
On the Rapid Calculation of Binding Affinities for Antigen and Antibody Design and Affinity Maturation Simulations


Abstract
:1. Introduction
2. Materials and Methods
2.1. Experimental HIV Antibody–Antigen Binding Affinities
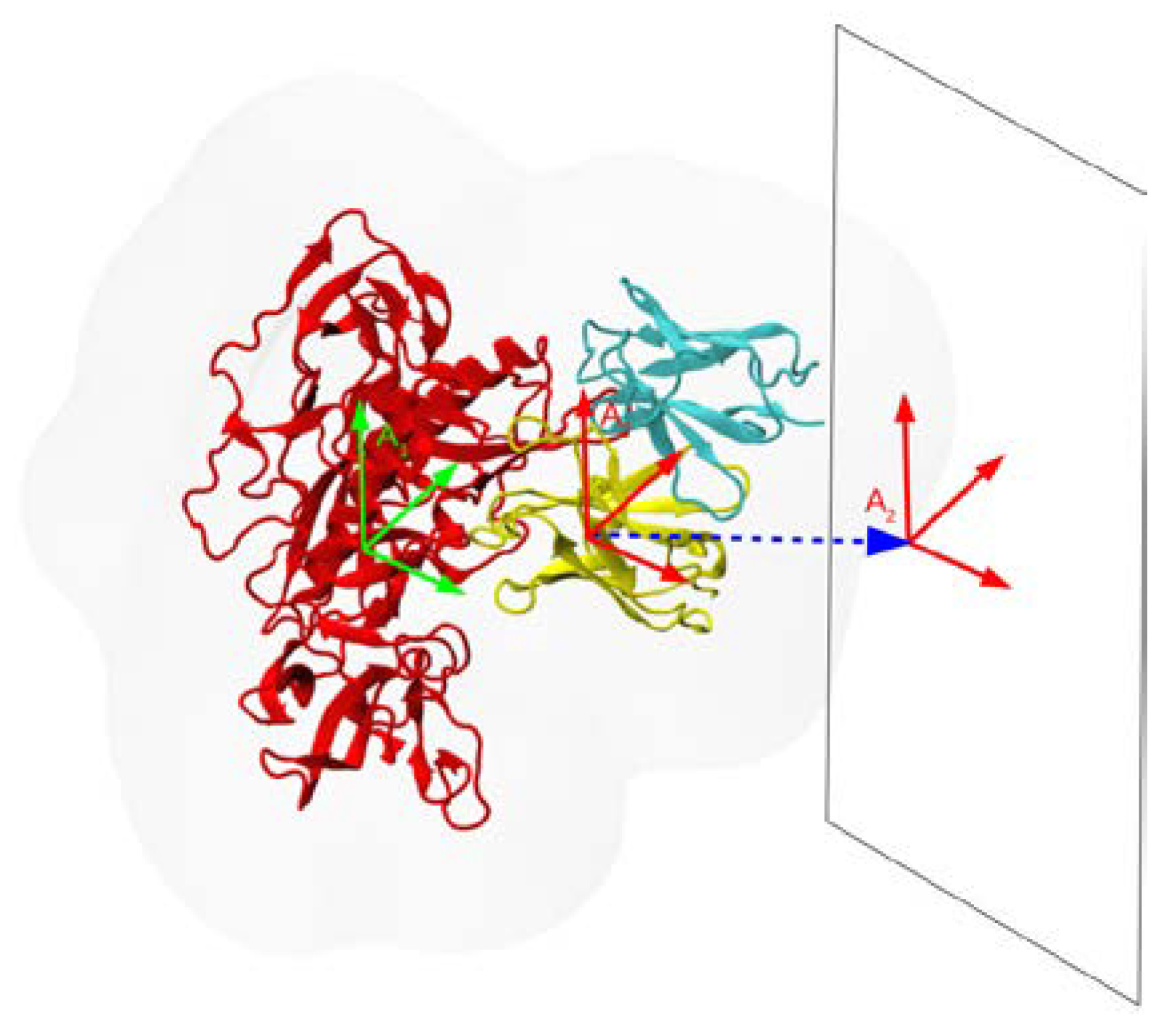
2.2. Binding Affinities from Potentials of Mean Force (PMF) Simulations
2.3. Rapid Scoring Functions
2.4. Implicit Models of Solvation
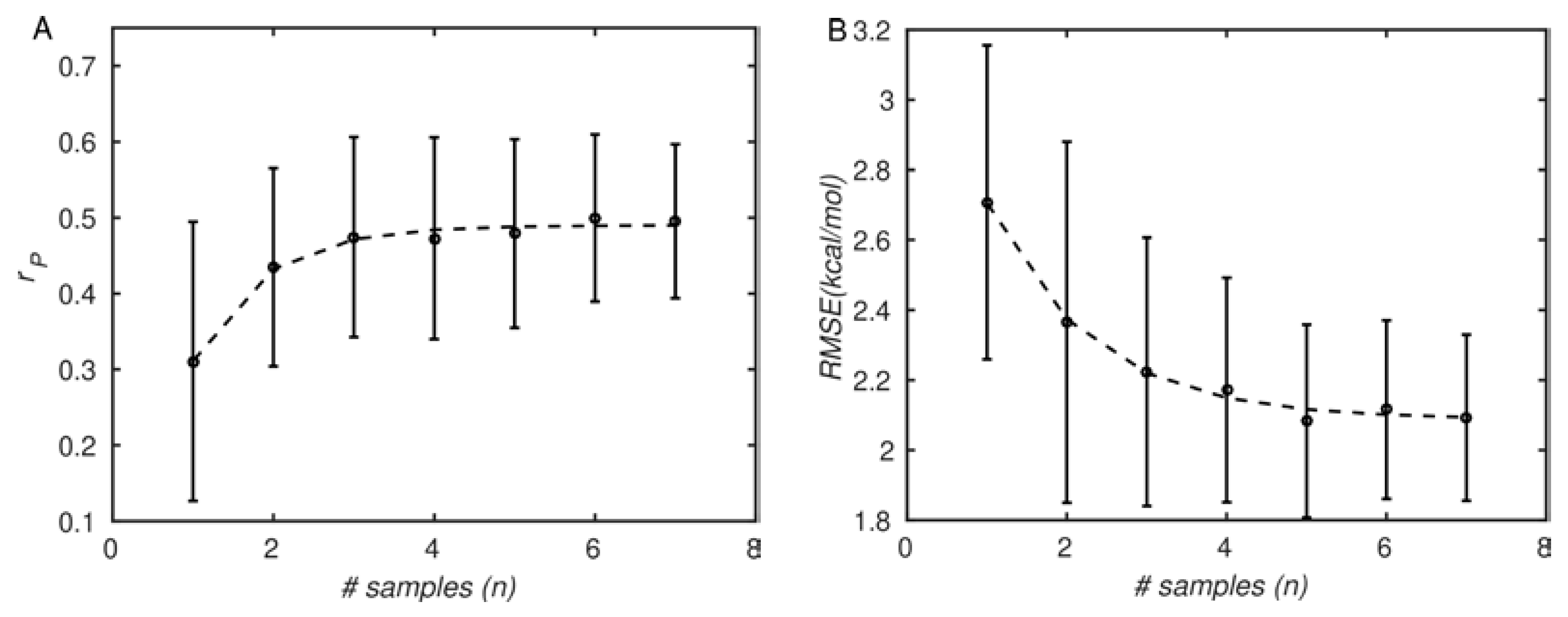
2.5. Comparison between Computed and Experimental Binding Affinities
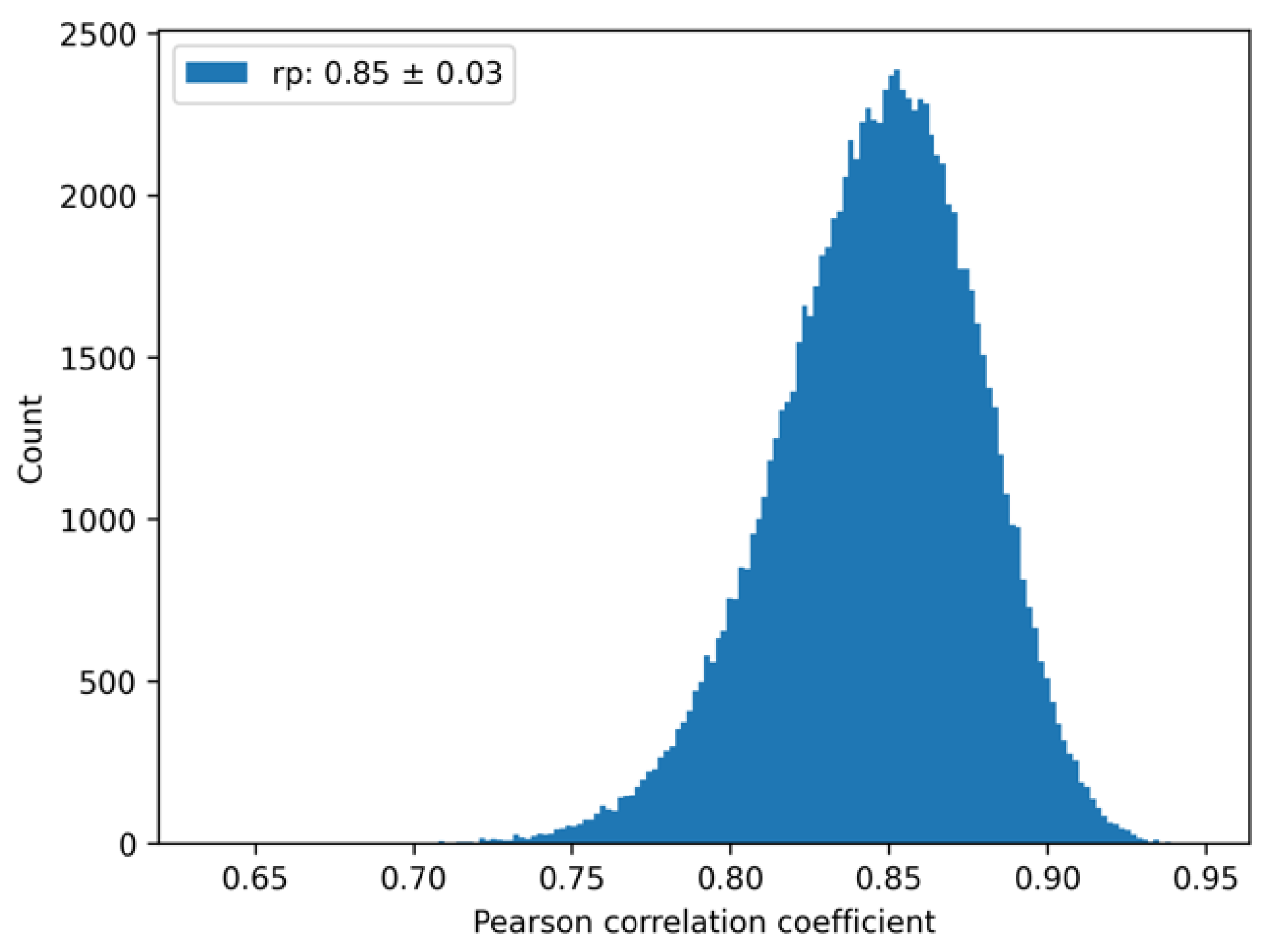
2.6. An Upper Bound for the Pearson Correlation
3. Results
3.1. Binding Free Energies (bFEs) from PMF Simulations
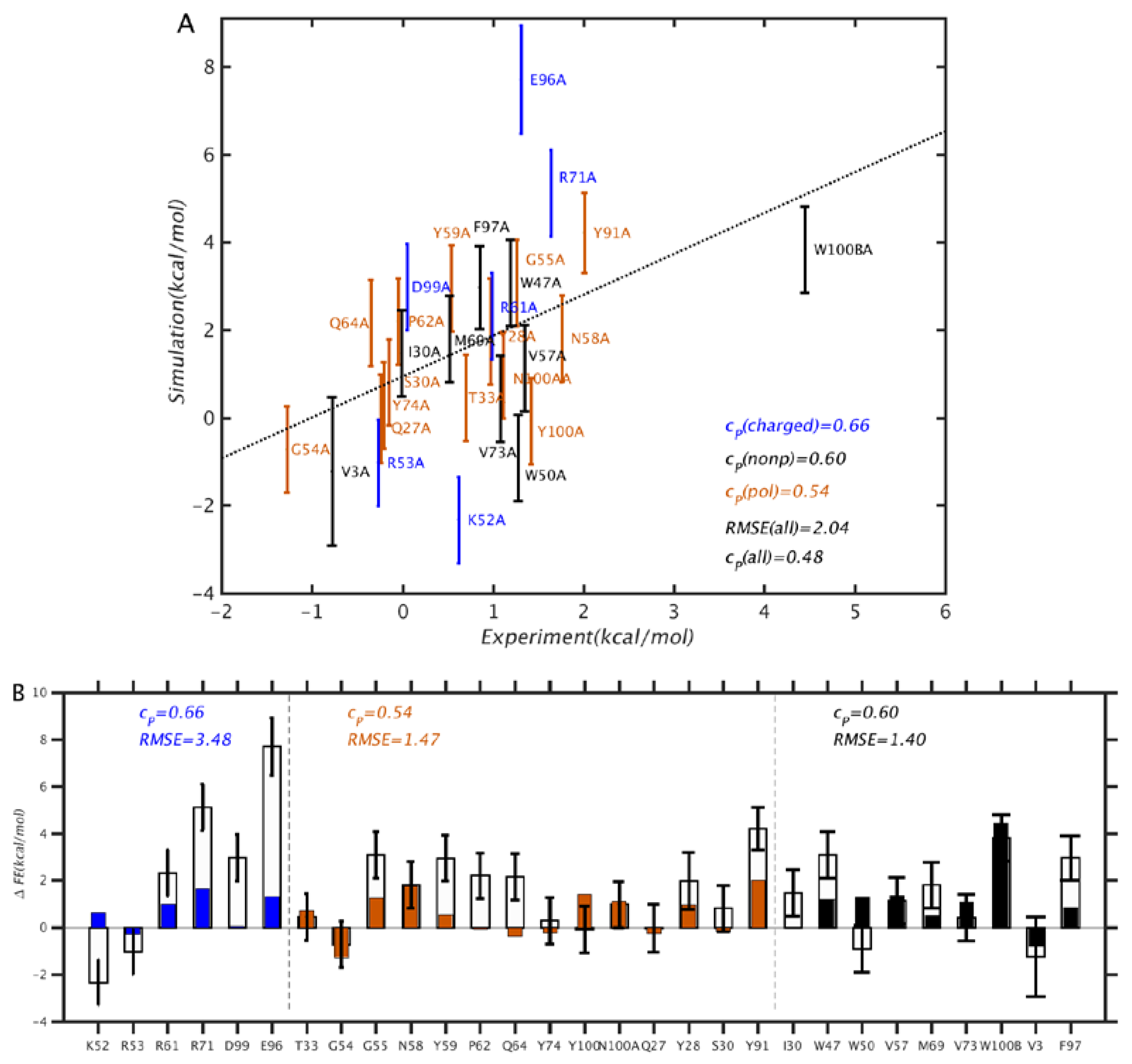
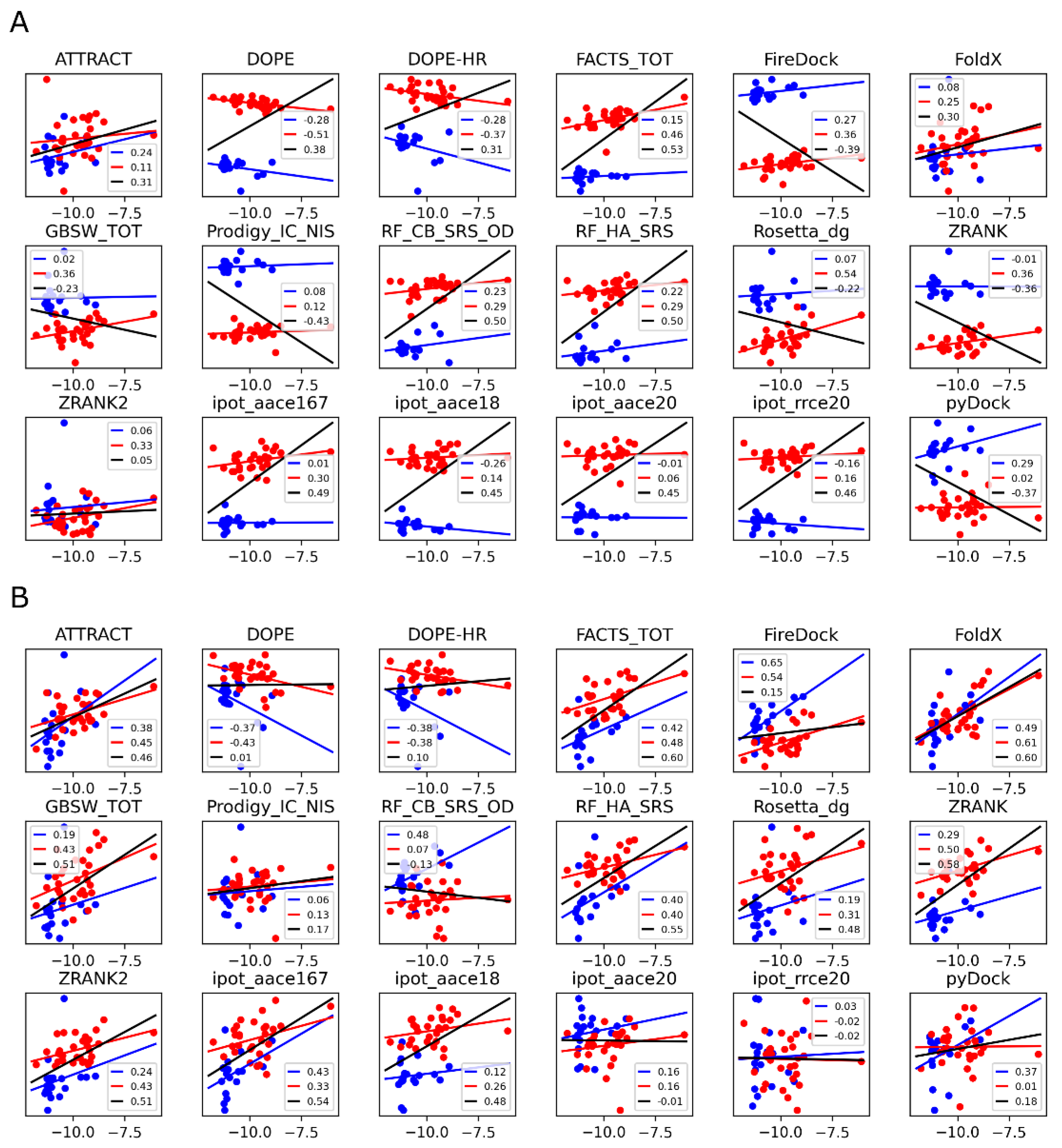
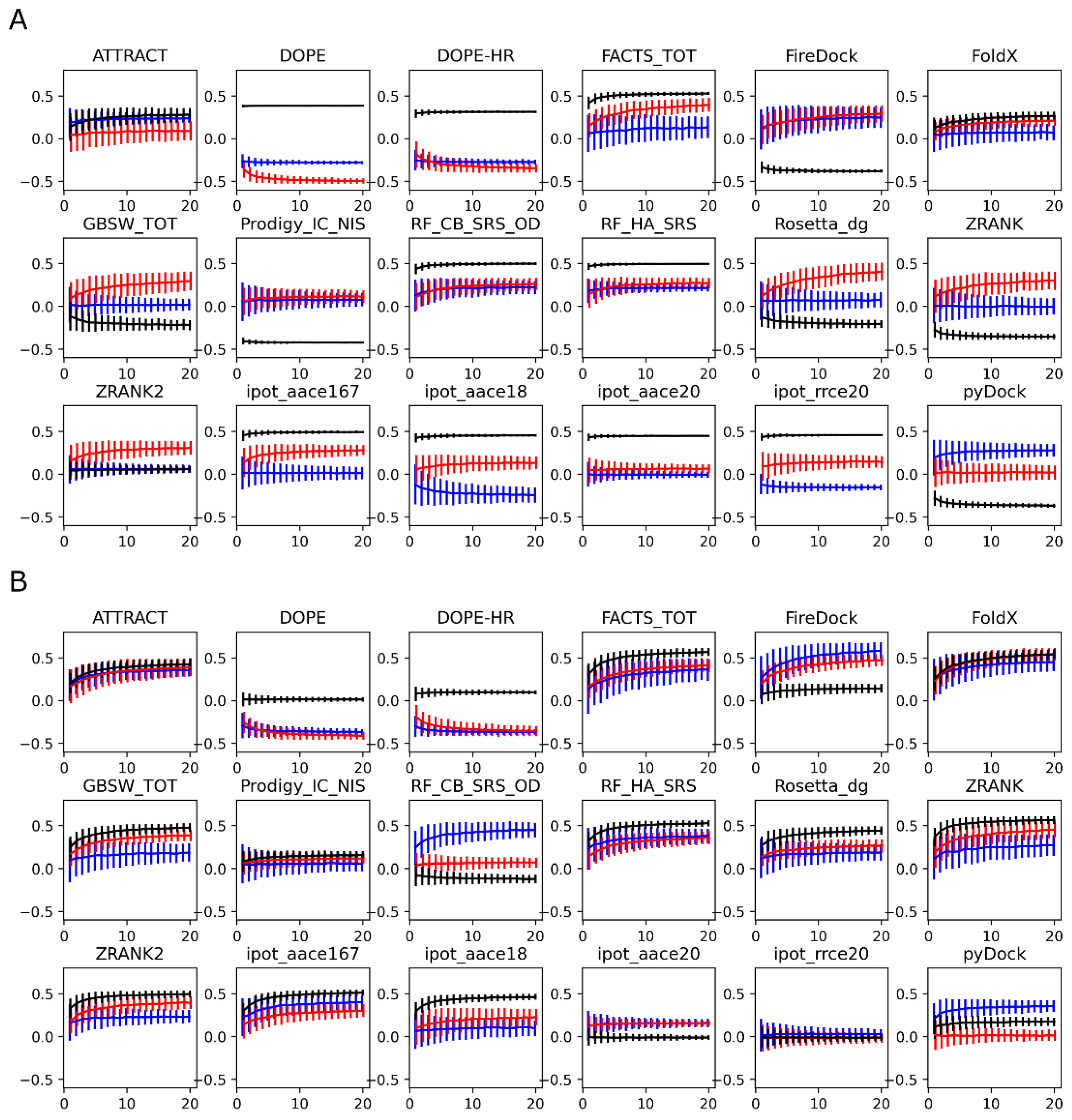
3.2. Scoring Functions
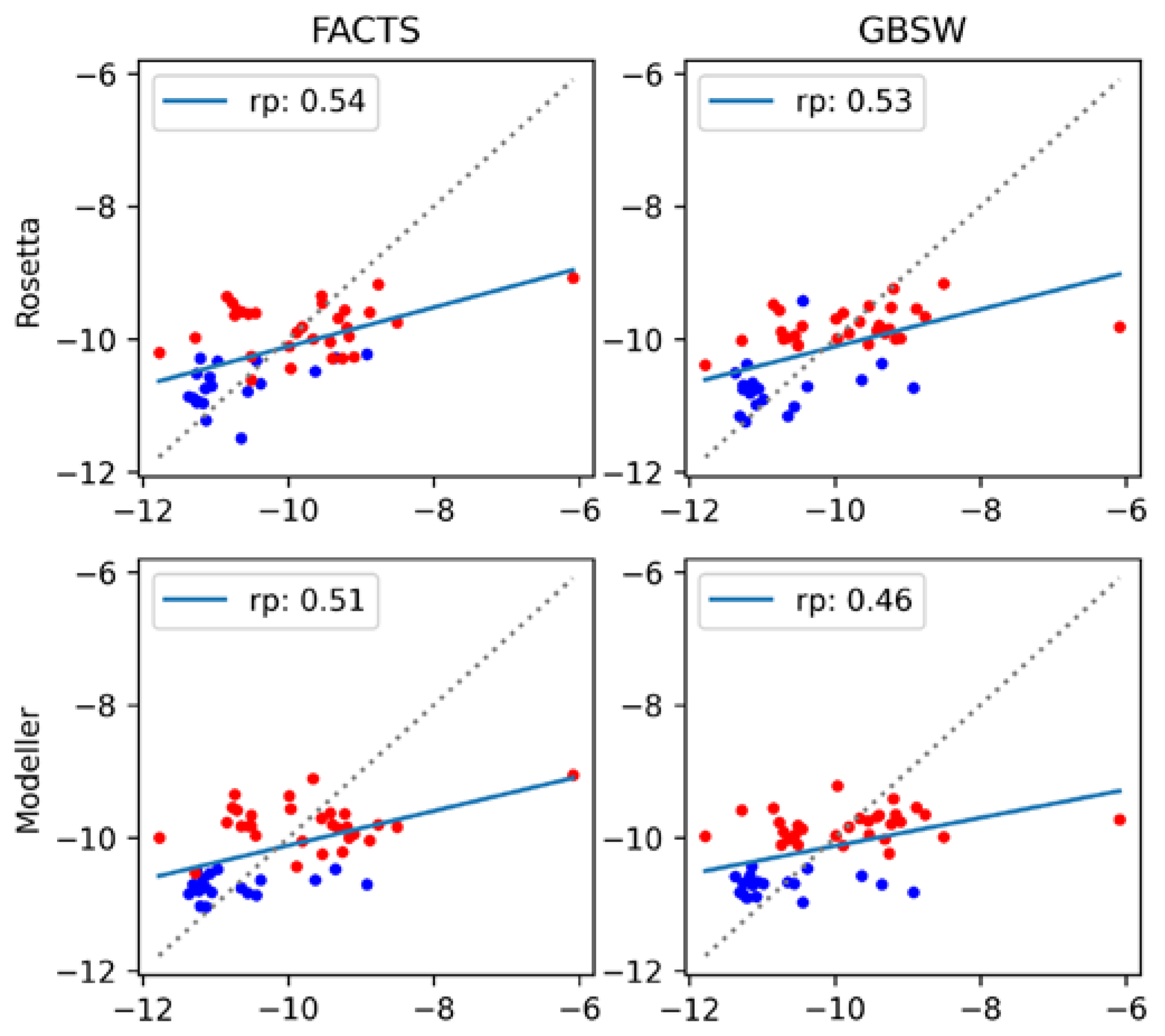
3.3. MM-GBSA with Optimized Coefficients
4. Concluding Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siebenmorgen, T.; Zacharias, M. Computational prediction of protein-protein binding affinities. WIREs Comput. Mol. Sci. 2019, 10, e1448. [Google Scholar] [CrossRef] [Green Version]
- Best, R.B.; Zhu, X.; Shim, J.; Lopes, P.E.M.; Mittal, J.; Feig, M. Optimization of the Additive CHARMM All-Atom Protein Force Field Targeting Improved Sampling of the Backbone ϕ, ψ and Side-Chain χ1 and χ2 Dihedral Angles. J. Chem. Theory Comput. 2012, 8, 3257–3273. [Google Scholar] [CrossRef] [Green Version]
- Guvench, O.; Mallajosyula, S.S.; Raman, E.P.; Hatcher, E.; Vanommeslaeghe, K.; Foster, T.J.; Jamison, I.F.W., 2nd; MacKerell, J.A.D., Jr. CHARMM Additive All-Atom Force Field for Carbohydrate Derivatives and Its Utility in Polysaccharide and Carbohydrate–Protein Modeling. J. Chem. Theory Comput. 2011, 7, 3162–3180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pearlman, D.A.; Case, D.A.; Caldwell, J.W.; Ross, W.S.; Cheatham, T.E., III; DeBolt, S.; Ferguson, D.; Seibel, G.; Kollman, P. AMBER, a package of computer programs for applying molecular mechanics, normal mode analysis, molecular dynamics and free energy calculations to simulate the structural and energetic properties of molecules. Comput. Phys. Commun. 1995, 91, 1–41. [Google Scholar] [CrossRef]
