
Mitochondrial Autoantibodies and the Role of Apoptosis in Pemphigus Vulgaris


Abstract
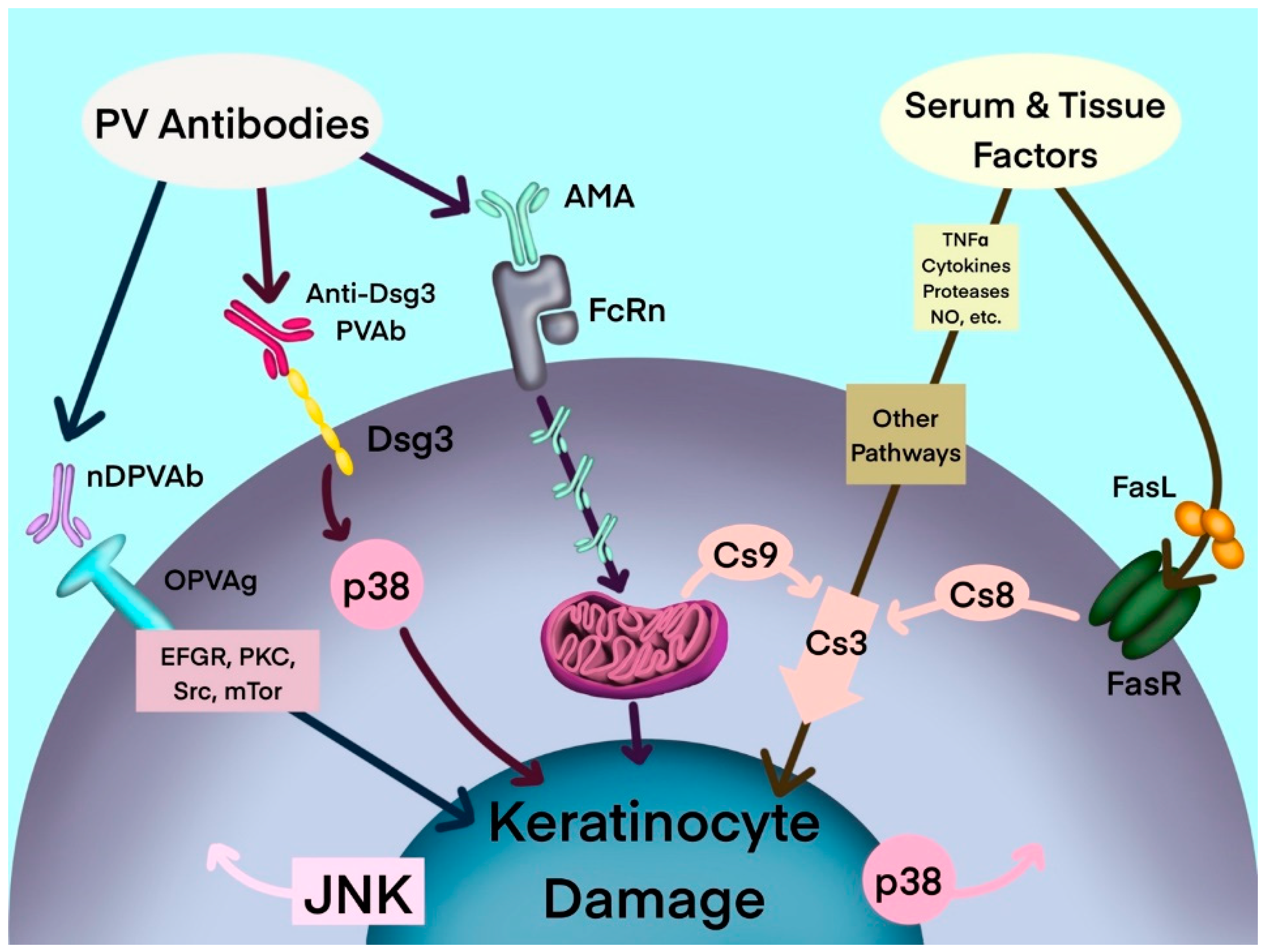
:1. Introduction
2. Apoptolysis
3. Mitochondrial Damage by AMA in PV
4. Efficacy of Mitochondrion Protective Agents in Pemphigus Patients
5. Conclusions and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Disclaimer
References
- Lever, W.F.; Talbott, J.H. Pemphigus: A historical study. Arch. Dermatol. Syphilol. 1942, 46, 800–823. [Google Scholar] [CrossRef]
- Dickson, S. Observations on Pemphigus. Lond. Med. J. 1788, 9, 309–324. [Google Scholar] [PubMed]
- Beutner, E.H.; Jordon, R.E. Demonstration of skin antibodies in sera of pemphigus vulgaris patients by indirect immunofluorescent staining. Proc. Soc. Exp. Biol. Med. 1964, 117, 505–510. [Google Scholar] [CrossRef]
- Ahmed, A.R.; Carrozzo, M.; Caux, F.; Cirillo, N.; Dmochowski, M.; Alonso, A.E.; Gniadecki, R.; Hertl, M.; López-Zabalza, M.J.; Lotti, R.; et al. Monopathogenic vs. multipathogenic explanations of pemphigus pathophysiology. Exp. Dermatol. 2016, 25, 839–846. [Google Scholar] [CrossRef] [PubMed]
- Grando, S.A. Pemphigus autoimmunity: Hypotheses and realities. Autoimmunity 2012, 45, 7–35. [Google Scholar] [CrossRef] [PubMed]
- Amber, K.T.; Valdebran, M.; Grando, S.A. Non-Desmoglein Antibodies in Patients with Pemphigus Vulgaris. Front. Immunol. 2018, 9, 1190. [Google Scholar] [CrossRef]
- Grando, S.A. Autoimmunity to keratinocyte acetylcholine receptors in pemphigus. Dermatology 2000, 201, 290–295. [Google Scholar] [CrossRef] [PubMed]
- Chernyavsky, A.; Amber, K.T.; Agnoletti, A.F.; Wang, C.; Grando, S.A. Synergy among non-desmoglein antibodies contributes to the immunopathology of desmoglein antibody-negative pemphigus vulgaris. J. Biol. Chem. 2019, 294, 4520–4528. [Google Scholar] [CrossRef]
- Sharma, P.; Mao, X.; Payne, A.S. Beyond steric hindrance: The role of adhesion signaling pathways in the pathogenesis of pemphigus. J. Dermatol. Sci. 2007, 48, 1–14. [Google Scholar] [CrossRef]
- Grando, S.A.; Bystryn, J.C.; Chernyavsky, A.I.; Frušić-Zlotkin, M.; Gniadecki, R.; Lotti, R.; Milner, Y.; Pittelkow, M.R.; Pincelli, C. Apoptolysis: A novel mechanism of skin blistering in pemphigus vulgaris linking the apoptotic pathways to basal cell shrinkage and suprabasal acantholysis. Exp. Dermatol. 2009, 18, 764–770. [Google Scholar] [CrossRef]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef] [PubMed]
- Yan, G.; Elbadawi, M.; Efferth, T. Multiple cell death modalities and their key features (Review). World Acad. Sci. J. 2020, 2, 39–48. [Google Scholar] [CrossRef]
