Challenges in Optimising the Successful Construction of Antibody Drug Conjugates in Cancer Therapy

Abstract
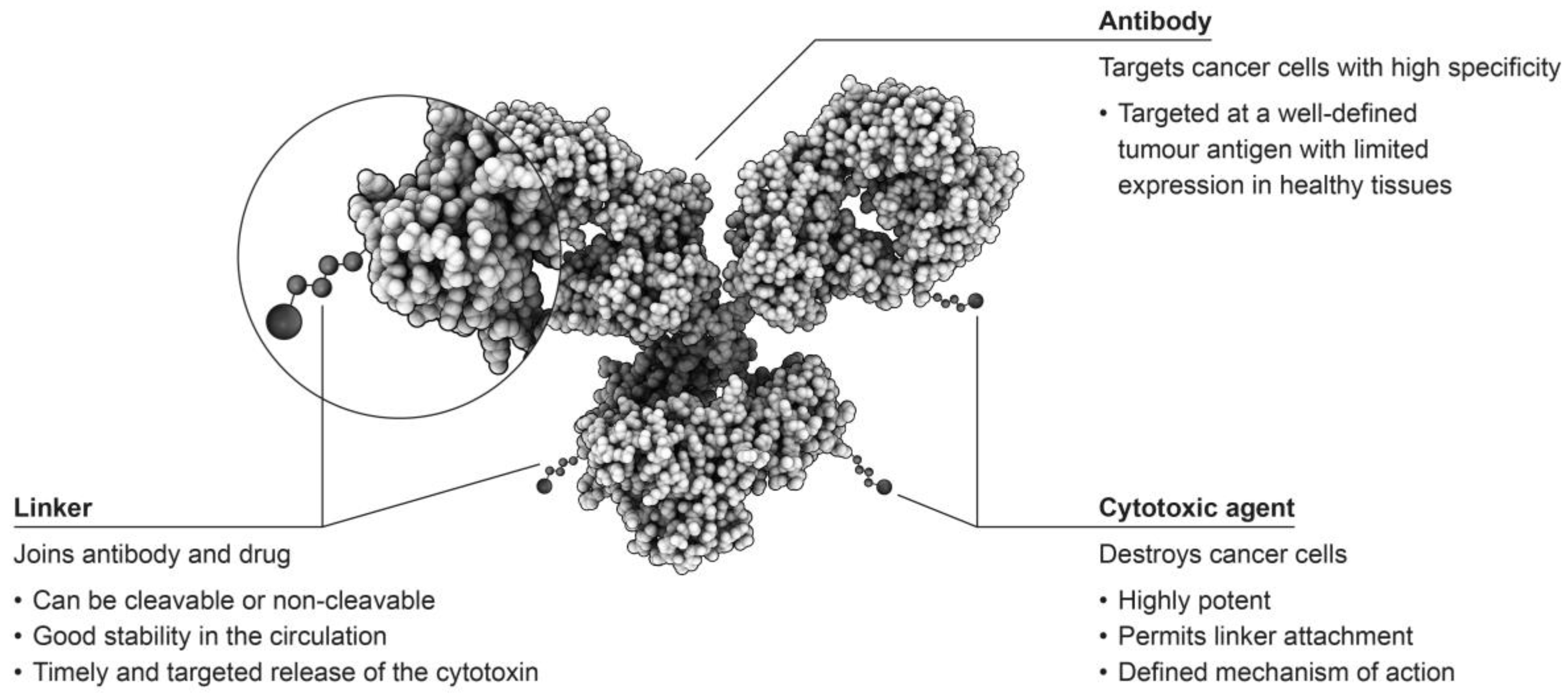
:1. Introduction
2. Smart Chemotherapy: The Future for Cancer Treatment?
3. Optimising Composition of ADCs
4. Assessing ADCs during Development
5. Preclinical Efficacy of ADCs in Development
6. Advances in ADC Development Technology
7. ADCs under Development by EDO
8. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Peters, C.; Brown, S. Antibody–drug conjugates as novel anti-cancer chemotherapeutics. Biosci. Rep. 2015, 35, e00225. [Google Scholar] [CrossRef] [PubMed]
- Tolcher, A.W. Antibody drug conjugates: Lessons from 20 years of clinical experience. Ann. Oncol. 2016, 27, 2168–2172. [Google Scholar] [CrossRef] [PubMed]
- Drake, P.M.; Rabuka, D. An emerging playbook for antibody–drug conjugates: Lessons from the laboratory and clinic suggest a strategy for improving efficacy and safety. Curr. Opin. Chem. Biol. 2015, 28, 174–180. [Google Scholar] [CrossRef] [PubMed]
- Ornes, S. Antibody–drug conjugates. Proc. Natl. Acad. Sci. USA 2013, 110, 13695. [Google Scholar] [CrossRef] [PubMed]
- Francisco, J.A.; Cerveny, C.G.; Meyer, D.L.; Mixan, B.J.; Klussman, K.; Chace, D.F.; Rejniak, S.X.; Gordon, K.A.; DeBlanc, R.; Toki, B.E.; et al. cAC10-vcMMAE, an anti-CD30-monomethyl auristatin E conjugate with potent and selective antitumor activity. Blood 2003, 102, 1458–1465. [Google Scholar] [CrossRef] [PubMed]
- Younes, A.; Gopal, A.K.; Smith, S.E.; Ansell, S.M.; Rosenblatt, J.D.; Savage, K.J.; Ramchandren, R.; Bartlett, N.L.; Cheson, B.D.; De Vos, S.; et al. Results of a pivotal phase II study of brentuximab vedotin for patients with relapsed or refractory Hodkgin’s lymphoma. J. Clin. Oncol. 2012, 30, 2183–2189. [Google Scholar] [CrossRef] [PubMed]
- Pro, B.; Advani, R.; Brice, P.; Bartlett, N.L.; Rosenblatt, J.D.; Illidge, T.; Matous, J.; Ramchandren, R.; Fanale, M.; Connors, J.M.; et al. Brentuximab vedotin (SGN-35) in patients with relapsed or refractory systemic anaplastic large-cell lymphoma: Results of a phase II study. J. Clin. Oncol. 2012, 30, 2190–2196. [Google Scholar] [CrossRef] [PubMed]
- Lambert, J.M.; Chari, R.V.J. Ado-trastuzumab Emtansine (T-DM1): An antibody−drug conjugate (ADC) for HER2-positive breast cancer. J. Med. Chem. 2014, 57, 6949–6964. [Google Scholar] [CrossRef] [PubMed]
