
Asymmetric Fc Engineering for Bispecific Antibodies with Reduced Effector Function

 and
and
Abstract
:1. Introduction
2. Results
2.1. Preparation and Expression of Antibody Constructs (Heteromultimers)
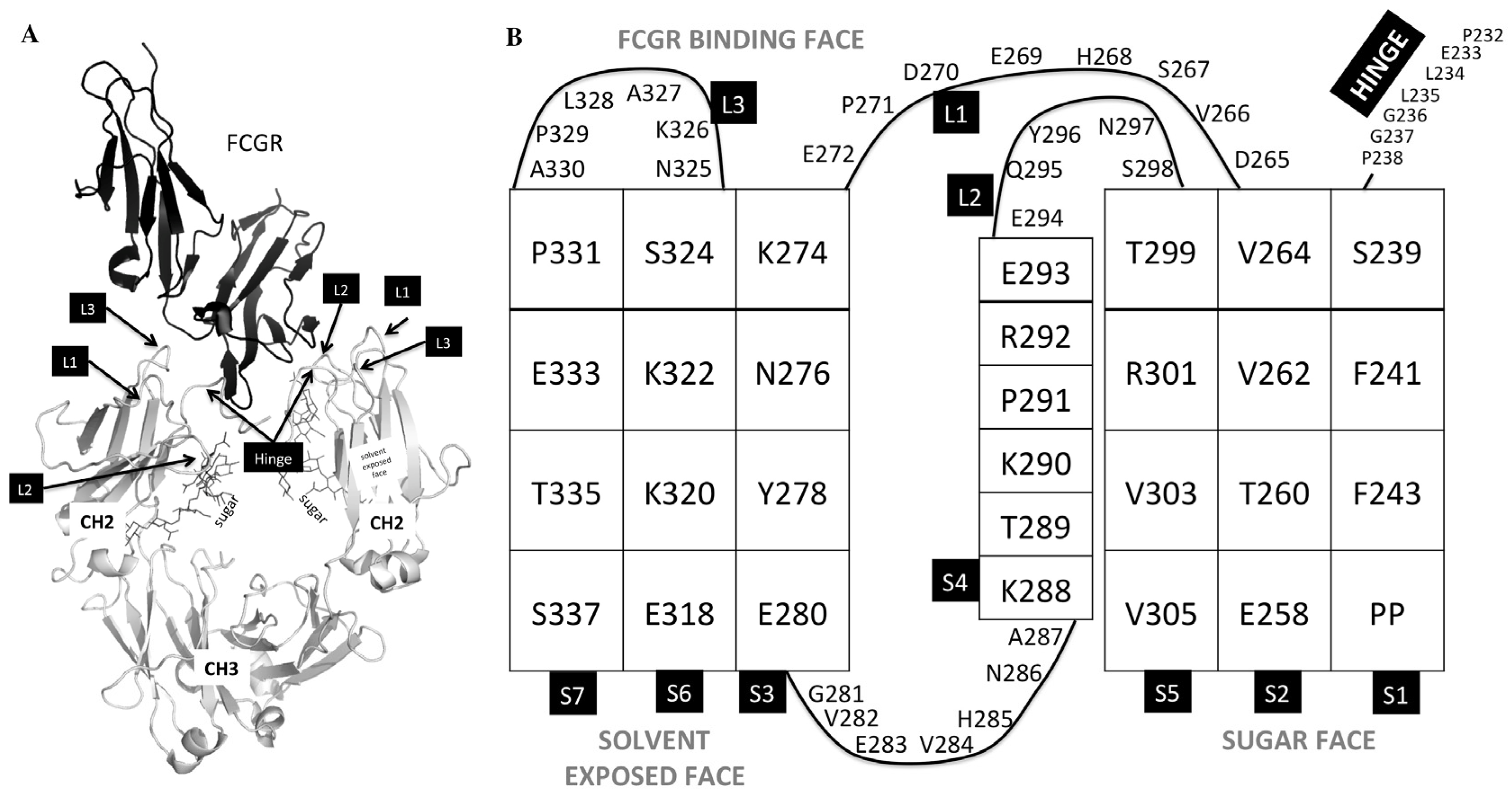
2.2. SPR Binding to Fcγ Receptors
2.3. Asymmetric Antibody Constructs Do Not Bind C1q
2.4. Asymmetric Antibody Constructs Bind to FcRn
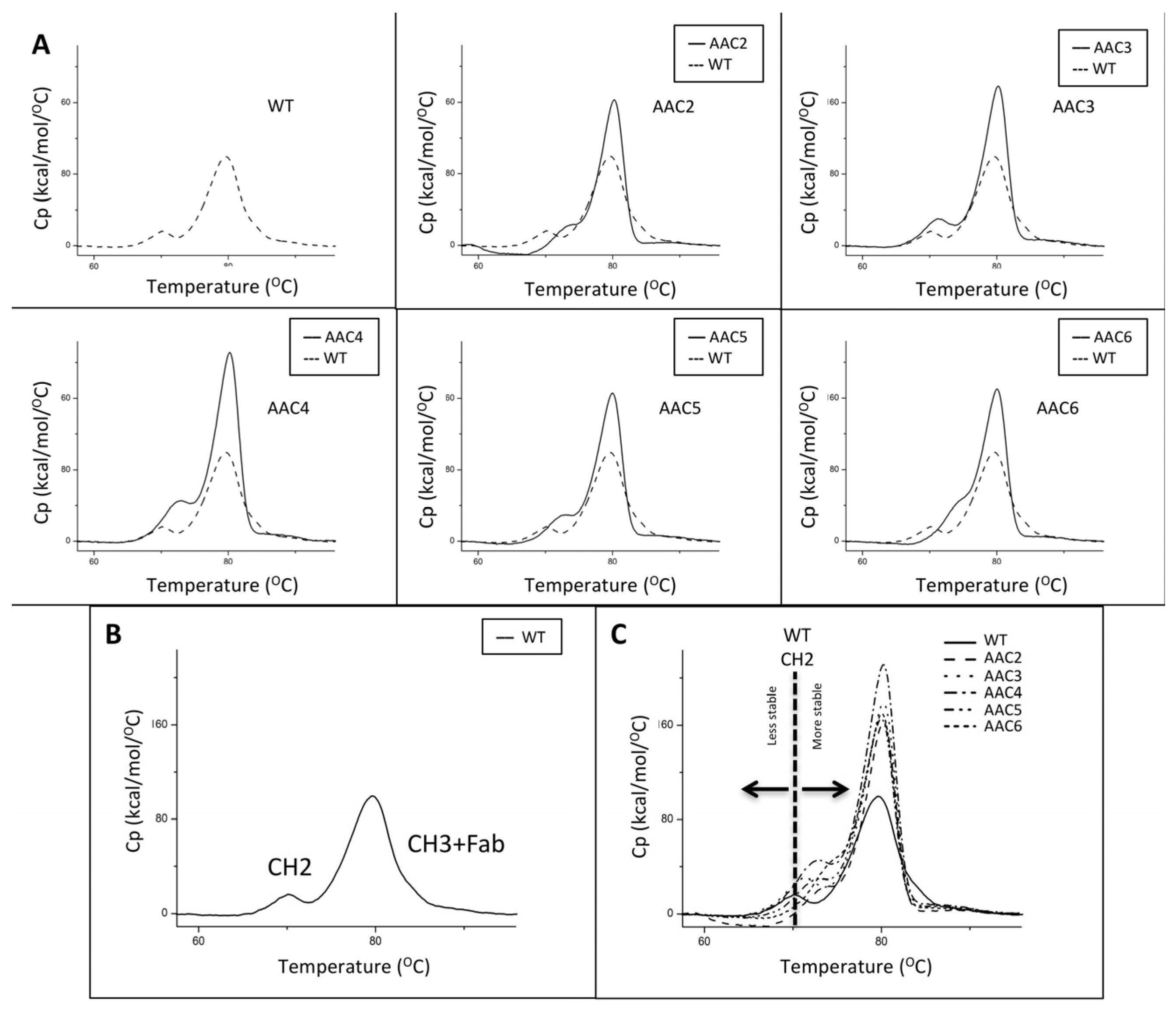
2.5. Asymmetric Antibody Constructs Are Thermally Stable
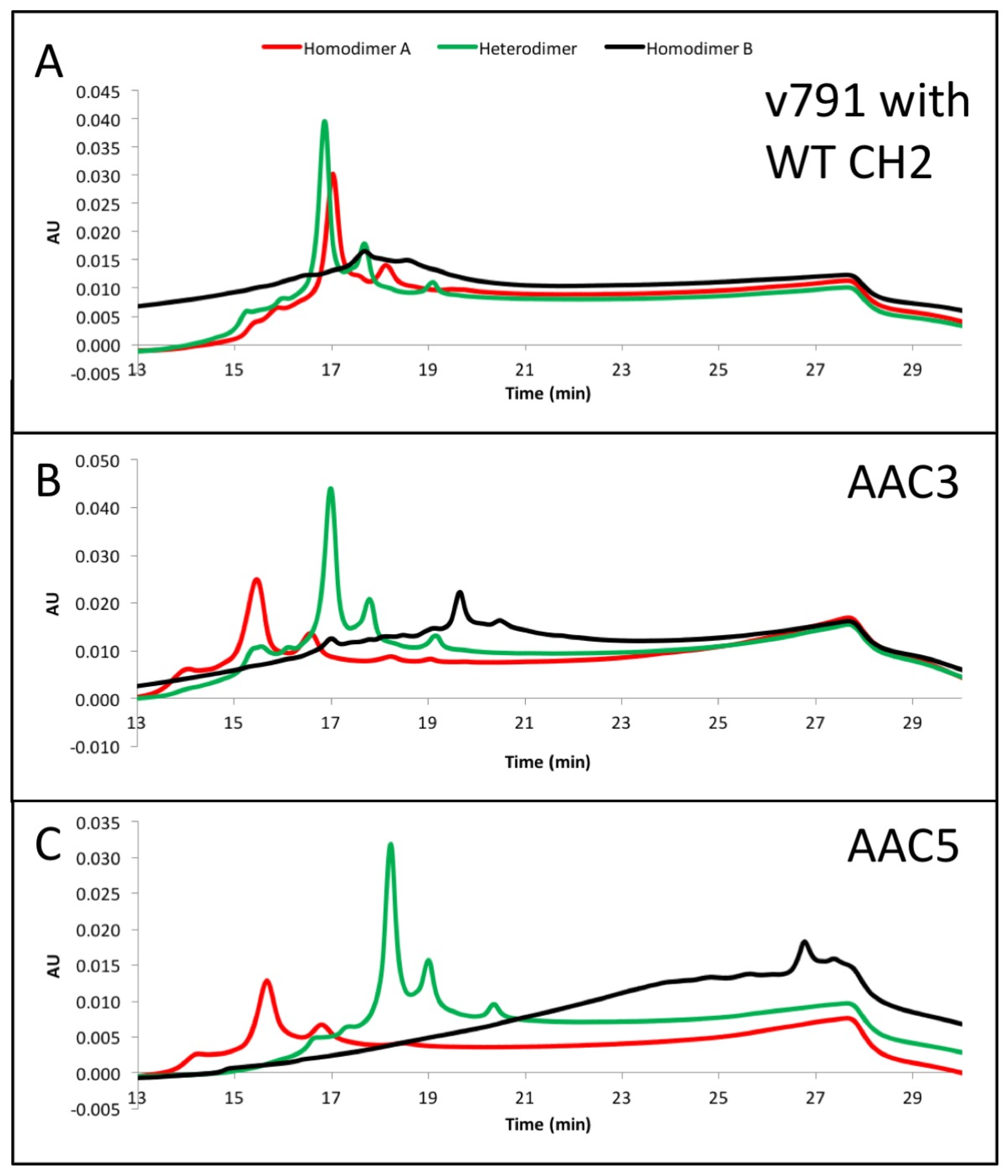
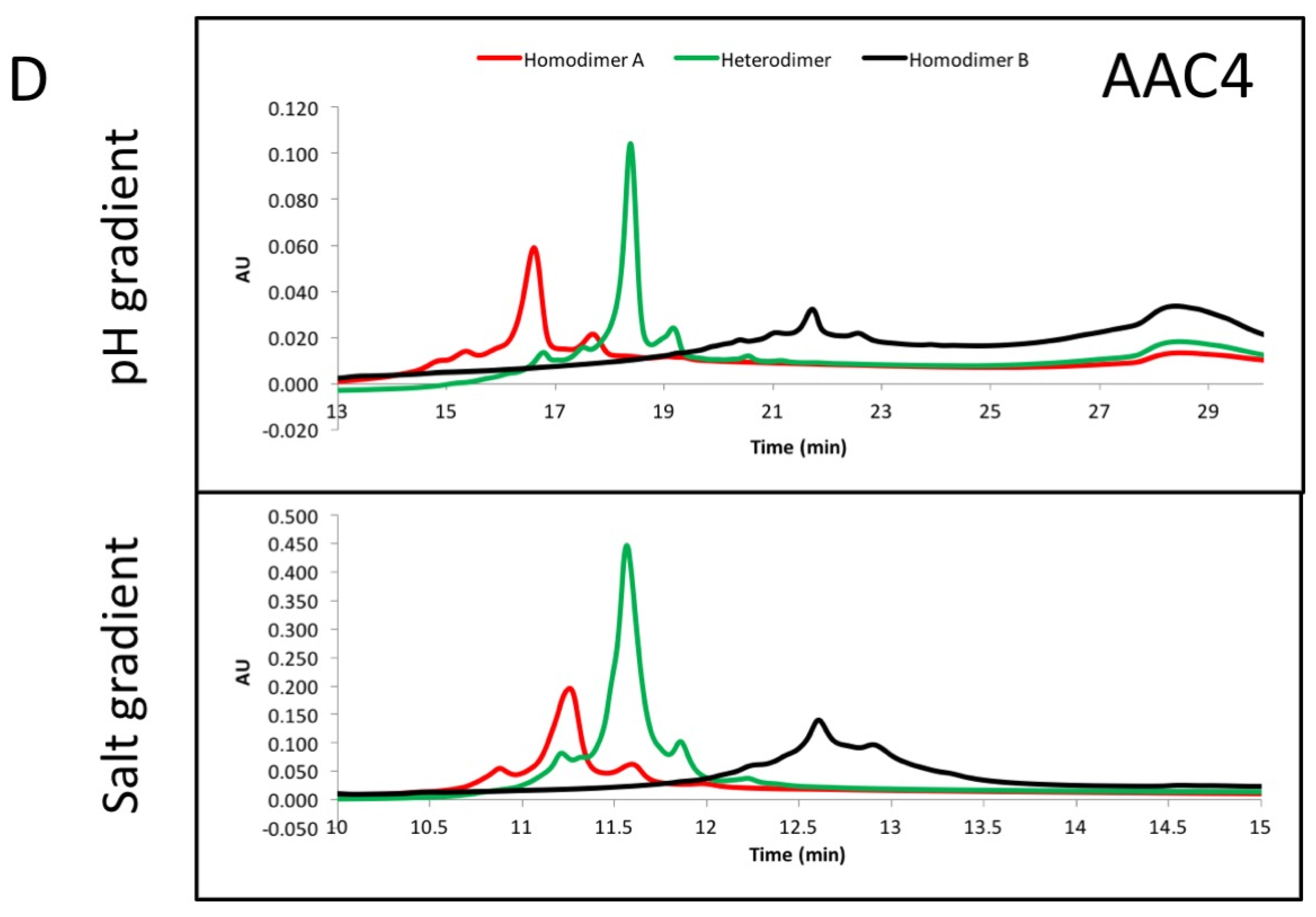
2.6. IEX Analysis of Asymmetric Antibody
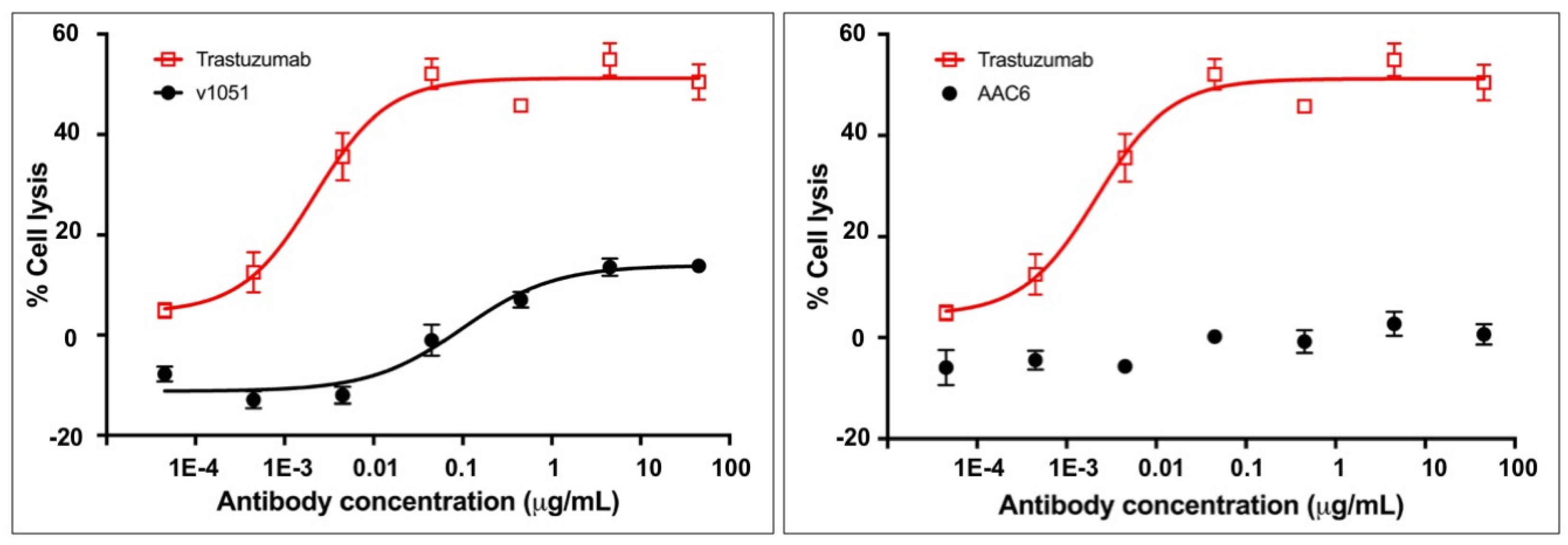
2.7. Asymmetric Antibody Construct 6 Based on Trastuzumab Do Not Stimulate ADCC (Antibody-Dependent Cell-Mediated Cytotoxicity) in SK-BR-3 Cells
2.8. Design from Asymmetric Antibody Construct 5 Is Selected as Lead
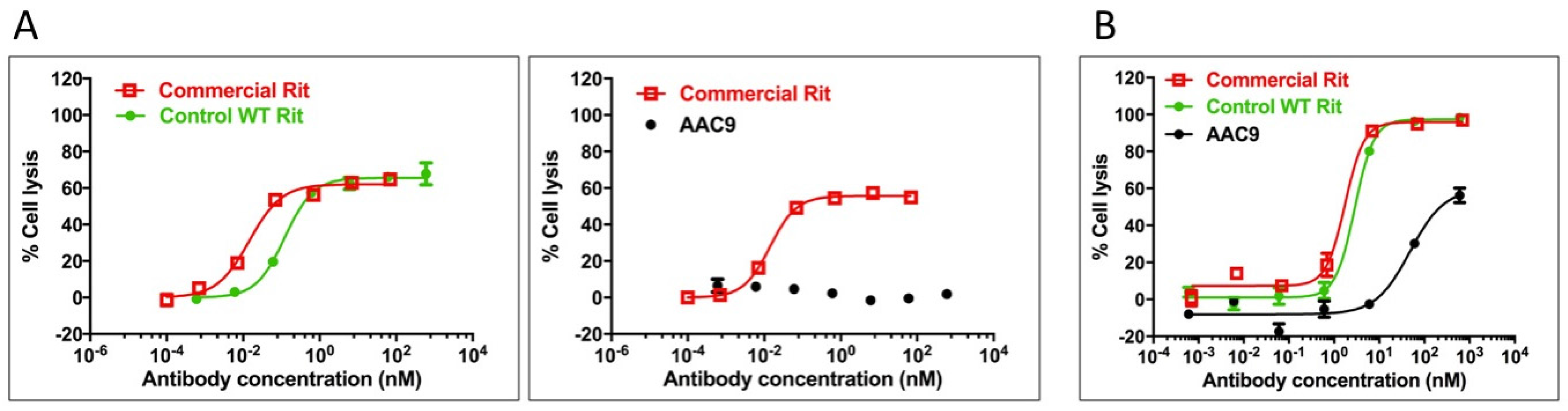
2.9. Asymmetric Antibody Construct Based on Rituximab Do Not Stimulate ADCC in Daudi Cells
2.10. Asymmetric Antibody Construct Based on Rituximab Reduce CDC (Complement-Dependent Cytotoxicity) in Daudi Cells
3. Discussion
4. Materials and Methods
4.1. Expression and Purification of the Samples
4.1.1. Antibody Constructs
4.1.2. FcγRs and FcRn
4.2. Surface Plasmon Resonance
4.2.1. FcγR Binding against Trastuzumab and Rituximab Constructs
4.2.2. C1q
4.2.3. FcRn
4.3. Differential Scanning Calorimetry
4.4. UPLC IEX
4.5. ADCC (Antibody-Dependent Cell-Mediated Cytotoxicity)
4.6. Asymmetric Antibody Constructs Based on Rituximab Reduce CDC (Complement-Dependent Cytotoxicity) in Daudi Cells