- Gupta, S.; Kass, G.E.; Szegezdi, E.; Joseph, B. The mitochondrial death pathway: A promising therapeutic target in diseases. J. Cell. Mol. Med. 2009, 13, 1004–1033. [Google Scholar] [CrossRef] [PubMed]
- Elmore, S. Apoptosis: A Review of Programmed Cell Death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef]
- Hasegawa, A.; Abe, R. Recent advances in managing and understanding Stevens-Johnson syndrome and toxic epidermal necrolysis [version 1; peer review: 2 approved]. F1000Research 2020, 9, 612. [Google Scholar] [CrossRef]
- Arredondo, J.; Chernyavsky, A.I.; Karaouni, A.; Grando, S.A. Novel mechanisms of target cell death and survival and of therapeutic action of IVIg in Pemphigus. Am. J. Pathol. 2005, 167, 1531–1544. [Google Scholar] [CrossRef]
- Lotti, R.; Shu, E.; Petrachi, T.; Marconi, A.; Palazzo, E.; Quadri, M.; Lin, A.; O’Reilly, L.A.; Pincelli, C. Soluble Fas Ligand Is Essential for Blister Formation in Pemphigus. Front. Immunol. 2018, 9, 370. [Google Scholar] [CrossRef] [PubMed]
- Kalantari-Dehaghi, M.; Chen, Y.; Deng, W.; Chernyavsky, A.; Marchenko, S.; Wang, P.H.; Grando, S.A. Mechanisms of mitochondrial damage in keratinocytes by pemphigus vulgaris antibodies. J. Biol. Chem. 2013, 288, 16916–16925. [Google Scholar] [CrossRef]
- Marchenko, S.; Chernyavsky, A.I.; Arredondo, J.; Gindi, V.; Grando, S.A. Antimitochondrial autoantibodies in pemphigus vulgaris: A missing link in disease pathophysiology. J. Biol. Chem. 2010, 285, 3695–3704. [Google Scholar] [CrossRef]
- Chernyavsky, A.; Chen, Y.; Wang, P.H.; Grando, S.A. Pemphigus vulgaris antibodies target the mitochondrial nicotinic acetylcholine receptors that protect keratinocytes from apoptolysis. Int. Immunopharmacol. 2015, 29, 76–80. [Google Scholar] [CrossRef]
- Chen, Y.; Chernyavsky, A.; Webber, R.J.; Grando, S.A.; Wang, P.H. Critical Role of the Neonatal Fc Receptor (FcRn) in the Pathogenic Action of Antimitochondrial Autoantibodies Synergizing with Anti-desmoglein Autoantibodies in Pemphigus Vulgaris. J. Biol. Chem. 2015, 290, 23826–23837. [Google Scholar] [CrossRef] [PubMed]
- Grando, S.A. Cholinergic control of epidermal cohesion. Exp. Dermatol. 2006, 15, 265–282. [Google Scholar] [CrossRef]
- Nguyen, V.T.; Ndoye, A.; Grando, S.A. Pemphigus vulgaris antibody identifies pemphaxin. A novel keratinocyte annexin-like molecule binding acetylcholine. J. Biol. Chem. 2000, 275, 29466–29476. [Google Scholar] [CrossRef] [PubMed]
- Chernyavsky, A.; Patel, K.G.; Grando, S.A. Mechanisms of synergy of autoantibodies to M3 muscarinic acetylcholine receptor and secretory pathway Ca(2+)/Mn(2+)-ATPase isoform 1 in patients with non-desmoglein pemphigus vulgaris. Int. Immunopharmacol. 2020, 80, 106149. [Google Scholar] [CrossRef]