- Shivakumar, D.; Harder, E.; Damm, W.; Friesner, R.A.; Sherman, W. Improving the Prediction of Absolute Solvation Free Energies Using the Next Generation OPLS Force Field. J. Chem. Theory Comput. 2012, 8, 2553–2558. [Google Scholar] [CrossRef]
- Hess, B.; Kutzner, C.; van der Spoel, D.; Lindahl, E. GROMACS 4: Algorithms for Highly Efficient, Load-Balanced, and Scalable Molecular Simulation. J. Chem. Theory Comput. 2008, 4, 435–447. [Google Scholar] [CrossRef] [Green Version]
- Clark, A.J.; Gindin, T.; Zhang, B.; Wang, L.; Abel, R.; Murret, C.S.; Xu, F.; Bao, A.; Lu, N.J.; Zhou, T.; et al. Free Energy Perturbation Calculation of Relative Binding Free Energy between Broadly Neutralizing Antibodies and the gp120 Glycoprotein of HIV-1. J. Mol. Biol. 2016, 429, 930–947. [Google Scholar] [CrossRef]
- Anderson, J.A.; Lorenz, C.; Travesset, A. General purpose molecular dynamics simulations fully implemented on graphics processing units. J. Comput. Phys. 2008, 227, 5342–5359. [Google Scholar] [CrossRef]
- Friedrichs, M.S.; Eastman, P.; Vaidyanathan, V.; Houston, M.; Legrand, S.; Beberg, A.L.; Ensign, D.L.; Bruns, C.M.; Pande, V.S. Accelerating molecular dynamic simulation on graphics processing units. J. Comput. Chem. 2009, 30, 864–872. [Google Scholar] [CrossRef]
- Harvey, M.; Giupponi, G.; De Fabritiis, G. ACEMD: Accelerating Biomolecular Dynamics in the Microsecond Time Scale. J. Chem. Theory Comput. 2009, 5, 1632–1639. [Google Scholar] [CrossRef] [Green Version]