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Daeron, M. Fc receptor biology. Annu. Rev. Immunol. 1997, 15, 203–234. [Google Scholar] [CrossRef] [PubMed]
- Ward, E.S.; Ghetie, V. The effector functions of immunoglobulins: Implications for therapy. Ther. Immunol. 1995, 2, 77–94. [Google Scholar] [PubMed]
- Ravetch, J.V.; Kinet, J.P. Fc receptors. Annu. Rev. Immunol. 1991, 9, 457–492. [Google Scholar] [CrossRef] [PubMed]
- Strohl, W.R. Optimization of fc-mediated effector functions of monoclonal antibodies. Curr. Opin. Biotechnol. 2009, 20, 685–691. [Google Scholar] [CrossRef] [PubMed]
- Labrijn, A.F.; Aalberse, R.C.; Schuurman, J. When binding is enough: Nonactivating antibody formats. Curr. Opin. Immunol. 2008, 20, 479–485. [Google Scholar] [CrossRef] [PubMed]
- Strohl, W.R.; Strohl, L.M. Therapeutic Antibody Engineering; Woodhead Publishing: Cambridge, UK, 2012; pp. 225–249. [Google Scholar]
- Schlothauer, T.; Herter, S.; Koller, C.F.; Grau-Richards, S.; Steinhart, V.; Spick, C.; Kubbies, M.; Klein, C.; Umana, P.; Mossner, E. Novel human igg1 and igg4 fc-engineered antibodies with completely abolished immune effector functions. Protein Eng. Des. Sel. 2016, 29, 457–466. [Google Scholar] [CrossRef] [PubMed]
- Jefferis, R.; Lund, J. Interaction sites on human igg-fc for fcgammar: Current models. Immunol. Lett. 2002, 82, 57–65. [Google Scholar] [CrossRef]
- Tao, M.H.; Morrison, S.L. Studies of aglycosylated chimeric mouse-human igg. Role of carbohydrate in the structure and effector functions mediated by the human igg constant region. J. Immunol. 1989, 143, 2595–2601. [Google Scholar] [PubMed]