- Okunade, G.W.; Miller, M.L.; Azhar, M.; Andringa, A.; Sanford, L.P.; Doetschman, T.; Prasad, V.; Shull, G.E. Loss of the Atp2c1 secretory pathway Ca(2+)-ATPase (SPCA1) in mice causes Golgi stress, apoptosis, and midgestational death in homozygous embryos and squamous cell tumors in adult heterozygotes. J. Biol. Chem. 2007, 282, 26517–26527. [Google Scholar] [CrossRef]
- Mancini, M.; Machamer, C.E.; Roy, S.; Nicholson, D.W.; Thornberry, N.A.; Casciola-Rosen, L.A.; Rosen, A. Caspase-2 is localized at the Golgi complex and cleaves golgin-160 during apoptosis. J. Cell Biol. 2000, 149, 603–612. [Google Scholar] [CrossRef] [PubMed]
- Qin, Y.; Vanden Hoek, T.L.; Wojcik, K.; Anderson, T.; Li, C.-Q.; Shao, Z.-H.; Becker, L.B.; Hamann, K.J. Caspase-dependent cytochrome c release and cell death in chick cardiomyocytes after simulated ischemia-reperfusion. Am. J. Physiol. Heart Circ. Physiol. 2004, 286, H2280–H2286. [Google Scholar] [CrossRef]
- Micaroni, M.; Giacchetti, G.; Plebani, R.; Xiao, G.G.; Federici, L. ATP2C1 gene mutations in Hailey-Hailey disease and possible roles of SPCA1 isoforms in membrane trafficking. Cell Death Dis. 2016, 7, e2259. [Google Scholar] [CrossRef]
- Cauza, K.; Hinterhuber, G.; Dingelmaier-Hovorka, R.; Brugger, K.; Klosner, G.; Horvat, R.; Wolff, K.; Foedinger, D. Expression of FcRn, the MHC Class I-Related Receptor for IgG, in Human Keratinocytes. J. Investig. Dermatol. 2005, 124, 132–139. [Google Scholar] [CrossRef]
- Wei, B.; Li, F. Mechanisms of Trx2/ASK1-Mediated Mitochondrial Injury in Pemphigus Vulgaris. BioMed Res. Int. 2021, 2021, 2471518. [Google Scholar] [CrossRef]
- Halestrap, A.P.; Connern, C.P.; Griffiths, E.J.; Kerr, P. Cyclosporin A binding to mitochondrial cyclophilin inhibits the permeability transition pore and protects hearts from ischaemia/reperfusion injury. Mol. Cell. Biochem. 1997, 174, 167–172. [Google Scholar] [CrossRef] [PubMed]
- Russell, G.; Graveley, R.; Seid, J.; Al-Humidan, A.-K.; Skjodt, H. Mechanisms of action of cyclosporine and effects on connective tissues. Semin. Arthritis Rheum. 1992, 21, 16–22. [Google Scholar] [CrossRef]
- Kang, H.T.; Hwang, E.S. Nicotinamide enhances mitochondria quality through autophagy activation in human cells. Aging Cell 2009, 8, 426–438. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhu, S.; Drozda, M.; Zhang, W.; Stavrovskaya, I.G.; Cattaneo, E.; Ferrante, R.J.; Kristal, B.S.; Friedlander, R.M. Minocycline inhibits caspase-independent and -dependent mitochondrial cell death pathways in models of Huntington’s disease. Proc. Natl. Acad. Sci. USA 2003, 100, 10483–10487. [Google Scholar] [CrossRef]
