0.00289 |
| 4.40 10−05 | 0.044 | 6.08 10−09 | 3.51 10−07 | 0.01732 | 2.90 10−09 | 6.71 10−07 | 0.00433 |
| 1.00 10−06 | 0.001 | 3.14 10−12 | 4.12 10−12 | 0.76200 | 1.50 10−12 | 7.87 10−12 | 0.19050 |
| 1.00 10−07 | 0.0001 | 3.14 10−14 | 4.12 10−15 | 7.62002 | 1.50 10−14 | 7.87 10−15 | 1.90500 |
| 1.00 10−08 | 0.00001 | 3.14 10−16 | 4.12 10−18 | 76.20015 | 1.50 10−16 | 7.87 10−18 | 19.05004 |
2.2.1. Negative activation energies
- Increasing the availability of OH− (i.e., increasing pH as the concentration of OH- (Mol L−1) = 10(14−pH)).
- Increasing the rate of degradation of HO2− to H2O + 0.5O2 (i.e., decreasing Eh, as Eh is a direct measure of the ratio [HO2−]:[OH− + HO2−]).
CO2 + 2H+ + 2e− = CO + H2O Eh = −0.53 V
CO2 + 6H+ + 6e− = CH3OH + H2O Eh = −0.38 V
2.3. Dissolved Organic Matter (DOM)

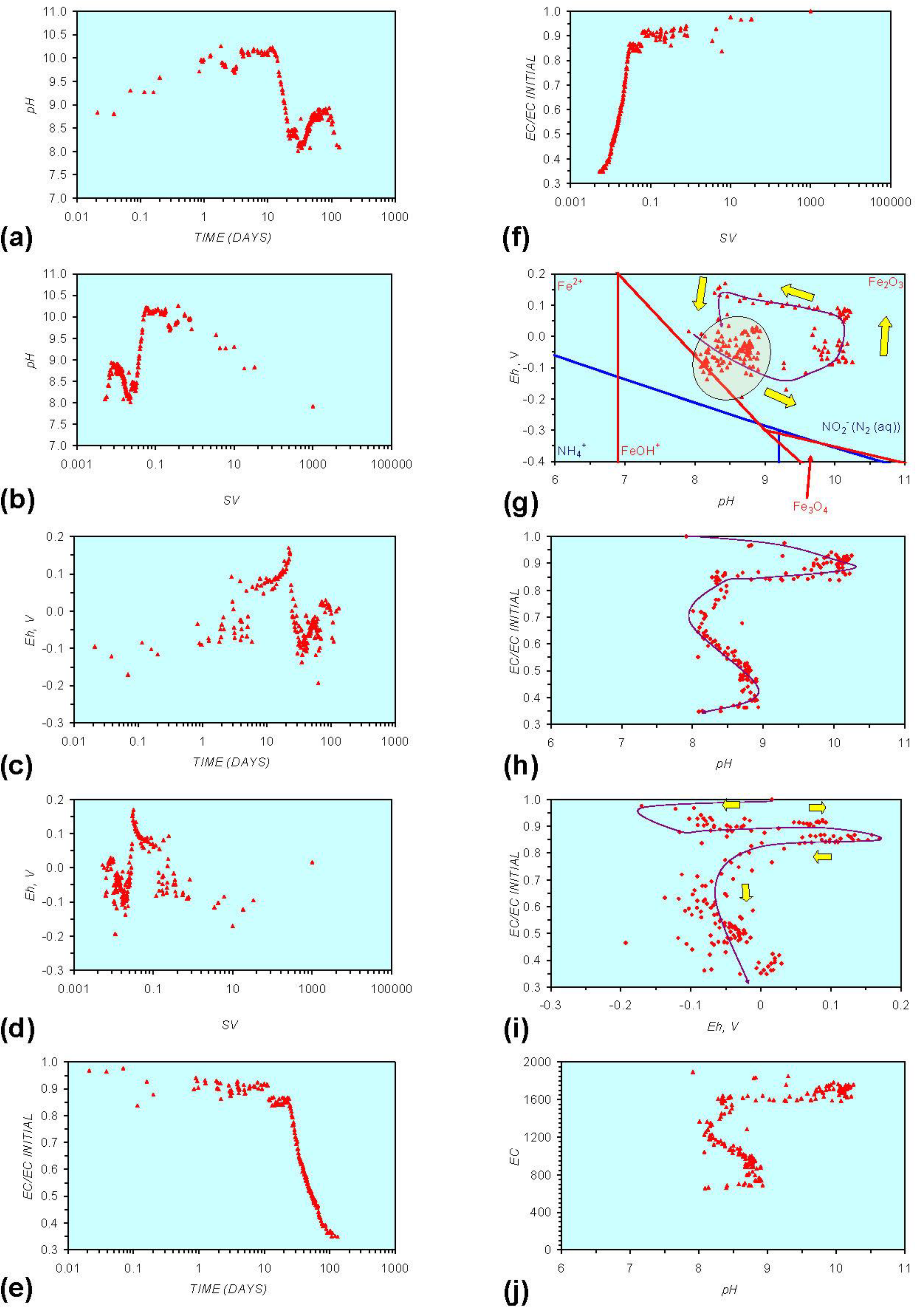
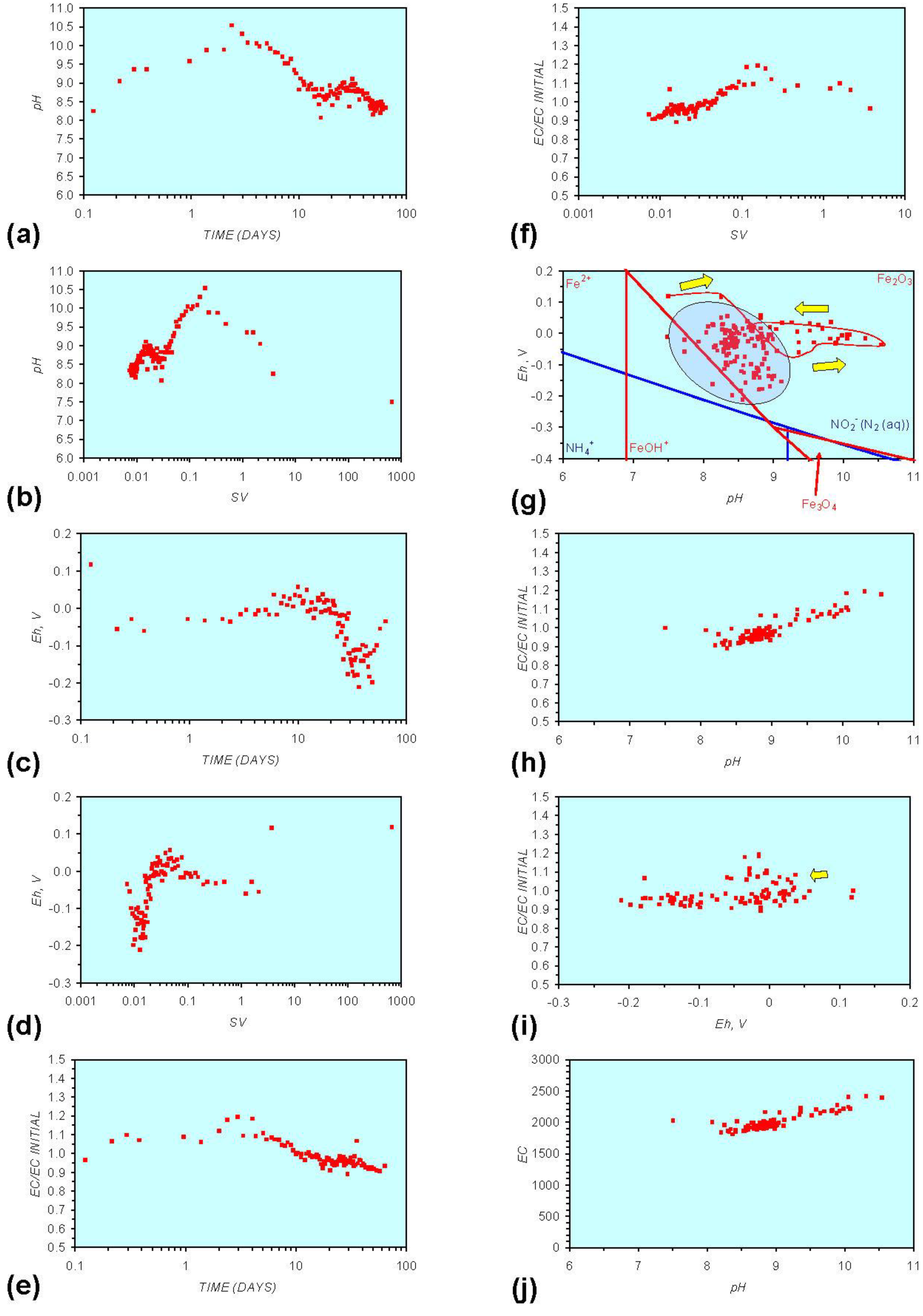
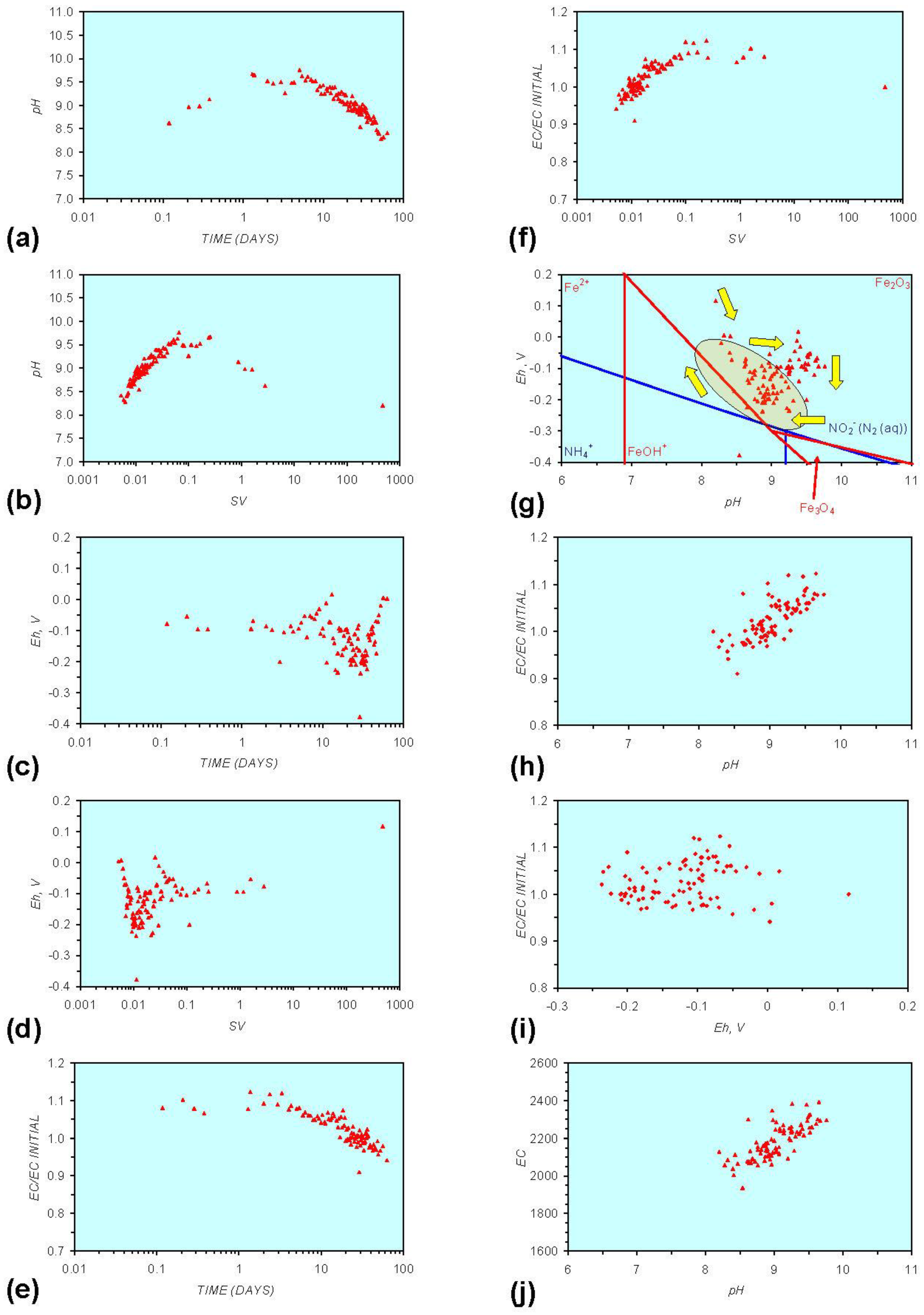
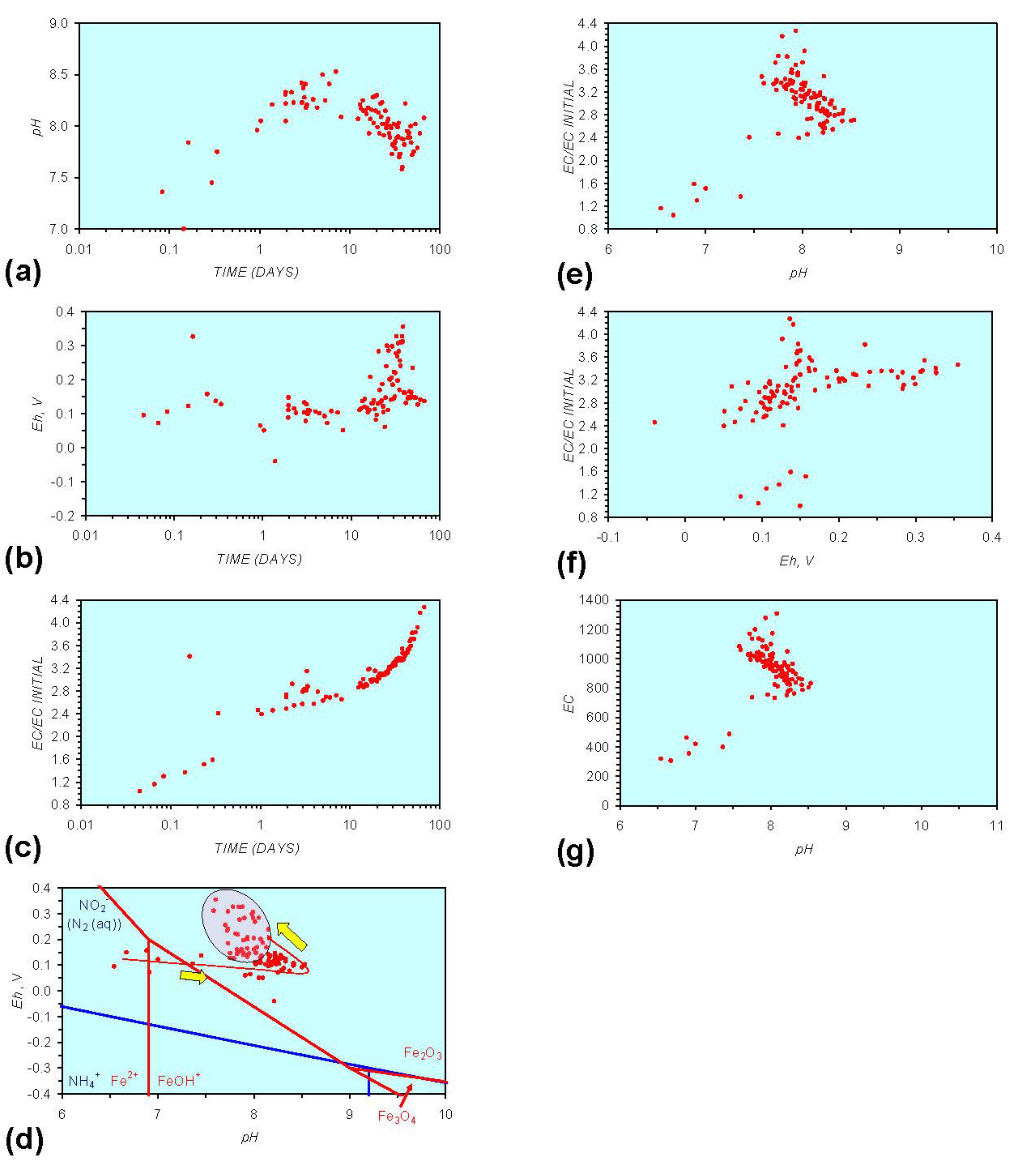
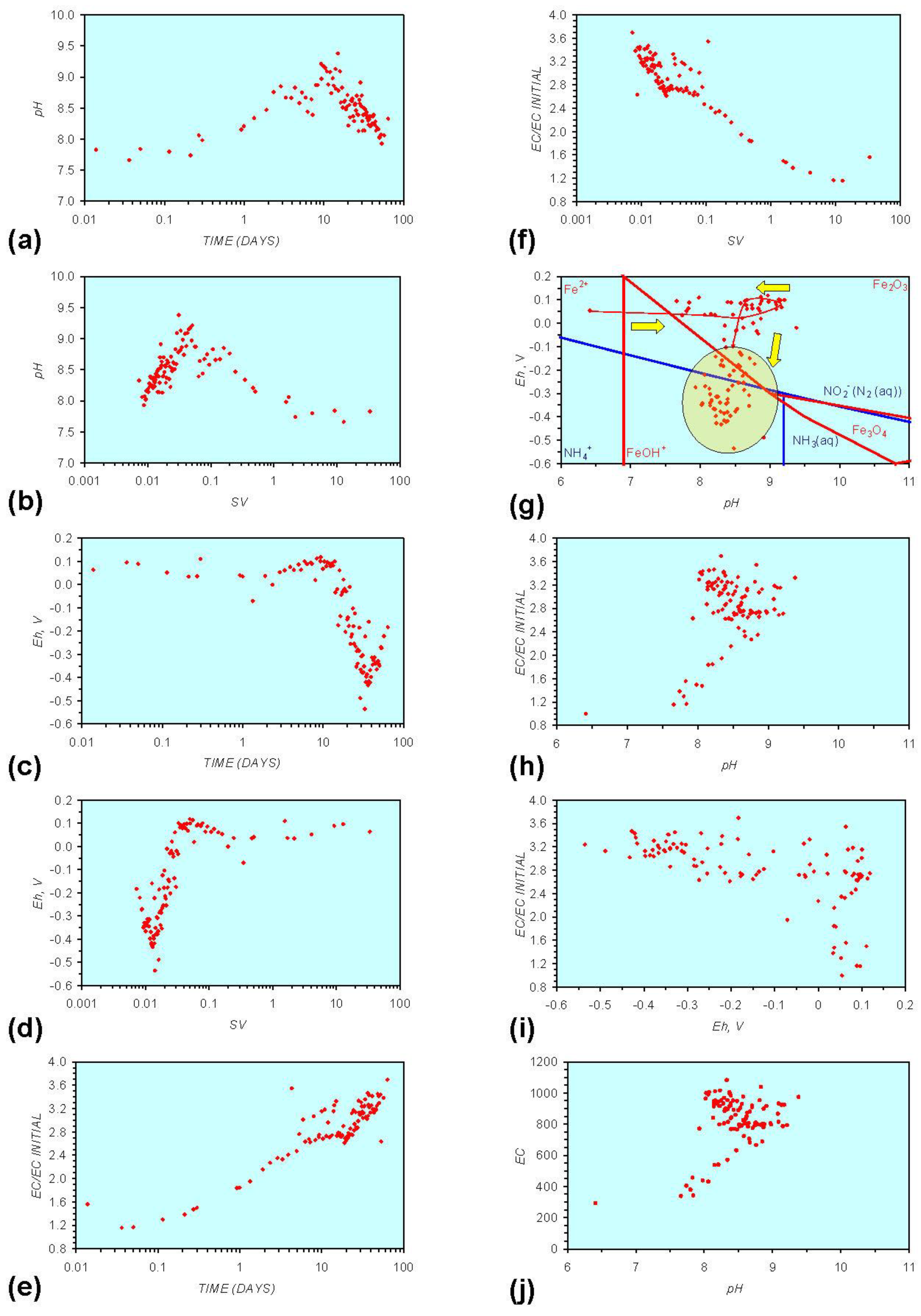
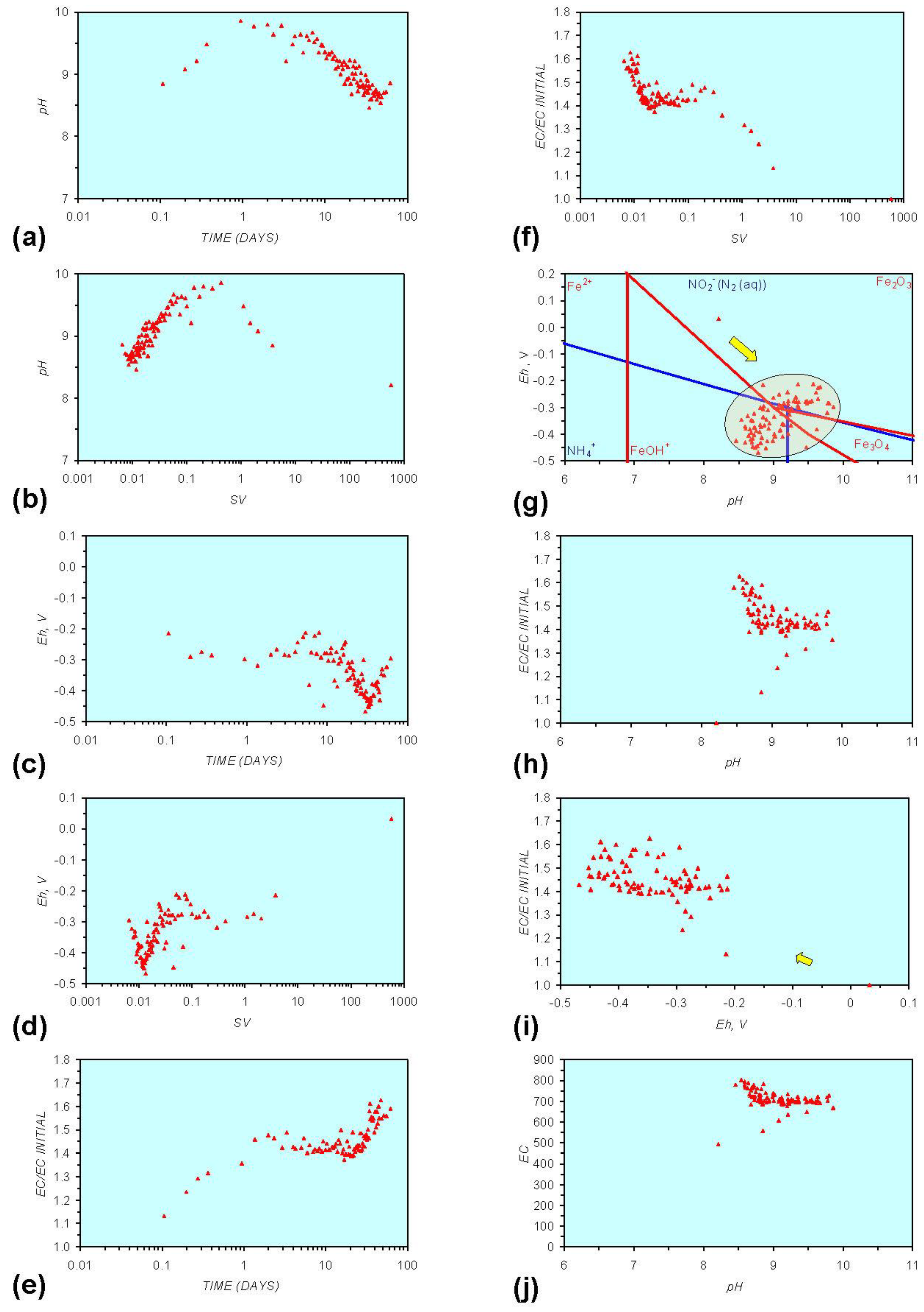
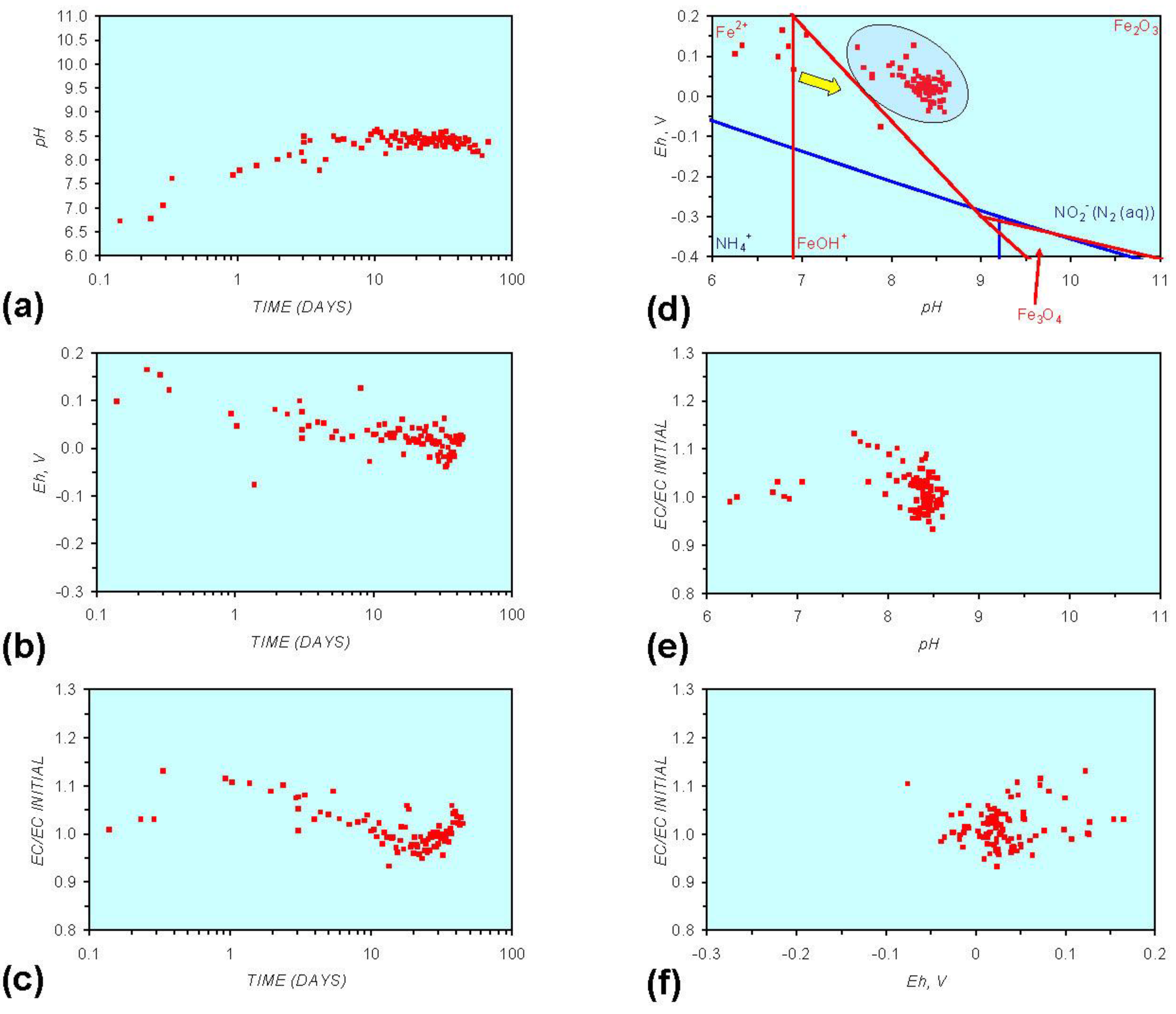
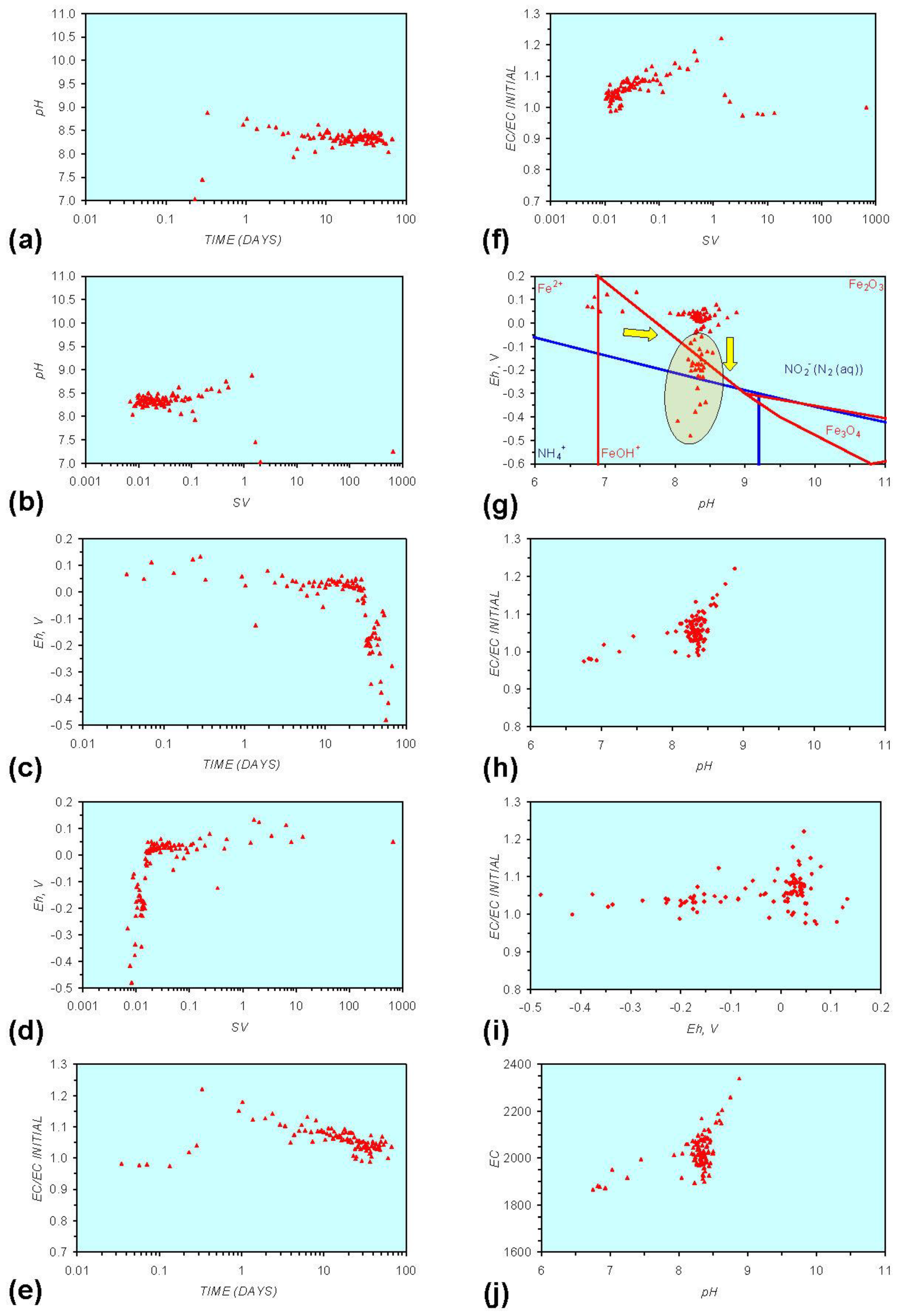
3. Experimental Results: n-ZVM + Water
3.1. Source Water
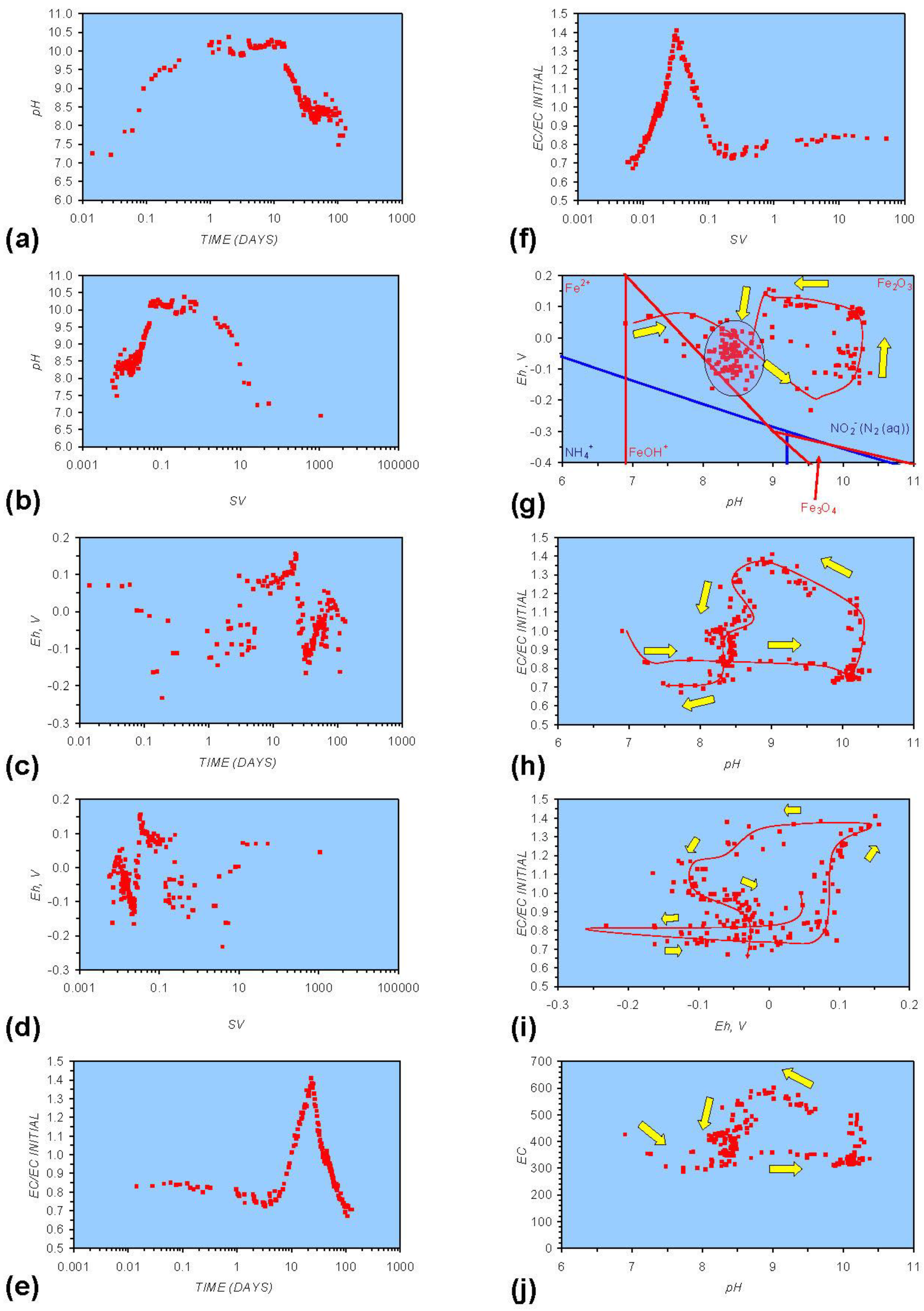
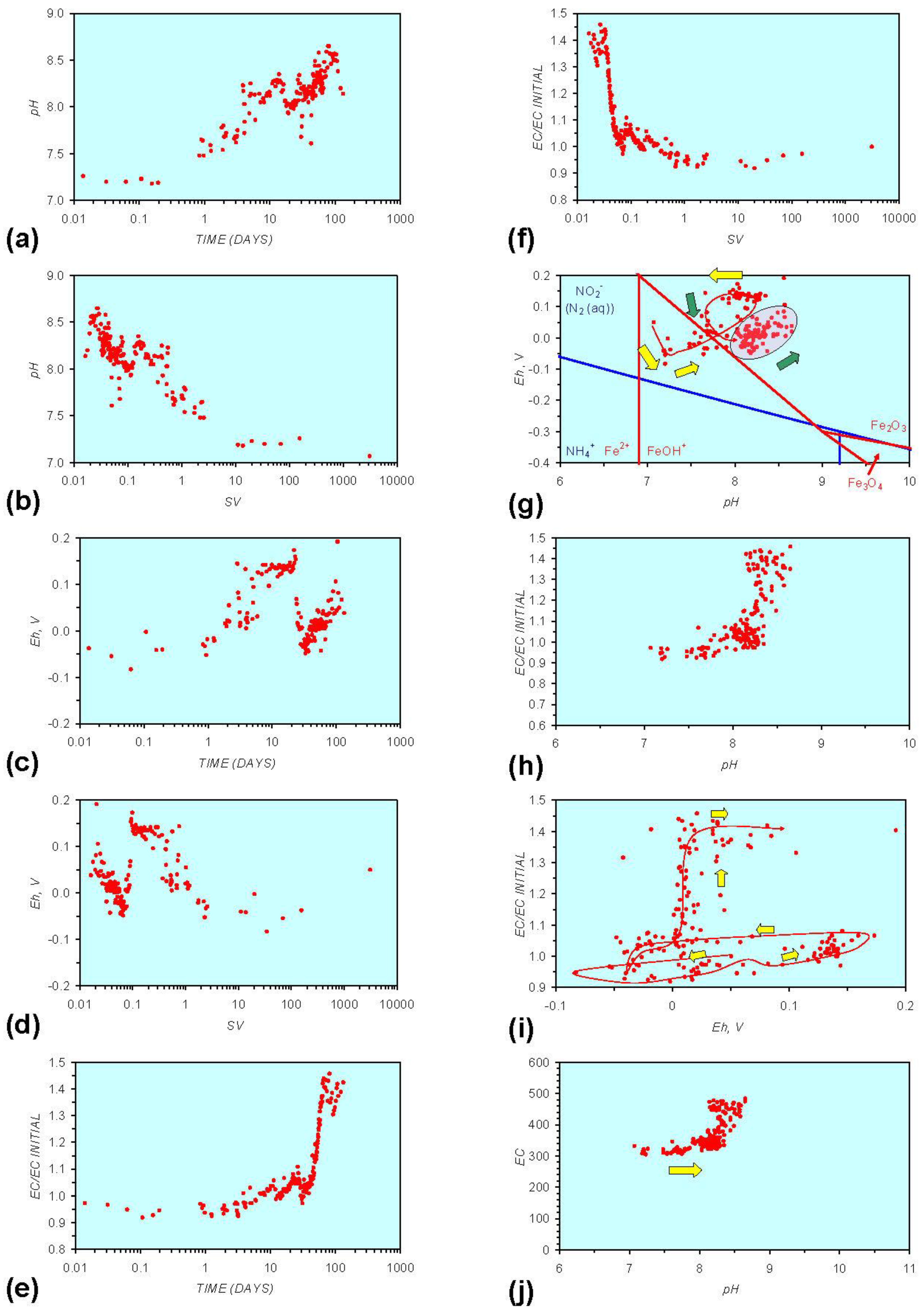
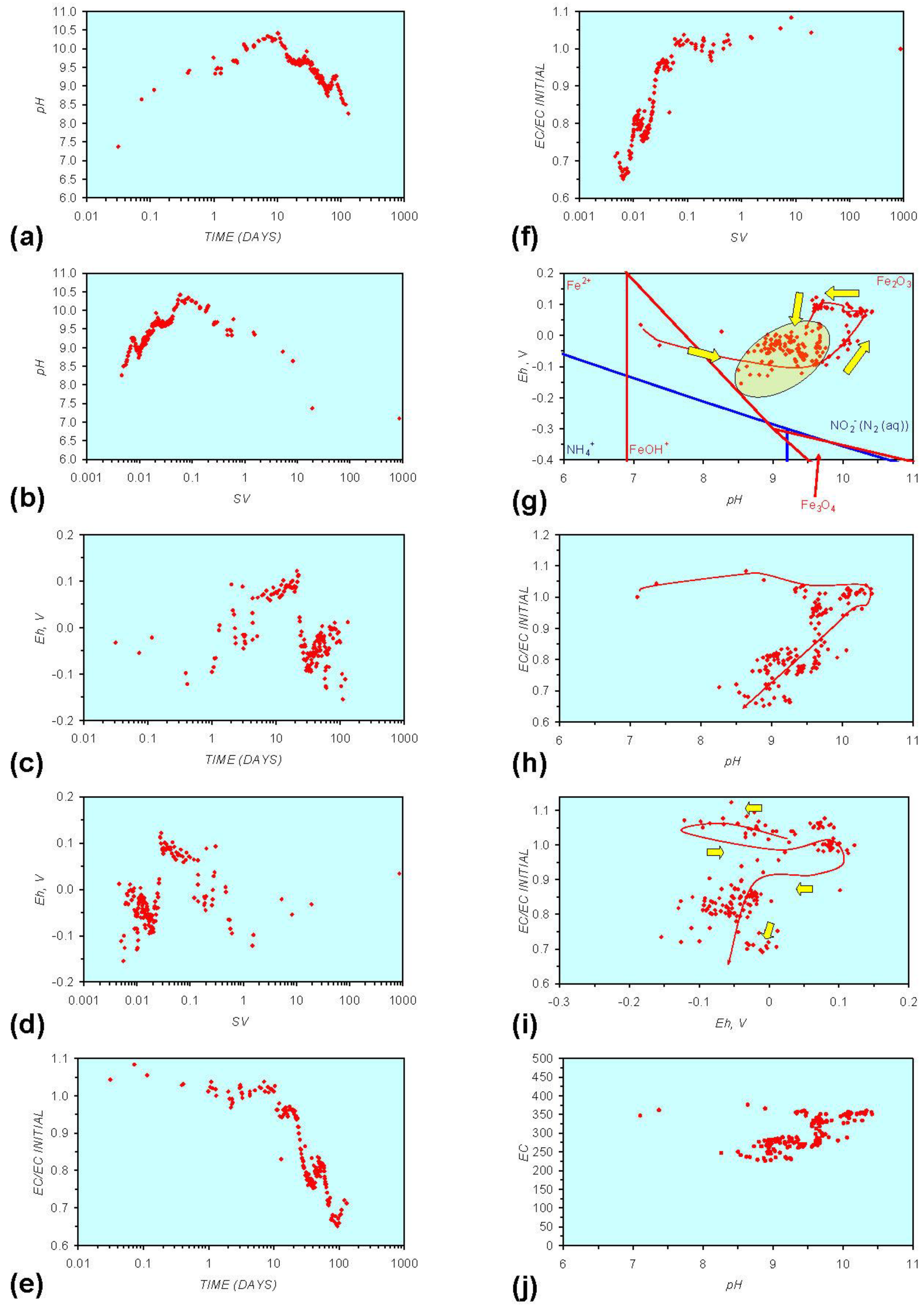
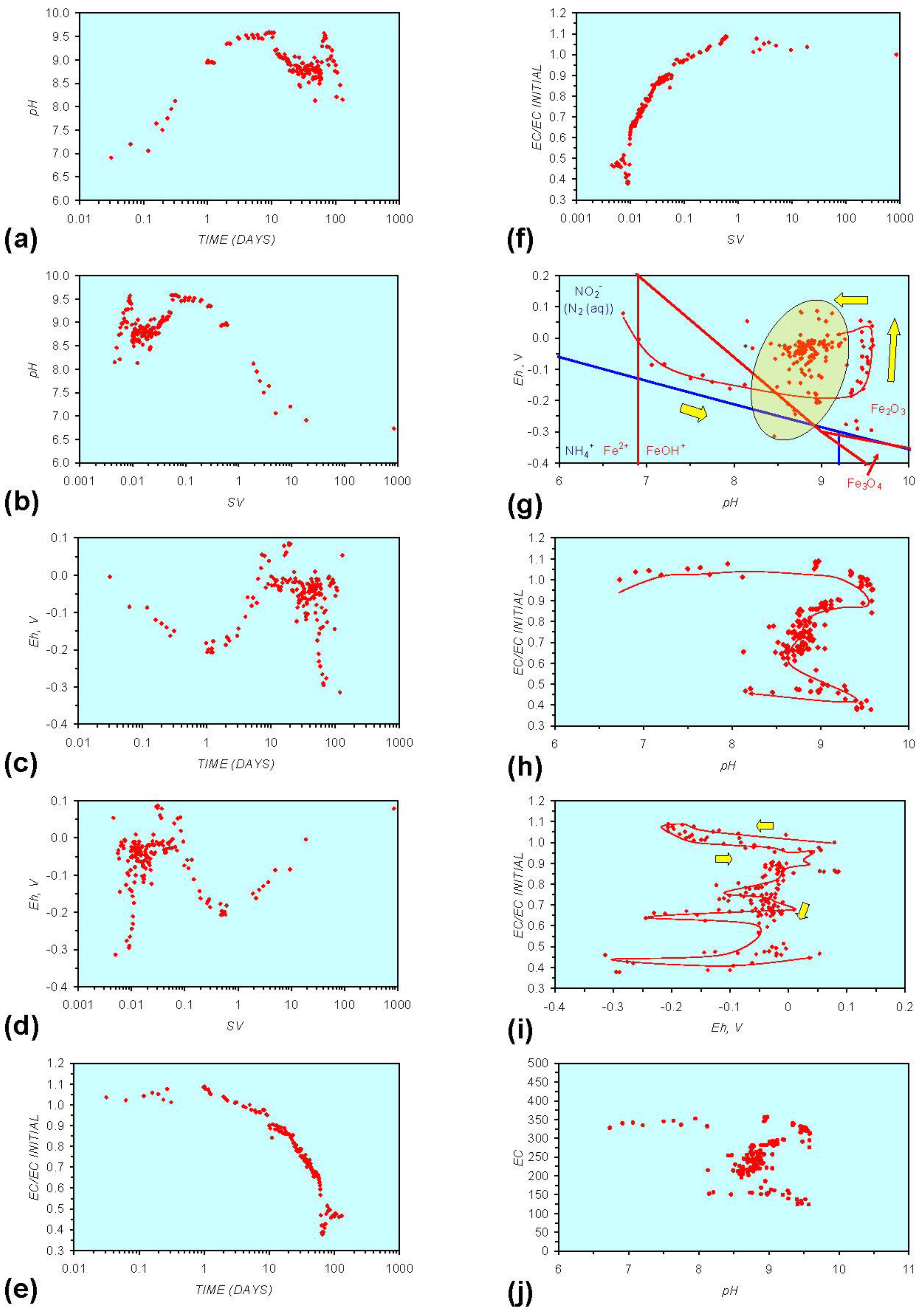
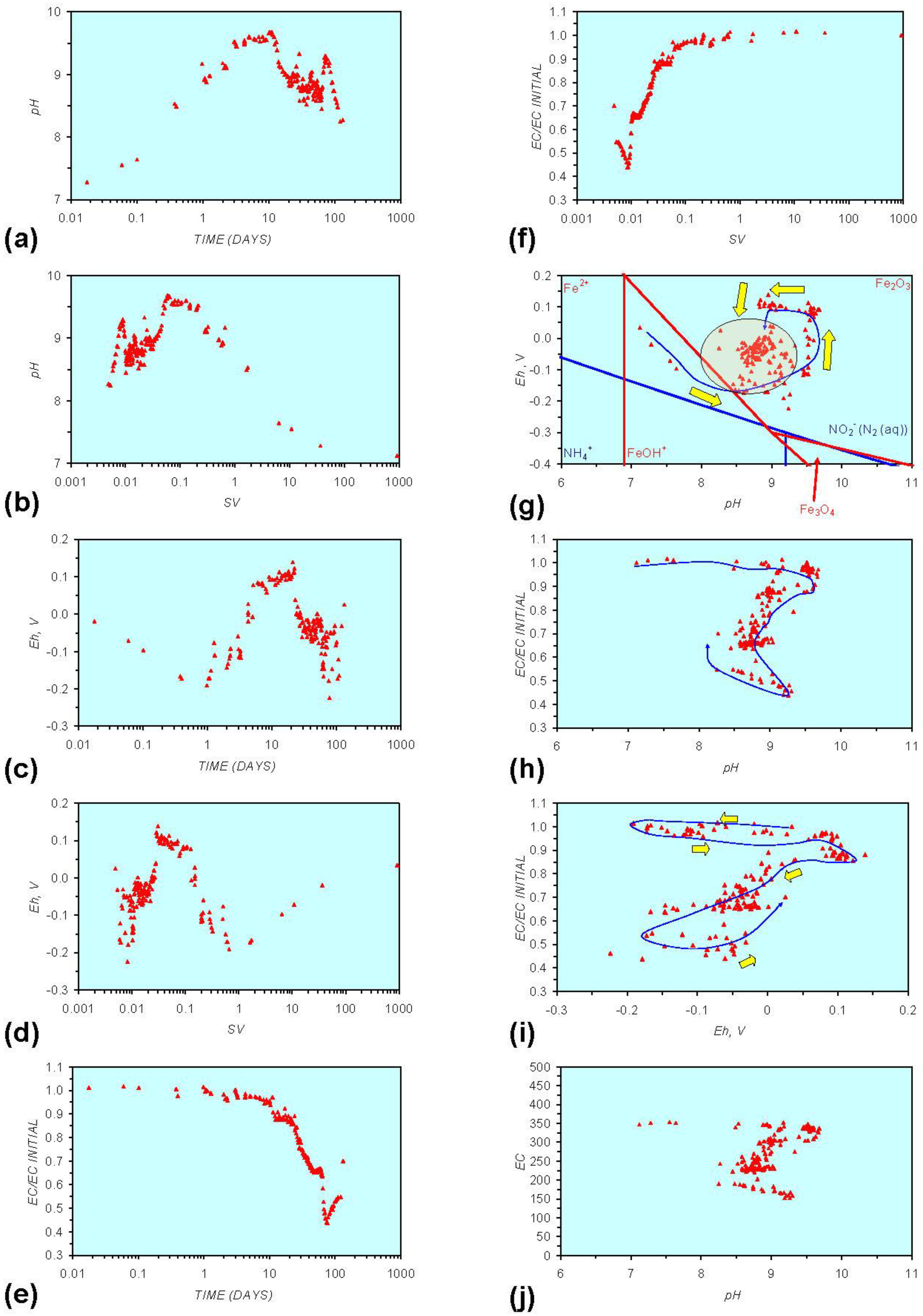