- Jacobsen, F.W.; Stevenson, R.; Li, C.; Salimi-Moosavi, H.; Liu, L.; Wen, J.; Luo, Q.; Daris, K.; Buck, L.; Miller, S.; et al. Engineering an igg scaffold lacking effector function with optimized developability. J. Biol. Chem. 2017, 292, 1865–1875. [Google Scholar] [CrossRef] [PubMed]
- Gillies, S.D.; Lan, Y.; Lo, K.M.; Super, M.; Wesolowski, J. Improving the efficacy of antibody-interleukin 2 fusion proteins by reducing their interaction with fc receptors. Cancer Res. 1999, 59, 2159–2166. [Google Scholar] [PubMed]
- Newman, R.; Hariharan, K.; Reff, M.; Anderson, D.R.; Braslawsky, G.; Santoro, D.; Hanna, N.; Bugelski, P.J.; Brigham-Burke, M.; Crysler, C.; et al. Modification of the fc region of a primatized igg antibody to human cd4 retains its ability to modulate cd4 receptors but does not deplete cd4(+) t cells in chimpanzees. Clin. Immunol. 2001, 98, 164–174. [Google Scholar] [CrossRef] [PubMed]
- Vafa, O.; Gilliland, G.L.; Brezski, R.J.; Strake, B.; Wilkinson, T.; Lacy, E.R.; Scallon, B.; Teplyakov, A.; Malia, T.J.; Strohl, W.R. An engineered fc variant of an igg eliminates all immune effector functions via structural perturbations. Methods 2014, 65, 114–126. [Google Scholar] [CrossRef] [PubMed]
- An, Z.; Forrest, G.; Moore, R.; Cukan, M.; Haytko, P.; Huang, L.; Vitelli, S.; Zhao, J.Z.; Lu, P.; Hua, J.; et al. Igg2m4, an engineered antibody isotype with reduced fc function. MAbs 2009, 1, 572–579. [Google Scholar] [CrossRef] [PubMed]
- Shields, R.L.; Namenuk, A.K.; Hong, K.; Meng, Y.G.; Rae, J.; Briggs, J.; Xie, D.; Lai, J.; Stadlen, A.; Li, B.; et al. High resolution mapping of the binding site on human igg1 for fc gamma ri, fc gamma rii, fc gamma riii, and fcrn and design of igg1 variants with improved binding to the fc gamma r. J. Biol. Chem. 2001, 276, 6591–6604. [Google Scholar] [CrossRef] [PubMed]