- Götz, A.W.; Williamson, M.J.; Xu, D.; Poole, D.; Le Grand, S.; Walker, R.C. Routine Microsecond Molecular Dynamics Simulations with AMBER on GPUs. 1. Generalized Born. J. Chem. Theory Comput. 2012, 8, 1542–1555. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Friesner, R.A.; Berne, B.J. Replica Exchange with Solute Scaling: A More Efficient Version of Replica Exchange with Solute Tempering (REST2). J. Phys. Chem. B 2011, 115, 9431–9438. [Google Scholar] [CrossRef] [Green Version]
- Vilseck, J.Z.; Armacost, K.A.; Hayes, R.L.; Goh, G.B.; Brooks, C.L. Predicting Binding Free Energies in a Large Combinatorial Chemical Space Using Multisite λ Dynamics. J. Phys. Chem. Lett. 2018, 9, 3328–3332. [Google Scholar] [CrossRef] [PubMed]
- Clark, A.J.; Negron, C.; Hauser, K.; Sun, M.; Wang, L.; Abel, R.; Friesner, R.A. Relative Binding Affinity Prediction of Charge-Changing Sequence Mutations with FEP in Protein–Protein Interfaces. J. Mol. Biol. 2019, 431, 1481–1493. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H. The Protein Data Bank. Nucl. Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eswar, N.; Webb, B.; Marti-Renom, M.A.; Madhusudhan, M.S.; Eramian, D.; Shen, M.-Y.; Pieper, U.; Sali, A. Comparative Protein Structure Modeling Using Modeller. Curr. Protoc. Bioinform. 2006, 15, 5.6.1–5.6.30. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Yan, R.; Roy, A.; Xu, D.; Poisson, J.; Zhang, Y. The I-TASSER Suite: Protein structure and function prediction. Nat. Methods 2015, 12, 7–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leaver-Fay, A.; Tyka, M.; Lewis, S.M.; Lange, O.F.; Thompson, J.; Jacak, R. Rosetta3: An Object-Oriented Software Suite for the Simulation and Design of Macromolecules. Methods Enzymol. 2011, 487, 545–574. [Google Scholar] [PubMed] [Green Version]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Applying and improving AlphaFold at CASP14. Protein Struct. Funct. Bioinf. 2021, 89, 1711–1721. [Google Scholar] [CrossRef] [PubMed]
- Weber, G. Energetics of Ligand Binding to Proteins. In Advances in Protein Chemistry; Anfinsen, C.B., Edsall, J.T., Richards, F.M., Eds.; Academic Press: Piscataway, NJ, USA, 1975; pp. 1–83. [Google Scholar]