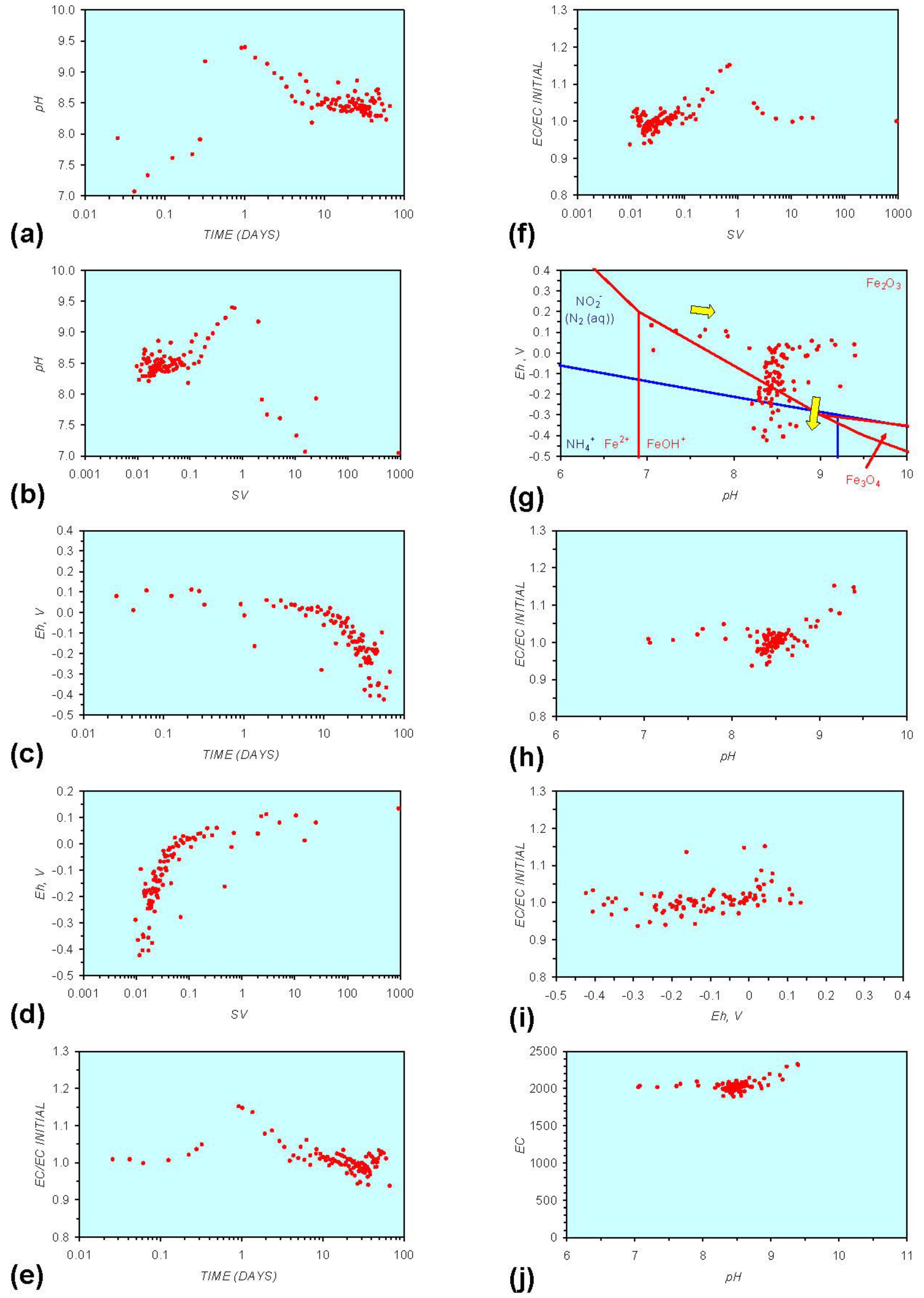
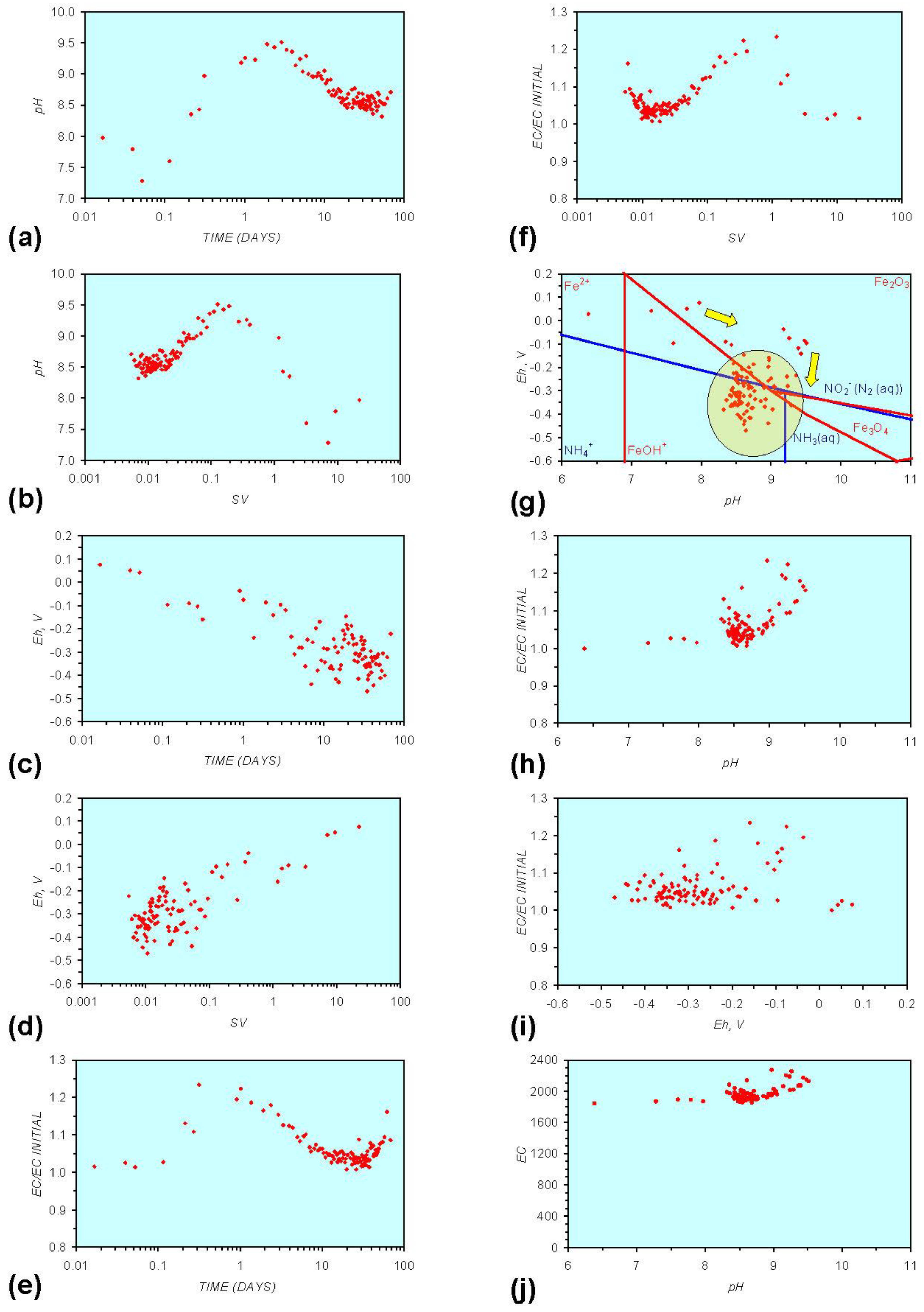
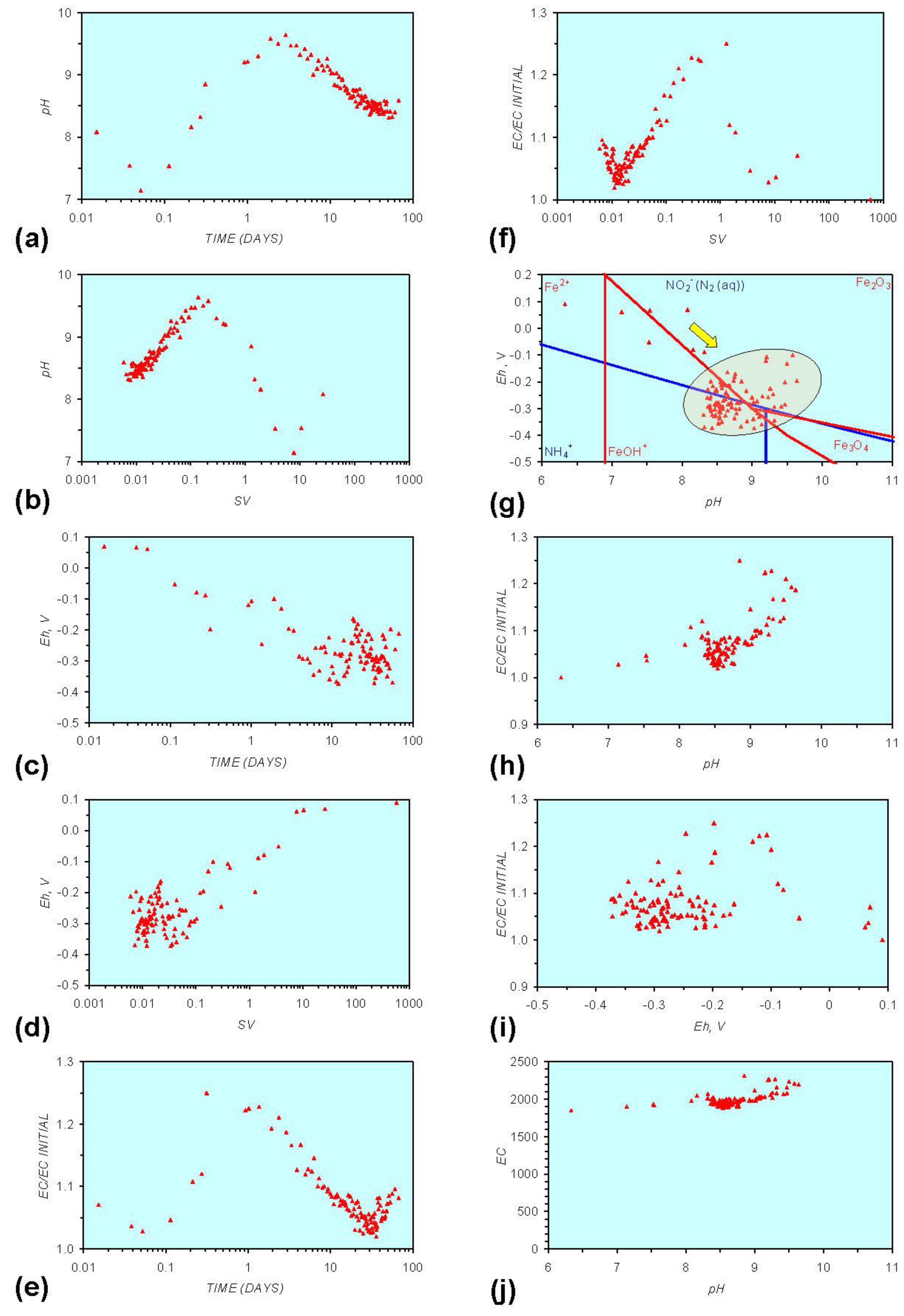
- Addition of n-ZVM resulted in a significant change in EC, pH or Eh.
- The change in Eh, EC, pH followed a specific (and predictable) pattern as a function of contact time with the ZVM (e.g., SV).
3.2. Experimental Results
- (i)
- An initial rapid increase in pH (to around pH = 10) is accompanied by a decrease in Eh.
- (ii)
- This is followed by a stabilization in pH (at around pH = 10) and a rise in Eh.
- (iii)
- This is then followed by a stabilization of Eh (e.g., Eh = 0.05 V) and a decrease in pH to a base level (e.g., pH = 8–9).
4. Interpretation