- Lund, J.; Pound, J.D.; Jones, P.T.; Duncan, A.R.; Bentley, T.; Goodall, M.; Levine, B.A.; Jefferis, R.; Winter, G. Multiple binding sites on the ch2 domain of igg for mouse fc gamma r11. Mol. Immunol. 1992, 29, 53–59. [Google Scholar] [CrossRef]
- Tamm, A.; Schmidt, R.E. Igg binding sites on human fc gamma receptors. Int. Rev. Immunol. 1997, 16, 57–85. [Google Scholar] [CrossRef] [PubMed]
- Ridgway, J.B.; Presta, L.G.; Carter, P. ‘Knobs-into-holes’ engineering of antibody ch3 domains for heavy chain heterodimerization. Protein Eng. 1996, 9, 617–621. [Google Scholar] [CrossRef] [PubMed]
- Gunasekaran, K.; Pentony, M.; Shen, M.; Garrett, L.; Forte, C.; Woodward, A.; Ng, S.B.; Born, T.; Retter, M.; Manchulenko, K.; et al. Enhancing antibody fc heterodimer formation through electrostatic steering effects: Applications to bispecific molecules and monovalent igg. J. Biol. Chem. 2010, 285, 19637–19646. [Google Scholar] [CrossRef] [PubMed]
- Von Kreudenstein, T.S.; Escobar-Carbrera, E.; Lario, P.I.; D'Angelo, I.; Brault, K.; Kelly, J.; Durocher, Y.; Baardsnes, J.; Woods, R.J.; Xie, M.H.; et al. Improving biophysical properties of a bispecific antibody scaffold to aid developability: Quality by molecular design. MAbs 2013, 5, 646–654. [Google Scholar] [CrossRef] [PubMed]
- Lund, J.; Winter, G.; Jones, P.T.; Pound, J.D.; Tanaka, T.; Walker, M.R.; Artymiuk, P.J.; Arata, Y.; Burton, D.R.; Jefferis, R.; et al. Human fc gamma ri and fc gamma rii interact with distinct but overlapping sites on human igg. J. Immunol. 1991, 147, 2657–2662. [Google Scholar] [PubMed]