- McCammon, J.A.; Gelin, B.R.; Karplus, M. Dynamics of folded proteins. Nature 1977, 267, 585–590. [Google Scholar] [CrossRef] [PubMed]
- Frauenfelder, H.; Sligar, S.G.; Wolynes, P.G. The energy landscapes and motions of proteins. Science 1991, 254, 1598–1603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shalloway, D. Macrostates of classical stochastic systems. J. Chem. Phys. 1996, 105, 9986–10007. [Google Scholar] [CrossRef]
- Iben, I.E.T.; Braunstein, D.; Doster, W.; Frauenfelder, H.; Hong, M.K.; Johnson, J.B.; Luck, S.; Ormos, P.; Schulte, A.; Steinbach, P.J.; et al. Glassy behavior of a protein. Phys. Rev. Lett. 1989, 62, 1916–1919. [Google Scholar] [CrossRef] [PubMed]
- Totrov, M.; Abagyan, R. Flexible ligand docking to multiple receptor conformations: A practical alternative. Curr. Opin. Struct. Biol. 2008, 18, 178–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barril, X.; Morley, S.D. Unveiling the Full Potential of Flexible Receptor Docking Using Multiple Crystallographic Structures. J. Med. Chem. 2005, 48, 4432–4443. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Lill, M.A. Utilizing Experimental Data for Reducing Ensemble Size in Flexible-Protein Docking. J. Chem. Inf. Model. 2011, 52, 187–198. [Google Scholar] [CrossRef]
- Gelin, B.R.; Karplus, M. Role of structural flexibility in conformational calculations. Application to acetylcholine and .beta.-methylacetylcholine. J. Am. Chem. Soc. 1975, 97, 6996–7006. [Google Scholar] [CrossRef]
- Luo, G.; Andricioaei, I.; Xie, X.S.; Karplus, M. Dynamic Distance Disorder in Proteins Is Caused by Trapping. J. Phys. Chem. B 2006, 110, 9363–9367. [Google Scholar] [CrossRef]
- Meroz, Y.; Ovchinnikov, V.; Karplus, M. Coexisting origins of subdiffusion in internal dynamics of proteins. Phys. Rev. E 2017, 95, 062403. [Google Scholar] [CrossRef] [Green Version]