- Barthelemy, H.; Frappaz, A.; Cambazard, F.; Mauduit, G.; Rouchouse, B.; Kanitakis, J.; Souteyrand, P.; Claudy, A.; Thivolet, J. Treatment of nine cases of pemphigus vulgaris with cyclosporine. J. Am. Acad. Dermatol. 1988, 18, 1262–1266. [Google Scholar] [CrossRef]
- Chaffins, M.L.; Collison, D.; Fivenson, D.P. Treatment of pemphigus and linear IgA dermatosis with nicotinamide and tetracycline: A review of 13 cases. J. Am. Acad. Dermatol. 1993, 28, 998–1000. [Google Scholar] [CrossRef]
- Sawai, T.; Kitazawa, K.; Danno, K.; Sugie, N.; Mochizuki, T.; Sugiura, H.; Uehara, M. Pemphigus vegetans with oesophageal involvement: Successful treatment with minocycline and nicotinamide. Br. J. Dermatol. 1995, 132, 668–670. [Google Scholar] [CrossRef]
- Mehta, J.N.; Martin, A.G. A Case of Pemphigus Vulgaris Improved by Cigarette Smoking. Arch. Dermatol. 2000, 136, 15–17. [Google Scholar] [CrossRef]
- Brenner, S.; Tur, E.; Shapiro, J.; Ruocco, V.; D’Avino, M.; Ruocco, E.; Tsankov, N.; Vassileva, S.; Drenovska, K.; Brezoev, P.; et al. Pemphigus vulgaris: Environmental factors. Occupational, behavioral, medical, and qualitative food frequency questionnaire. Int. J. Dermatol. 2001, 40, 562–569. [Google Scholar] [CrossRef]
- Sullivan, T.P.; Elgart, G.W.; Kirsner, R.S. Pemphigus and smoking. Int. J. Dermatol. 2002, 41, 528–530. [Google Scholar] [CrossRef]
- Valikhani, M.; Kavusi, S.; Chams-Davatchi, C.; Hallaji, Z.; Esmaili, N.; Ghandi, N.; Farahani, F.; Lajevardi, V. Impact of smoking on pemphigus. Int. J. Dermatol. 2008, 47, 567–570. [Google Scholar] [CrossRef] [PubMed]
- Pretel, M.; España, A.; Marquina, M.; Pelacho, B.; López-Picazo, J.M.; López-Zabalza, M.J. An imbalance in Akt/mTOR is involved in the apoptotic and acantholytic processes in a mouse model of pemphigus vulgaris. Exp. Dermatol. 2009, 18, 771–780. [Google Scholar] [CrossRef] [PubMed]
- Cheema, N.; Cameron, J.M.; Hood, D.A. Effect of rapamycin on mitochondria and lysosomes in fibroblasts from patients with mtDNA mutations. Am. J. Physiol.-Cell Physiol. 2021, 321, C176–C186. [Google Scholar] [CrossRef] [PubMed]
- Grando, S.A. Retrospective analysis of a single-center clinical experience toward development of curative treatment of 123 pemphigus patients with a long-term follow-up: Efficacy and safety of the multidrug protocol combining intravenous immunoglobulin with the cytotoxic immunosuppressor and mitochondrion-protecting drugs. Int. J. Dermatol. 2019, 58, 114–125. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Symbol | Antigen | Localization on Mitochondria | Frequency | |
|---|---|---|---|---|
| PV (%) | Control (%) | |||
| ABAT-V1 | 4-Aminobutyrate aminotransferase, mitochondrial; 50 kDa | Matrix | 19 | 4 |