- Ecker, D.M.; Jones, S.D.; Levine, H.L. The therapeutic monoclonal antibody market. MAbs 2015, 7, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Kontermann, R.E.; Brinkmann, U. Bispecific antibodies. Drug Discov. Today 2015, 20, 838–847. [Google Scholar] [CrossRef] [PubMed]
- Leaver-Fay, A.; Froning, K.J.; Atwell, S.; Aldaz, H.; Pustilnik, A.; Lu, F.; Huang, F.; Yuan, R.; Hassanali, S.; Chamberlain, A.K.; et al. Computationally designed bispecific antibodies using negative state repertoires. Structure 2016, 24, 641–651. [Google Scholar] [CrossRef] [PubMed]
- Mimoto, F.; Kadono, S.; Katada, H.; Igawa, T.; Kamikawa, T.; Hattori, K. Crystal structure of a novel asymmetrically engineered fc variant with improved affinity for fcgammars. Mol. Immunol. 2014, 58, 132–138. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Gunasekaran, K.; Wang, W.; Razinkov, V.; Sekirov, L.; Leng, E.; Sweet, H.; Foltz, I.; Howard, M.; Rousseau, A.M.; et al. Asymmetrical fc engineering greatly enhances antibody-dependent cellular cytotoxicity (adcc) effector function and stability of the modified antibodies. J. Biol. Chem. 2014, 289, 3571–3590. [Google Scholar] [CrossRef] [PubMed]
- Sondermann, P.; Huber, R.; Oosthuizen, V.; Jacob, U. The 3.2-a crystal structure of the human igg1 fc fragment-fc gammariii complex. Nature 2000, 406, 267–273. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, C.; Grau, S.; Jager, C.; Sondermann, P.; Brunker, P.; Waldhauer, I.; Hennig, M.; Ruf, A.; Rufer, A.C.; Stihle, M.; et al. Unique carbohydrate-carbohydrate interactions are required for high affinity binding between fcgammariii and antibodies lacking core fucose. Proc. Natl. Acad. Sci. USA 2011, 108, 12669–12674. [Google Scholar] [CrossRef] [PubMed]