- Conti, S.; Ovchinnikov, V.; Faris, J.G.; Chakraborty, A.K.; Karplus, M.; Sprenger, K.G. Multiscale affinity maturation simulations to elicit broadly neutralizing antibodies against HIV. PLOS Comput. Biol. 2022, 18, e1009391. [Google Scholar] [CrossRef]
- Zhou, T.; Georgiev, I.; Wu, X.; Yang, Z.-Y.; Dai, K.; Finzi, A.; Kwon, Y.D.; Scheid, J.F.; Shi, W.; Xu, L.; et al. Structural Basis for Broad and Potent Neutralization of HIV-1 by Antibody VRC01. Science 2010, 329, 811–817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karplus, M.; McCammon, J.A. Molecular dynamics simulations of biomolecules. Nat. Struct. Mol. Biol. 2002, 9, 646–652. [Google Scholar] [CrossRef]
- Hill, T.L.; Gillis, J. An Introduction to Statistical Thermodynamics; Addison-Wesley Publishing Company, Inc.: Reading, MA, USA, 1960. [Google Scholar]
- Frenkel, D.; Smit, B. Understanding Molecular Simulation: From Algorithms to Applications, 2nd ed.; Academic Press: San Diego, CA, USA, 2001. [Google Scholar]
- Wang, J.; Deng, Y.; Roux, B. Absolute Binding Free Energy Calculations Using Molecular Dynamics Simulations with Restraining Potentials. Biophys. J. 2006, 91, 2798–2814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gumbart, J.C.; Roux, B.; Chipot, C. Efficient Determination of Protein–Protein Standard Binding Free Energies from First Principles. J. Chem. Theory Comput. 2013, 9, 3789–3798. [Google Scholar] [CrossRef] [Green Version]
- Shirts, M.R. Best Practices in Free Energy Calculations for Drug Design. In Computational Drug Discovery and Design; Baron, R., Ed.; Springer: New York, NY, USA, 2012; pp. 425–467. [Google Scholar]
- Ovchinnikov, V.; Conti, S.; Lau, E.Y.; Lightstone, F.C.; Karplus, M. Microsecond Molecular Dynamics Simulations of Proteins Using a Quasi-Equilibrium Solvation Shell Model. J. Chem. Theory Comput. 2020, 16, 1866–1881. [Google Scholar] [CrossRef] [PubMed]
- Impagliazzo, A.; Milder, F.; Kuipers, H.; Wagner, M.V.; Zhu, X.; Hoffman, R.M.; Van Meersbergen, R.; Huizingh, J.; Wanningen, P.; Verspuij, J.; et al. A stable trimeric influenza hemagglutinin stem as a broadly protective immunogen. Science 2015, 349, 1301–1306. [Google Scholar] [CrossRef] [Green Version]