4.1. Equilibrium Oscillation
[O2 degassing, Eh reduction, and pH increase or decrease]
[H+ ion or H2(g) formation, Eh and pH increase or decrease]
ZVM-H+ + CO2 = ZVM-COOH+
ZVM-H+ + CO2 + xH+ = ZVM-CHn + 2H2O
ZVM-H+ + CH4 = ZVM-CH3 + 2H+
ZVM-H+ + CO2 + 6H+ = ZVM-CH3 + 2H2O
ZVM-H+ + CxH(2x+2) = ZVM-(CH2)(x-1)CH3 + 2H+
ZVM-H+ + CxHyCln = ZVM-CHCl or ZVM-CHCl2 or ZVM-CHCl3 or ZVM-CH2Cl
ZVM-H+ + CxHyOz + mH+ = ZVM-CpHq + nH2O
4.2. Impact of CO2
Fe0 + H3O+ = Fe-H+ + H2O − catalytic nuclei formation
Fe-H+ + CO2 + 6H3O+ = Fe-CH3 + 8H2O − chain formation
Fe-CH3+ CO2 + 6H3O+ = Fe-CH2CH3 + 8H2O − chain growth
Fe-CH3+ CO2 + 6H3O+ = Fe-(CH2)2CH3 + 8H2O − chain growth