- Radaev, S.; Motyka, S.; Fridman, W.H.; Sautes-Fridman, C.; Sun, P.D. The structure of a human type iii fcgamma receptor in complex with fc. J. Biol. Chem. 2001, 276, 16469–16477. [Google Scholar] [CrossRef]
- Kiyoshi, M.; Caaveiro, J.M.; Kawai, T.; Tashiro, S.; Ide, T.; Asaoka, Y.; Hatayama, K.; Tsumoto, K. Structural basis for binding of human igg1 to its high-affinity human receptor fcgammari. Nat. Commun. 2015, 6, 6866. [Google Scholar] [CrossRef]
- Lu, J.; Chu, J.; Zou, Z.; Hamacher, N.B.; Rixon, M.W.; Sun, P.D. Structure of fcgammari in complex with fc reveals the importance of glycan recognition for high-affinity igg binding. Proc. Natl. Acad. Sci. USA 2015, 112, 833–838. [Google Scholar] [CrossRef] [PubMed]
- Oganesyan, V.; Mazor, Y.; Yang, C.; Cook, K.E.; Woods, R.M.; Ferguson, A.; Bowen, M.A.; Martin, T.; Zhu, J.; Wu, H.; et al. Structural insights into the interaction of human igg1 with fcgammari: No direct role of glycans in binding. Acta Crystallogr. D Biol. Crystallogr. 2015, 71, 2354–2361. [Google Scholar] [CrossRef] [PubMed]
- Fekete, S.; Beck, A.; Fekete, J.; Guillarme, D. Method development for the separation of monoclonal antibody charge variants in cation exchange chromatography, part ii: Ph gradient approach. J. Pharm. Biomed. Anal. 2015, 102, 282–289. [Google Scholar] [CrossRef]
- Fekete, S.; Beck, A.; Fekete, J.; Guillarme, D. Method development for the separation of monoclonal antibody charge variants in cation exchange chromatography, part i: Salt gradient approach. J. Pharm. Biomed. Anal. 2015, 102, 33–44. [Google Scholar] [CrossRef] [PubMed]