- Webb, B.; Sali, A. Comparative Protein Structure Modeling Using MODELLER. In Current Protocols in Bioinformatics; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2002. [Google Scholar]
- Brooks, B.R.; Brooks, C.L.; Mackerell, A.D.; Nilsson, L.; Petrella, R.J.; Roux, B. CHARMM: The biomolecular simulation program. J. Comput. Chem. 2009, 30, 1545–1614. [Google Scholar] [CrossRef]
- Olsson, M.H.M.; Søndergaard, C.R.; Rostkowski, M.; Jensen, J.H. PROPKA3: Consistent Treatment of Internal and Surface Residues in Empirical pKa Predictions. J. Chem. Theory Comput. 2011, 7, 525–537. [Google Scholar] [CrossRef]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef] [Green Version]
- Eastman, P.; Swails, J.; Chodera, J.D.; McGibbon, R.T.; Zhao, Y.; Beauchamp, K.A.; Wang, L.-P.; Simmonett, A.C.; Harrigan, M.P.; Stern, C.D.; et al. OpenMM 7: Rapid development of high performance algorithms for molecular dynamics. PLOS Comput. Biol. 2017, 13, e1005659. [Google Scholar] [CrossRef] [PubMed]
- Ovchinnikov, V.; Nam, K.; Karplus, M. A Simple and Accurate Method To Calculate Free Energy Profiles and Reaction Rates from Restrained Molecular Simulations of Diffusive Processes. J. Phys. Chem. B 2016, 120, 8457–8472. [Google Scholar] [CrossRef]
- Boresch, S.; Tettinger, F.; Leitgeb, M.; Karplus, M. Absolute Binding Free Energies: A Quantitative Approach for Their Calculation. J. Phys. Chem. B 2003, 107, 9535–9551. [Google Scholar] [CrossRef]
- Song, Y.; DiMaio, F.; Wang, R.Y.-R.; Kim, D.; Miles, C.; Brunette, T.; Thompson, J.; Baker, D. High-Resolution Comparative Modeling with RosettaCM. Structure 2013, 21, 1735–1742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conti, S.; Ovchinnikov, V.; Karplus, M. ppdx: Automated modeling of protein-protein interaction descriptors for use with machine learning. J. Comput. Chem. 2022, in press.