Fe-(CH2)2CH3 + 2H3O+ = Fe-H+ + 2H2O + CH3CH2CH3 − chain termination
ZVM + CxHyClz = ZVM-CaHbClc + … − catalytic nuclei formation
ZVM-CCl3 + CH4 = ZVM-CCl2CH3 + Cl− + H+ − chain growth
ZVM-CHCl2 + CH4 = ZVM-CH2CH3 + 2Cl− − chain growth
ZVM-CH2CH3 + CH4 = ZVM-CH2CH2CH3 + 2H+ − chain growth
ZVM-CHCl2 + 4H+ = ZVM-H+ + CH4 + 2Cl− − chain termination
ZVM-CH2CH2CH3 + 2H+ = ZVM-H+ + CH3CH2CH3 − chain termination
ZVM-CH2CH2CH3 + H+ = ZVM + CH3CH2CH3 − chain termination
ZVM-CH2CH3 + ZVM-CH2CH3 + 2H+ = 2ZVM-H+ + CH3 CH2CH2CH3 − chain termination
ZVM-CH2CH3 + ZVM-CH2CH3 = 2ZVM + CH3 CH2CH2CH3 − chain termination
CO2(aq) + H2O (l) = HCO3−(aq) + H+ (aq) − ΔGo = +36.305 kJ mol−1
Fe0 + 2H+ (aq) = Fe2+ (aq) + H2(g) − ΔGo = −91.525 kJ mol−1