| ALDH4A1 | Aldehyde dehydrogenase 4 family, member A1 | Matrix | 23 | 5 |
| CPT1B | Carnitine O-palmitoyltransferase 1B | Outer membrane | 18 | 5 |
| CRAT | Carnitine O-acetyltransferase | Inner membrane | 28 | 7 |
| CYB5B | Cytochrome b5 type B; 21 kDa | Outer membrane | 19 | 1 |
| ETFA | Electron transfer flavoprotein, α protein | Matrix | 19 | 4 |
| ETFB | Electron transfer flavoprotein, β protein | Matrix | 21 | 3 |
| FDXR-V2 | NADPH:adrenodoxin oxidoreductase | Matrix | 25 | 6 |
| FH | Fumarate hydratase (fumarase) | Mitochondrion | 29 | 3 |
| MAOB | Amine oxidase (flavin-containing) B | Outer membrane | 27 | 5 |
| ME2 | NAD-dependent malic enzyme | Matrix | 18 | 6 |
| ME3 | NADP-dependent malic enzyme, mitochondrial | Matrix | 23 | 8 |
| MLYCD | Malonyl-CoA decarboxylase | Mitochondrion | 29 | 4 |
| NDUFA9 | NADH dehydrogenase [ubiquinone] 1α subcomplex subunit 9; 39 kDa | Matrix | 20 | 3 |
| NDUFA13 | NADH dehydrogenase [ubiquinone] 1α subcomplex subunit 13; 16 kDa | Inner membrane | 24 | 6 |
| NDUFB10 | NADH dehydrogenase [ubiquinone] 1β subcomplex subunit 10 | Matrix | 17 | 2 |
| NDUFV3 | NADH dehydrogenase [ubiquinone] flavoprotein 3; 9 kDa | Inner membrane | 19 | 4 |
| NDUFS6 | NADH dehydrogenase [ubiquinone] iron-sulfur protein 6; 13 kDa | Inner membrane | 24 | 6 |
| PC | Pyruvate carboxylase | Matrix | 32 | 5 |
| PDK4 | Pyruvate dehydrogenase kinase, isozyme 4 | Matrix | 24 | 4 |
| PDHA1 | Pyruvate dehydrogenase E1 component α subunit, somatic form | Glycolysis | 30 | 3 |
| PMPCB | Mitochondrial processing peptidase β subunit | Mitochondrial organization | 31 | 4 |
| PRODH | Proline oxidase | Matrix | 25 | 6 |
| SOD2 | Superoxide dismutase [Mn] | Matrix | 23 | 2 |
| TIMM44 | Mitochondrial import inner membrane translocase subunit | Inner membrane | 20 | 4 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hutchison, D.M.; Hosking, A.-M.; Hong, E.M.; Grando, S.A. Mitochondrial Autoantibodies and the Role of Apoptosis in Pemphigus Vulgaris. Antibodies 2022, 11, 55. https://doi.org/10.3390/antib11030055
Hutchison DM, Hosking A-M, Hong EM, Grando SA. Mitochondrial Autoantibodies and the Role of Apoptosis in Pemphigus Vulgaris. Antibodies. 2022; 11(3):55. https://doi.org/10.3390/antib11030055
Chicago/Turabian StyleHutchison, Dana M., Anna-Marie Hosking, Ellen M. Hong, and Sergei A. Grando. 2022. "Mitochondrial Autoantibodies and the Role of Apoptosis in Pemphigus Vulgaris" Antibodies 11, no. 3: 55. https://doi.org/10.3390/antib11030055
APA StyleHutchison, D. M., Hosking, A. -M., Hong, E. M., & Grando, S. A. (2022). Mitochondrial Autoantibodies and the Role of Apoptosis in Pemphigus Vulgaris. Antibodies, 11(3), 55. https://doi.org/10.3390/antib11030055