- Pierce, B.; Weng, Z. ZRANK: Reranking protein docking predictions with an optimized energy function. Proteins: Struct. Funct. Bioinform. 2007, 67, 1078–1086. [Google Scholar] [CrossRef] [PubMed]
- Pierce, B.; Weng, Z. A combination of rescoring and refinement significantly improves protein docking performance. Proteins Struct. Funct. Bioinform. 2008, 72, 270–279. [Google Scholar] [CrossRef] [Green Version]
- Mintseris, J.; Pierce, B.; Wiehe, K.; Anderson, R.; Chen, R.; Weng, Z. Integrating statistical pair potentials into protein complex prediction. Proteins Struct. Funct. Bioinform. 2007, 69, 511–520. [Google Scholar] [CrossRef] [PubMed]
- Cheng, T.M.-K.; Blundell, T.L.; Fernandez-Recio, J. pyDock: Electrostatics and desolvation for effective scoring of rigid-body protein-protein docking. Proteins Struct. Funct. Bioinform. 2007, 68, 503–515. [Google Scholar] [CrossRef] [PubMed]
- Zacharias, M. ATTRACT: Protein-protein docking in CAPRI using a reduced protein model. Proteins: Struct. Funct. Bioinform. 2005, 60, 252–256. [Google Scholar] [CrossRef] [PubMed]
- Andrusier, N.; Nussinov, R.; Wolfson, H.J. FireDock: Fast interaction refinement in molecular docking. Proteins: Struct. Funct. Bioinform. 2007, 69, 139–159. [Google Scholar] [CrossRef] [PubMed]
- Shen, M.Y.; Sali, A. Statistical potential for assessment and prediction of protein structures. Protein Sci. 2006, 15, 2507–2524. [Google Scholar] [CrossRef] [Green Version]
- Rykunov, D.; Fiser, A. Effects of amino acid composition, finite size of proteins, and sparse statistics on distance-dependent statistical pair potentials. Protein Struct. Funct. Bioinform. 2007, 67, 559–568. [Google Scholar] [CrossRef] [PubMed]
- Rykunov, D.; Fiser, A. New statistical potential for quality assessment of protein models and a survey of energy functions. BMC Bioinform. 2010, 11, 128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anishchenko, I.; Kundrotas, P.J.; Vakser, I.A. Contact Potential for Structure Prediction of Proteins and Protein Complexes from Potts Model. Biophys. J. 2018, 115, 809–821. [Google Scholar] [CrossRef] [Green Version]
- Schymkowitz, J.; Borg, J.; Stricher, F.; Nys, R.; Rousseau, F.; Serrano, L. The FoldX web server: An online force field. Nucleic Acids Res. 2005, 33, W382–W388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guerois, R.; Nielsen, J.E.; Serrano, L. Predicting Changes in the Stability of Proteins and Protein Complexes: A Study of More Than 1000 Mutations. J. Mol. Biol. 2002, 320, 369–387. [Google Scholar] [CrossRef]
- Stranges, P.B.; Kuhlman, B. A comparison of successful and failed protein interface designs highlights the challenges of designing buried hydrogen bonds. Protein Sci. 2012, 22, 74–82. [Google Scholar] [CrossRef] [Green Version]
- Nivón, L.G.; Moretti, R.; Baker, D. A Pareto-Optimal Refinement Method for Protein Design Scaffolds. PLoS ONE 2013, 8, e59004. [Google Scholar] [CrossRef] [Green Version]
- Vangone, A.; Bonvin, A.M. Contacts-based prediction of binding affinity in protein-protein complexes. eLife 2015, 4, e07454. [Google Scholar] [CrossRef]
- Roux, B.; Simonson, T. Implicit solvent models. Biophys. Chem. 1999, 78, 1–20. [Google Scholar] [CrossRef]
- Onufriev, A.V.; Case, D.A. Generalized Born Implicit Solvent Models for Biomolecules. Annu. Rev. Biophys. 2019, 48, 275–296. [Google Scholar] [CrossRef] [PubMed]
- Im, W.; Lee, M.S.; Brooks, C.L. Generalized born model with a simple smoothing function. J. Comput. Chem. 2003, 24, 1691–1702. [Google Scholar] [CrossRef]
- Haberthür, U.; Caflisch, A. FACTS: Fast analytical continuum treatment of solvation. J. Comput. Chem. 2008, 29, 701–715. [Google Scholar] [CrossRef]
- Onufriev, A.; Bashford, D.; Case, D.A. Exploring protein native states and large-scale conformational changes with a modified generalized born model. Protein Struct. Funct. Bioinform. 2004, 55, 383–394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, M.; Petukh, M.; Alexov, E.; Panchenko, A.R. Predicting the Impact of Missense Mutations on Protein–Protein Binding Affinity. J. Chem. Theory Comput. 2014, 10, 1770–1780. [Google Scholar] [CrossRef] [PubMed]
- Murphy, K. Janeway’s Immunobiology, 8th ed.; Garland Science: New York, NY, USA, 2012. [Google Scholar]
- Becker, O.M.; Karplus, M. The topology of multidimensional potential energy surfaces: Theory and application to peptide structure and kinetics. J. Chem. Phys. 1997, 106, 1495–1517. [Google Scholar] [CrossRef] [Green Version]