- Delafosse, L.; Xu, P.; Durocher, Y. Comparative study of polyethylenimines for transient gene expression in mammalian hek293 and cho cells. J. Biotechnol. 2016, 227, 103–111. [Google Scholar] [CrossRef] [PubMed]
- Dorion-Thibaudeau, J.; Raymond, C.; Lattova, E.; Perreault, H.; Durocher, Y.; de Crescenzo, G. Towards the development of a surface plasmon resonance assay to evaluate the glycosylation pattern of monoclonal antibodies using the extracellular domains of cd16a and cd64. J. Immunol. Methods 2014, 408, 24–34. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CH2 Mutations 1 | Yield 2 [mg/L] | SPR Kamut/KaWT 4 | SPR Binding C1q 5 | SPR Binding FcRn 6 | DSC Tm (CH2 Transition) 7 [°C] | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Variant | HC-A | HC-B | 2aH | 2aR | 2b | 3aF | 3aV | 1a | ||||
| WT 8 | - | - | 30 | 30 | 1 | 1 | 1 | 1 | 1 | Yes | Yes | 71 |
| 791 9 | - | - | 73 10 | 1.7 | 1.7 | 1.2 | n/d 3 | 1.3 | n/d | Yes | n/d | 72 |
| 1051 (control) | L234A/L235A | L234A/L235A | 48 | 0.06 | 0.06 | 0.52 | 0.29 | 0.10 | 0.01 | NB | Yes | 72 |
| AAC1 | L234A/L235A | - | n/d | n/d | n/d | n/d | 0.87 | 0.71 | 0.48 | partial | n/d | n/d |
| AAC2 | L234A/L235A | L234K/L235K | 63 | NB | NB | NB | LOW | LOW | LOW | NB | Yes | 74 |
| AAC3 | L234D/L235E | L234K/L235K | 39 | NB | NB | NB | LOW | LOW | LOW | NB | Yes | 72 |
| AAC4 | E233A/L234D/L235E | E233A/L234R/L235R | 42 | NB | NB | NB | LOW | LOW | LOW | NB | Yes | 73 |
| AAC5 | L234D/L235E | E233K/L234R/L235R | 44 | NB | NB | NB | LOW | LOW | LOW | NB | Yes | 73 |
| AAC6 | E233A/L234K/L235A | E233K/L234A/L235K | 31 | NB | NB | NB | NB | LOW | LOW | NB | Yes | 75 |