Fe0 + 2CO2(g) = 2HCO3− (aq) + Fe2+ (aq) + H2 (g) − ΔGo = −2.245 kJ mol−1
4.3. Fenton Reactions
OMoxidized 1 + Fe0 = OMreduced + Fe2+
OMreduced + O2 + 2H+ = H2O2 (aq) + OMoxidized 2
2H2O2 (aq) = 2H2O (l) + O2 (g)
2Fe0 + 2H2O = 2Fe-H+ + 2OH−
2Fe0 + O2 + 2H2O = 2Fe2+ + 4OH−
2Fe0 + 2H2O[O] = 2Fe2+ + 4OH−
2OH− = H2O2
Fe2++H2O2 = Fe3+ + 2OH−
4.4. Sulphur and Oxylate Removal
ZVM-CH3+ H2S = CH3-ZVM-SH+ + 0.5H2
4.5. General Patterns
- (i)
- (ii)
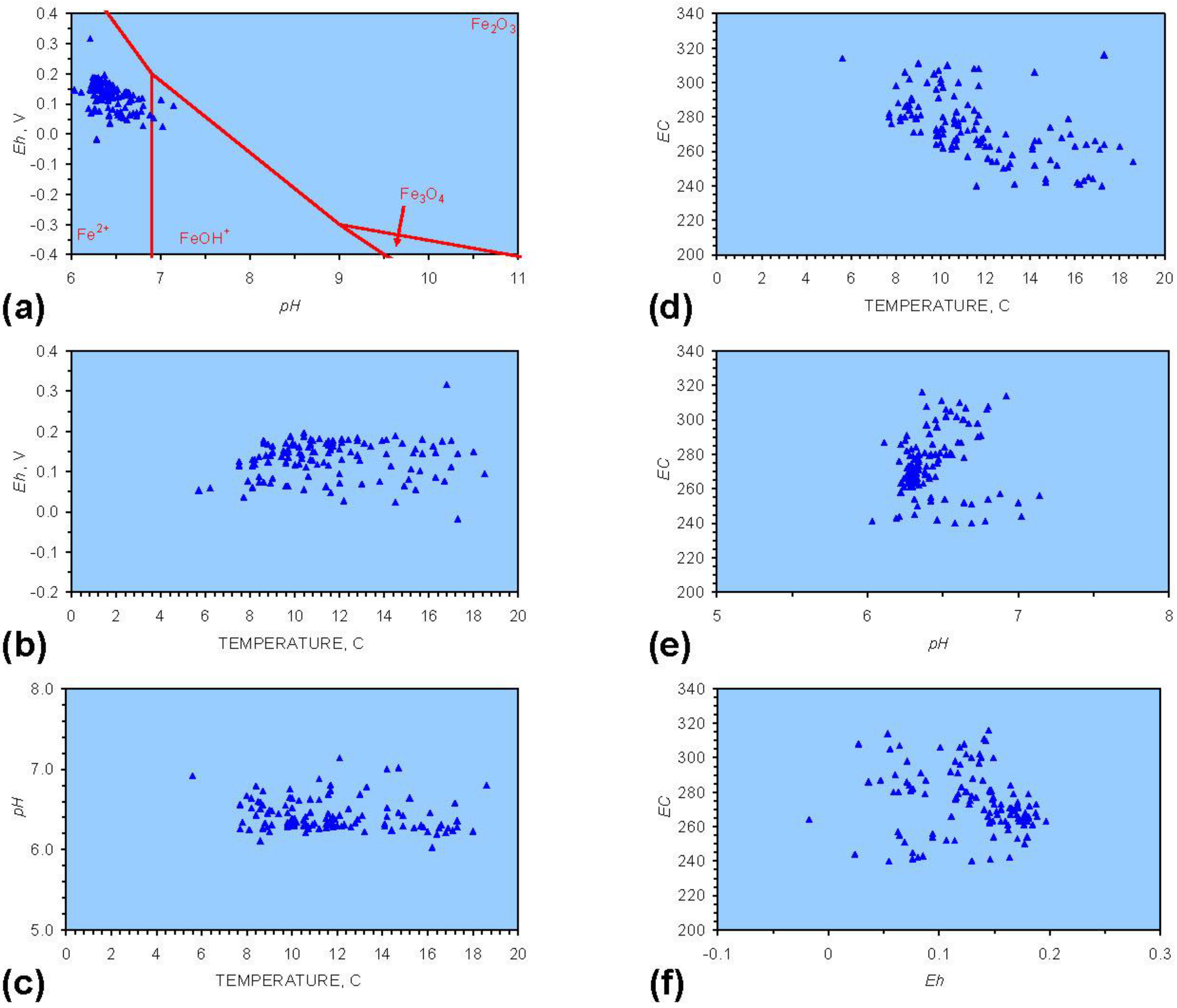
- The formation of Al3+ (aq) ions, resulting in a substantial increase in EC (Figure A2 in Appendix) is linked directly to a decrease in Eh and an increase in pH, e.g., H+ + HO2− = 2OH− (or H2O2).