- Gainza, P.; Roberts, K.E.; Georgiev, I.; Lilien, R.H.; Keedy, D.A.; Chen, C.Y. Chapter Five-osprey: Protein Design with Ensembles, Flexibility, and Provable Algorithms. In Methods in Enzymology; Keating, A.E., Ed.; Academic Press: Piscataway, NJ, USA, 2013; pp. 87–107. [Google Scholar]
- Liu, P.; Kim, B.; Friesner, R.A.; Berne, B.J. Replica exchange with solute tempering: A method for sampling biological systems in explicit water. Proc. Natl. Acad. Sci. USA 2005, 102, 13749–13754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ollikainen, N.; De Jong, R.M.; Kortemme, T. Coupling Protein Side-Chain and Backbone Flexibility Improves the Re-design of Protein-Ligand Specificity. PLOS Comput. Biol. 2015, 11, e1004335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davey, J.A.; Chica, R.A. Improving the accuracy of protein stability predictions with multistate design using a variety of backbone ensembles. Protein Struct. Funct. Bioinform. 2013, 82, 771–784. [Google Scholar] [CrossRef] [PubMed]
- Marze, N.A.; Burman, S.S.R.; Sheffler, W.; Gray, J.J. Efficient flexible backbone protein-protein docking for challenging targets. Bioinformatics 2018, 34, 3461–3469. [Google Scholar] [CrossRef] [Green Version]
- Ovchinnikov, V.; Louveau, J.E.; Barton, J.P.; Karplus, M.; Chakraborty, A.K. Role of framework mutations and antibody flexibility in the evolution of broadly neutralizing antibodies. eLife 2018, 7, e33038. [Google Scholar] [CrossRef] [Green Version]
- Dominguez, C.; Boelens, R.; Bonvin, A.M.J.J. HADDOCK: A Protein−Protein Docking Approach Based on Biochemical or Biophysical Information. J. Am. Chem. Soc. 2003, 125, 1731–1737. [Google Scholar] [CrossRef] [Green Version]
- Mitchell, J.B.O. Machine learning methods in chemoinformatics. WIREs Comput. Mol. Sci. 2014, 4, 468–481. [Google Scholar] [CrossRef] [Green Version]
- LeCun, Y.; Bengio, Y.; Hinton, G. Deep learning. Nature 2015, 521, 436–444. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 93TH057/VRC01 | RSC3/VRC01 | ||
|---|---|---|---|
| Mutation | ΔG | Mutation | ΔG |
| - | −11.31 | - | −10.51 |
| H-T33Y | −11.23 | H-I30A | −10.51 |
| H-G54S | −10.56 | H-T33A | −9.81 |
| H-A56G | −11.37 | H-W47A | −9.32 |
| H-V57T | −11.17 | H-W50A | −9.23 |
| H-P62K | −11.08 | H-K52A | −9.89 |
| H-R61Q | −11.21 | H-R53A | −10.77 |
| H-K52N | −11.13 | H-G54A | −11.78 |
| H-R53N | −11.26 | H-G55A | −9.26 |
| H-V73T | −11.14 | H-V57A | −9.17 |
| H-Y74S | −10.98 | H-N58A | −8.76 |
| H-I30T | −11.26 | H-Y59A | −9.97 |
| H-4rev | −9.63 | H-R61A | −9.53 |
| H-7rev | −9.36 | H-P62A | −10.55 |
| HL-11rev | −10.44 | H-Q64A | −10.85 |
| L-Y28S | −10.65 | H-M69A | −9.99 |
| H-C32S+H-C98A | −11.05 | H-R71A | −8.88 |
| L-iAA | −8.92 | H-V73A | −9.43 |
| L-iSY | −10.38 | H-Y74A | −10.71 |
| H-D99A | −10.45 | ||
| H-Y100A | −9.10 | ||
| H-N100AA | −9.40 | ||
| H-W100BA | −6.09 | ||
| L-V3A | −11.28 | ||
| L-Q27A | −10.74 | ||
| L-Y28A | −9.54 | ||
| L-S30A | −10.65 | ||
| L-Y91A | −8.51 | ||
| L-E96A | −9.20 | ||
| L-F97A | −9.66 | ||
| Rosetta | Modeller | |||
|---|---|---|---|---|
| FACTS | GBSW | FACTS | GBSW | |
| a (Eelec) | 0.0478 | −0.0157 | 0.0225 | −0.0111 |
| b (Evdw) | 0.0961 | −0.0651 | 0.0036 | −0.0687 |
| c (EGB) | 0.0541 | −0.0131 | 0.0307 | −0.0053 |
| d (SASA) | 0.1375 | 0.3614 | 0.2736 | 0.1618 |
| e | −3.65 | −5.39 | −4.77 | −13.60 |
| Pearson: | 0.54 | 0.53 | 0.51 | 0.46 |
| Slope: | 0.29 | 0.28 | 0.26 | 0.21 |
| Intercept: | −7.17 | −7.32 | −7.52 | −8.01 |
| p-value: | 5.63 × 10−5 | 9.48 × 10−5 | 1.85 × 10−4 | 8.79 × 10−4 |
| RMSE: | 0.87 | 0.88 | 0.89 | 0.92 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Conti, S.; Lau, E.Y.; Ovchinnikov, V. On the Rapid Calculation of Binding Affinities for Antigen and Antibody Design and Affinity Maturation Simulations. Antibodies 2022, 11, 51. https://doi.org/10.3390/antib11030051
Conti S, Lau EY, Ovchinnikov V. On the Rapid Calculation of Binding Affinities for Antigen and Antibody Design and Affinity Maturation Simulations. Antibodies. 2022; 11(3):51. https://doi.org/10.3390/antib11030051
Chicago/Turabian StyleConti, Simone, Edmond Y. Lau, and Victor Ovchinnikov. 2022. "On the Rapid Calculation of Binding Affinities for Antigen and Antibody Design and Affinity Maturation Simulations" Antibodies 11, no. 3: 51. https://doi.org/10.3390/antib11030051
APA StyleConti, S., Lau, E. Y., & Ovchinnikov, V. (2022). On the Rapid Calculation of Binding Affinities for Antigen and Antibody Design and Affinity Maturation Simulations. Antibodies, 11(3), 51. https://doi.org/10.3390/antib11030051