| AAC7 | E269Q/D270N | E269K/D270R | n/d | n/d | n/d | n/d | LOW | LOW | 0.15 | NB | n/d | n/d |
| AAC8 | - | L235K/A327K | n/d | n/d | n/d | n/d | 0.19 | 0.10 | 0.13 | NB | n/d | n/d |
| CH2 mutations 1 | SPR Kamut/KaWT 2 | DSC Tm 3 (CH2 Transition) (°C) | ADCC | CDC | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Variant | Chain A | Chain B | 2aH | 2aR | 2b | 3aF | 3aV | EC50 (nM) | Maximum Lysis | EC50 (nM) | Maximum Lysis (%) | |
| Control Rituximab v1261 | - | - | 1.9 | 1.5 | 1.9 | 1.4 | 1.34 | 73 | 0.1 | 66 | 2.9 | 96 |
| AAC9 | L234D/L235E | E233K/L234R/L235R | NB | NB | NB | NB | 0.08 | 75 | Non-lytic | Non-lytic | >10 * | <60 * |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Escobar-Cabrera, E.; Lario, P.; Baardsnes, J.; Schrag, J.; Durocher, Y.; Dixit, S. Asymmetric Fc Engineering for Bispecific Antibodies with Reduced Effector Function. Antibodies 2017, 6, 7. https://doi.org/10.3390/antib6020007
Escobar-Cabrera E, Lario P, Baardsnes J, Schrag J, Durocher Y, Dixit S. Asymmetric Fc Engineering for Bispecific Antibodies with Reduced Effector Function. Antibodies. 2017; 6(2):7. https://doi.org/10.3390/antib6020007
Chicago/Turabian StyleEscobar-Cabrera, Eric, Paula Lario, Jason Baardsnes, Joseph Schrag, Yves Durocher, and Surjit Dixit. 2017. "Asymmetric Fc Engineering for Bispecific Antibodies with Reduced Effector Function" Antibodies 6, no. 2: 7. https://doi.org/10.3390/antib6020007
APA StyleEscobar-Cabrera, E., Lario, P., Baardsnes, J., Schrag, J., Durocher, Y., & Dixit, S. (2017). Asymmetric Fc Engineering for Bispecific Antibodies with Reduced Effector Function. Antibodies, 6(2), 7. https://doi.org/10.3390/antib6020007