4.6. Removal of Dissolved Organic Matter
- a)
- the rate HA removal increases with increasing n-Fe0 concentration (though k remains constant) [61], indicating that the removal may be linked to surface area.
- b)
- HA removal is associated with a significant decrease in pH [61].
- c)
- HA removal is associated over time with a change in n-Fe0 elemental ratio (Fe:O:C) from 85:13.5:0 (at onset) to 31:52:17 (after 90 days) [61].
- a)
- Reduced cost. n-ZVM (n-ZVMA) derived from a ground metal is typically about 10% of the cost of n-ZVM (n-ZVMB) produced by precipitation from a nitrate, chloride or sulphate.
- b)
- Increased active life. n-ZVMA is less prone to fouling by DOM/HA than n-ZVMB [61].
- c)
- Increased activity. n-ZVMA produces larger shifts in both pH and Eh than n-ZVMB (Figures A1–A16) or m-ZVM [1].
- d)
- Increased control on product selectivity. Figures A1 to A16 in Appendix demonstrate that changing the concentrations of ZVM components can alter the redox trajectory, Eh, pH and consequently the degree and level of remediation. The use of ground metal particles, rather than bi-metal or tri-metal precipitated complexes, allows the relative ratios of Fe:Cu:Al to be rapidly adjusted during the course of the remediation program in order to compensate for redox changes/interactions associated with the aquifer microbiota, aquifer chemistry, interactions with the host rock minerals and changes in the aquifer flow rate.
5. Conclusions
Acknowledgements
References
- Antia, D.D.J. Sustainable zero-valent metal (ZVM) water treatment associated with diffusion, infiltration, abstraction and recirculation. Sustainability 2010, 2, 2988–3073. [Google Scholar] [CrossRef]
- Chuang, F-W.; Larson, R.A.; Wessman, M.S. Zero-valent iron-promoted dechlorination of polychlorinated biphenyls. Environ. Sci. Technol. 1995, 29, 2460–2463. [Google Scholar]
- Orth, R.; Daudo, T.; McKenzie, D.E. Reductive dechlorination of DNAPL Trichloroethylene by zero-valent iron. Prac. Period. Hazard. Toxic. Radioact. Waste Manag. 1998, 2, 123–128. [Google Scholar] [CrossRef]
- Chen, J.L.; Al-Abed, S.R.; Ryan, J.A.; Li, Z. Effects of pH on dechlorination of trichloroethylene by zero-valent iron. J. Hazard Mater. 2001, 83, 243–254. [Google Scholar] [CrossRef] [PubMed]
- Lowry, G.V.; Johnson, K.M. Congener-specific dechlorination of dissolved PCBs by microscale and nanoscale zerovalent iron in water/methanol solution. Environ. Sci. Technol. 2004, 38, 5208–5216. [Google Scholar] [CrossRef] [PubMed]
- Shin, M-C.; Choi, H-D.; Kim, D-H.; Baek, K. Effect of surfactant on reductive dechlorination of trichloroethylene by zero-valent iron. Desalination 2008, 223, 299–307. [Google Scholar]
- Kim, G.; Jeong, W.; Choe, S. Dechlorination of atrazine using zero-valent iron (Fe0) under neutral pH conditions. J. Hazard. Mater. 2008, 155, 502–506. [Google Scholar] [CrossRef] [PubMed]
- Ghazali, M.; McBean, E.; Shen, H.; Dastous, P-A. Impact of iron concentration and ph on zero-valent iron dechlorination of DDT for brownfields. Remediat. J. 2010, 20, 97–107. [Google Scholar] [CrossRef]
- Thompson, J.M.; Chisholm, B.J.; Bezbaruah, A.N. Reductive dechlorination of Chloroactetanilide herbicide (Alachlor) using zero-valent iron nanoparticles. Environ. Engrg. Sci. 2010, 27, 227–232. [Google Scholar] [CrossRef]
- McElroy, B.; Keith, A.; Glasgow, J.; Dasappa, S. The use of zero-valent iron injection to remediate groundwater: Results of a pilot test at Marshall space flight center. Remediation 2003, 13, 145–153. [Google Scholar] [CrossRef]
- Demonstration of in situ Dehalogenation of DNAPL through Injection of Emulsified Zero-Valent Iron at Launch Complex 34 in Cape Canaveral Air Force Station, Florida; Innovative Technology Evaluation Report, EPA/540/R-07/006; U.S. Environmental Protection Agency: Cincinnati, OH, USA, September 2004.
- Gavaskar, A.; Tatar, L.; Condit, W. Cost and Performance Report: Nanoscale Zero-Valent Iron Technologies for Source Remediation; Contract Report CR-05-007-ENV; Naval Facilities Engineering Command/Engineering Service Center (NAVFAC ESC): Port Hueneme, CA, USA, 2005; p. 44. [Google Scholar]
- Pupeza, M.; Cernik, M.; Greco, M. Dechlorination of chlorinated hydrocarbons by zero-valent iron nano-particles. NATO Sci. Ser. 2007, 75, 111–118. [Google Scholar]
- Gavaskar, A.; Bhargava, M.; Condit, W. Cost and Performance Report for a Zero-Valent Iron (ZVI) Treatability Study at Naval Air Station North Island; Technical Report TR-2307-ENV; Naval Facilities Engineering Command/Engineering Service Center (NAVFAC ESC): Port Hueneme, CA, USA, 2008. [Google Scholar]
- Alvarado, J.S.; Rose, C.; LaFreniere, L. Degradation of carbon tetrachloride in the presence of zero-valent iron. J. Environ. Monit. 2010, 12, 1524–1530. [Google Scholar] [CrossRef] [PubMed]
- Sunkara, B.; Zhan, J.; He, J.; McPherson, G.L.; Piringer, G.; John, V.T. Nanoscale zerovalent iron supported on uniform carbon microspheres for the in situ remediation of chlorinated hydrocarbons. ACS Appl. Mater. Interfaces, 2010, 2, 285–2862. [Google Scholar] [CrossRef]
- Sorel, D.; Warner, S.D.; Longino, B.L.; Honniball, J.H.; Hamilton, L.A. Performance monitoring and dissolved hydrogen measurements at a permeable zero valent iron reactive barrier. ACS Symp. Ser. 2002, 837, 278–285. [Google Scholar]
- Choi, J-H.; Kim, Y-H.; Choi, S.J. Reductive dechlorination and bidegradation of 2,4,6-trichlorophenol using sequential permeable reactive barriers: laboratory studies. Chemosphere 2007, 67, 1551–1557. [Google Scholar] [CrossRef] [PubMed]
- Sass, B.M. Reduction of TNT and RDX by core material from an iron permeable reactive barrier. Paper M-009. In Proceedings of the 6th International Conference on Remediation of Chlorinated and Recalcitrant Compounds, Monterey, CA, USA, May 2008; Battelle: Columbus, OH, USA, 2008. [Google Scholar]
- Higgins, M.R.; Olson, T.M. Life-cycle case study comparison of permeable reactive barrier versus pump-and-treat remediation. Environ. Sci. Technol. 2009, 43, 9432–9438. [Google Scholar] [CrossRef] [PubMed]
- Phillips, D.H.; Van Nooten, T.; Bastiaens, L.; Russell, M.I.; Dickson, K.; Plant, S.; Ahad, J.M.; Newton, T.; Elliot, T.; Kalin, R.M. Ten year performance evaluation of a field-scale zero-valent iron permeable reactive barrier installed to remediate trichloroethene contaminated groundwater. Environ. Sci. Technol. 2010, 15, 3861–3869. [Google Scholar] [CrossRef]
- Gilbert, O.; de Pablo, J.; Cortina, J.-L.; Ayora, C. In situ removal of arsenic from groundwater by using permeable reactive barriers of organic matter/limestone/zero-valent iron mixtures. Environ. Geochem. Health 2010, 32, 373–378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, H.; Jin, S.; Fallgren, P.H.; Colberg, P.J.S.; Johnson, P.A. Prevention of iron passivation and enhancement of nitrate reduction by electron supplementation. Chem. Eng. J. 2010, 160, 185–189. [Google Scholar] [CrossRef]
- Antia, D.D.J. Prediction of overland flow and seepage zones associated with the interaction of multiple Infiltration Devices (Cascading Infiltration Devices). Hydrol. Proc. 2008, 22, 2595–2614. [Google Scholar] [CrossRef]
- Antia, D.D.J. Formation and control of self-sealing high permeability groundwater mounds implications for SUDS and sustainable pressure mound management. Sustainability 2009, 1, 855–923. [Google Scholar] [CrossRef]
- Antia, D.D.J. Interacting Infiltration Devices (Field Analysis, Experimental Observation and Numerical Modeling): Prediction of Seepage (Overland Flow) Locations, Mechanisms and Volumes–Implications for SUDS, Groundwater Raising Projects. In Hydraulic Engineering: Structural Applications, Numerical Modeling and Environmental Impacts, 1st ed.; Hirsch, G., Kappel, B., Eds.; Nova Science Publishers: Hauppauge, NY, USA, 2010; pp. 85–156. [Google Scholar]
- Antia, D.D.J. Interpretation of Overland Flow Associated with Infiltration Devices Placed in Boulder Clay and Construction Fill. In Overland Flow and Surface Runoff, 1st ed.; Wong, T.S.W., Ed.; Nova Science Publishers: Hauppauge, NY, USA, 2011. (In Press) [Google Scholar]
- Fiore, S.; Zanetti, M.C. Preliminary tests concerning zero-valent iron efficiency in organic pollutants removal. Am. J. Environ. Sci. 2009, 5, 555–560. [Google Scholar] [CrossRef]
- Hao, Z.W.; Xu, X.H.; Wang, D.H. Reductive denitrification of nitrate by scrap iron filings. J. Zhejang Univ. Sci. B. 2005, 6, 182–186. [Google Scholar] [CrossRef]
- Hao, Z.W.; Xu, X.H.; Jin, J.; He, P.; Liu, Y.; Wang, D.H. Simultaneous removal of nitrate and heavy metals by iron metal. J. Zhejang Univ. Sci. B. 2005, 6, 307–310. [Google Scholar] [CrossRef]
- Zhang, W-X. Nanoscale iron particles for environmental remediation: An overview. J. Nanoparticle Res. 2003, 5, 323–332. [Google Scholar]
- Wang, C-B.; Zhang, W-X. Synthesising nano-scale iron particles for rapid and complete dechlorination of TCE and PCBs. Environ. Sci. Technol. 1997, 31, 2154–2156. [Google Scholar]
- Liu, Y.; Majetich, S.A.; Tilton, R.D.; Sholl, D.S.; Lowry, G.V. TCE dechlorination rtaes, pathways and efficiency of nanoscale particles with different properties. Environ. Sci. Technol. 2005, 39, 1338–1345. [Google Scholar] [CrossRef] [PubMed]
- Tokoro, H.; Nakabayashi, T.; Fujii, S.; Zhao, H.; Hafeli, U.O. Magnetic iron particles with high magnetization useful for immunoassay. J. Magnetism Magnetic Mat. 2009, 321, 1676–1678. [Google Scholar] [CrossRef]
- Antony, J.; Nutting, J.; Baer, D.R.; Meyer, D.; Sharma, A.; Qiang, Y. Size-dependent specific surface area of nanoporous film assembled by core-shell iron nanoclusters. J. Nanomat. 2006, 1, 1–4. [Google Scholar] [CrossRef]
- Krasnoperov, L.N.; Peng, J.; Marshall, P. Modified transition state theory and negative apparent activation energies of simple metathesis reactions: Application to the reaction CH3 + HBr = CH4 + Br. J. Phys. Chem. A 2006, 110, 3110–3120. [Google Scholar] [CrossRef] [PubMed]
- Wei, J. Adsorption and cracking of n-alkanes over ZSM-5: negative activation energy of reaction. Chem. Engrg. Sci. 1996, 51, 2995–2999. [Google Scholar] [CrossRef]
- Alvarez-Idaboy, J.R.; Mora-Diez, N.; Vivier-Bunge, A. A quantum chemical and classical transition state theory explanation of negative activation energies in OH addition to substituted ethenes. J. Am. Chem. Soc. 2000, 122, 3715–3720. [Google Scholar] [CrossRef]
- Alvarez-Idaboy, J.R.; Mora-Diez, N.; Boyd, R.J.; Vivier-Bunge, A. On the importance of prereactive complexes in molecule-radical reactions: hydrogen abstraction from aldehydes by OH. J. Am. Chem. Soc. 2001, 123, 2018–2024. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, D.A.H.; Vogel, W.; Haruta, M. Negative activation energies in CO oxidation over an icosahedral Au/Mg(OH)2 catalyst. Cat. Let. 1999, 63, 43–47. [Google Scholar] [CrossRef]
- Brookhaven National Laboratory Carbon Dioxide (Reduction); Report BNL-67077(Produced for the U.S. Department of Energy), Contract DE-AC02-98CH10886; Brookhaven National Laboratory: Long Island, NY, USA, 1998; p. 10.
- Perkins, H.F.; Parker, M.B.; Walker, M.L. Chicken Manure—Its Production, Composition and Use as a Fertilizer; Georgia Agricultural Experiment Stations, University of Georgia College of Agriculture: Athens, GA, USA, 1964. [Google Scholar]
- Dikinya, O.; Mufwanzala, N. Chicken manure—enhanced soil fertility and productivity: Effects of application rates. J. Soil Sci. Environ. Manag. 2010, 1, 46–54. [Google Scholar]
- Materechera, S.A.; Mkhabela, T.S. The effectiveness of lime, chicken manure and leaf litter ash in ameliorating acidity in a soil previously under black wattle (Acacia mearnsii) plantation. Biores. Technol. 2002, 85, 9–16. [Google Scholar] [CrossRef]
- Himathongkham, S.; Bahari, S.; Riemann, H.; Cliver, D. Survivial of Escherichia coli O157:H7 and Salmonella typhimurium in cow manure and cow slurry. FEMS Mircobiol. Let. 1999, 178, 251–257. [Google Scholar] [CrossRef]
- Kunz, A.; Steinmetz, R.L.R; Ramme, M.A.A.; Coldebella, A. Effect of storage time on swine manure solid separation by screening. Biores. Technol. 2009, 100, 1815–1818. [Google Scholar] [CrossRef]
- Xing, Y.; Li, Z.; Fan, Y.; Hou, H. Biohydrogen production from dairy manures with acidification pretreatment by anaerobic fermentation. Environ. Sci. Pollut. Res. Int. 2010, 17, 392–399. [Google Scholar] [CrossRef] [PubMed]
- Antia, D.D.J. Oil polymerisation and fluid expulsion from low temperature, low maturity, over pressured sediments. J. Petrol. Geol. 2008, 31, 263–282. [Google Scholar] [CrossRef]
- Elek, J. Homogenous Catalytic Reduction of CO2 in Aqueous Medium. PhD Thesis, University of Debrecen, Debrecen, Hajdu-Bihar, Hungary, 2003. [Google Scholar]
- Guan, G.; Kida, T.; Ma, T.; Kimura, K.; Abe, E.; Yoshida, A. Reduction of aqueous CO2 at ambient temperature using zero-valent iron-based composites. Green Chemistry 2003, 5, 630–634. [Google Scholar] [CrossRef]
- Kang, S.H.; Choi, W. Oxidative degradation of organic compounds using zero-valent iron in the presence of natural organic matter serving as an electron shuttle. Environ. Sci. Technol. 2009, 43, 878–883. [Google Scholar] [CrossRef] [PubMed]
- Rahmani, A.R.; Zarrabi, M.; Samarghandi, M.R.; Afkhami, A.; Ghaffari, H.R. Degradation of Azo Dye Reactive Black 5 and Acid Orange 7 by Fenton like mechanim. Iranian J. Chem. Eng. 2010, 7, 87–94. [Google Scholar]
- Wang, F. Modelling of Aqueous Carbon Dioxide Corrosion in Turbulent Pipe Flow. PhD Thesis, University of Saskatchewan, Saskatoon, Canada, 1999. [Google Scholar]
- Schmitt-Kopplin, P.; Gabelica, Z.; Gougeon, R.D.; Fekete, A.; Kanawati, B.; Harir, M.; Gebefugi, I.; Eckel, G.; Hertkorn, N. High molecular diversity of extraterrestrial organic matter in Murchison meteorite revealed 40 years after its fall. PNAS 2010, 107, 2763–2768. [Google Scholar] [CrossRef] [PubMed]
- Drobner, E.; Huber, H.; Wachterhauser, G.; Rose, D.; Stetter, K.O. Pyrite formation linked with hydrogen evolution under anaerobic conditions. Nature 1990, 346, 742–744. [Google Scholar] [CrossRef] [Green Version]
- Chai, L.; Navrotsky, A. Enthalpy of formation of siderite and its application in phase equilibria calculation. Am. Mineral. 1994, 79, 921–929. [Google Scholar]
- Angamuthi, R.; Byers, P.; Lutz, M.; Spek, A.L.; Bouwman, E. Electrocatalytic CO2 conversion to oxylate by a copper complex. Science 2010, 327, 313–315. [Google Scholar] [CrossRef] [PubMed]
- Baur, R.F.; Smith, W.M. The mono-oxalato complexes of iron (III). Can. J. Chem. 1965, 43, 2755–2762. [Google Scholar] [CrossRef]
- Dries, J.; Bastiaens, L.; Springael, D.; Kuypers, S.; Agathos, S.N.; Diels, L. Effect of humic acids on heavy metal removal by zero-valent iron in batch and continuous flow column systems. Water Res. 2005, 39, 3531–3540. [Google Scholar] [CrossRef] [PubMed]
- Geological Survey Japan. Atlas of Eh-pH Diagrams; Open File Report No. 419; National Institute of Advanced Industrial Science and Technology: Naoto, Japan, 2005.
- Giasuddin, A.B.M.; Kanel, S.R.; Choi, H. Adsorption of humic acid onto nanoscale zerovalent iron and its effect on arsenic removal. Environ. Sci. Technol. 2007, 41, 2022–2027. [Google Scholar] [CrossRef] [PubMed]
- Doong, R-A.; Lai, Y-J. Dechlorination of tetrachloroethylene by palladized iron in the presence of humic acid. Water Res. 2005, 39, 2309–2318. [Google Scholar]
- Liu, X.; Wazne, M.; Han, Y.; Christodoulatos, C.; Jasinkiewicz, K.L. Effects of natural organic matter on aggregation kinetics of boron nanoparticles in monovalent and divalent electrolytes. J. Colloid. Interface Sci. 2010, 348, 101–107. [Google Scholar] [CrossRef] [PubMed]
- Cole, E.B.; Lakkaraju, P.S.; Rampulla, D.M.; Morris, A.J.; Abelev, E.; Bocarsly, A.B. Using a one-electron shuttle for the multielectron reduction of CO2 to methanol: Kinetic, mechanistic and structural insights. J. Am. Chem. Soc. 2010, 132, 11539–11551. [Google Scholar] [CrossRef] [PubMed]
- Royer, R.A.; Burgos, W.D.; Fischer, A.S.; Unz, R.F.; Dempsey, B.A. Enhancement of biological reduction of hematite by electron shuttling and Fe(II) complexation. Environ. Sci. Technol. 2002, 36, 1939–1946. [Google Scholar] [CrossRef] [PubMed]
- Kappler, A.; Benz, M.; Schink, B.; Brune, A. Electron shuttling via humic acids in microbial iron (III) reduction in a freshwater sediment. FEMS Microbiol. Ecol. 2004, 47, 85–92. [Google Scholar] [CrossRef] [PubMed]
Appendix
















© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Antia, D.D.J. Modification of Aquifer Pore-Water by Static Diffusion Using Nano-Zero-Valent Metals. Water 2011, 3, 79-112. https://doi.org/10.3390/w3010079
Antia DDJ. Modification of Aquifer Pore-Water by Static Diffusion Using Nano-Zero-Valent Metals. Water. 2011; 3(1):79-112. https://doi.org/10.3390/w3010079
Chicago/Turabian StyleAntia, David D. J. 2011. "Modification of Aquifer Pore-Water by Static Diffusion Using Nano-Zero-Valent Metals" Water 3, no. 1: 79-112. https://doi.org/10.3390/w3010079
APA StyleAntia, D. D. J. (2011). Modification of Aquifer Pore-Water by Static Diffusion Using Nano-Zero-Valent Metals. Water, 3(1), 79-112. https://doi.org/10.3390/w3010079