- Phillips, G.D.L.; Li, G.; Dugger, D.L.; Crocker, L.M.; Parsons, K.L.; Mai, E.; Blättler, W.A.; Lambert, J.M.; Chari, R.V.; Lutz, R.J.; et al. Targeting HER2-positive breast cancer with trastuzumab-DM1, an antibody−cytotoxic drug conjugate. Cancer Res. 2008, 68, 9280–9290. [Google Scholar] [CrossRef] [PubMed]
- Verma, S.; Miles, D.; Gianni, L.; Krop, I.E.; Welslau, M.; Baselga, J.; Pegram, M.; Oh, D.Y.; Diéras, V.; Guardino, E.; et al. Trastuzumab emtansine for HER2-positive advanced breast cancer. New Engl. J. Med. 2012, 367, 1783–1791. [Google Scholar] [CrossRef] [PubMed]
- Donaghy, H. Effects of antibody, drug and linker on the preclinical and clinical toxicities of antibody-drug conjugates. mAbs 2016, 8, 659–671. [Google Scholar] [CrossRef] [PubMed]
- Besse, L.; Kraus, M.; Besse, A.; Bader, J.; Silzle, T.; Mehrling, T.; Driessen, C. The first-in-class alkylating HDAC inhibitor EDO-S101 is highly synergistic with proteasome inhibition against multiple myeloma through activation of multiple pathways. Blood Cancer J. 2017, 7, e589. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, U.; Bernt, K.M.; Lange, B.; Wenkel, J.; Jikai, J.; Shabat, D.; Amir, R.; Huebener, N.; Niethammer, A.G.; Hagemeier, C.; et al. Hydrolytically activated etoposide prodrugs inhibit MDR-1 function and eradicate established MDR-1 multidrug-resistant T-cell leukemia. Blood 2003, 102, 246–253. [Google Scholar] [CrossRef] [PubMed]
- Dal Corso, A.; Gébleux, R.; Murer, P.; Soltermann, A.; Neri, D. A non-internalizing antibody-drug conjugate based on an anthracycline payload displays potent therapeutic activity in vivo. J. Control. Release 2017, 264, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Lambert, J.M.; Morris, C.Q. Antibody–drug conjugates (ADCs) for personalized treatment of solid tumors: A review. Adv. Ther. 2017, 34, 1015–1035. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.G.; Kim, K.M. Strategies and advancement in antibody-drug conjugate optimization for targeted cancer therapeutics. Biomol. Ther. 2015, 23, 493–509. [Google Scholar] [CrossRef] [PubMed]
- Beck, A.; Lambert, J.; Sun, M.; Lin, K. Fourth World Antibody-Drug Conjugate Summit: February 29–March 1, 2012, Frankfurt, Germany. mAbs 2012, 4, 637–647. [Google Scholar] [CrossRef] [PubMed]
- Burris, H.A., III; Rugo, H.S.; Vukelja, S.J.; Vogel, C.L.; Borson, R.A.; Limentani, S.; Tan-Chiu, E.; Krop, I.E.; Michaelson, R.A.; Girish, S.; et al. Phase II study of the antibody drug conjugate trastuzumab-DM1 for the treatment of human epidermal growth factor receptor 2 (HER2)-positive breast cancer after prior HER2-directed therapy. J. Clin. Oncol. 2011, 29, 398–405. [Google Scholar] [CrossRef] [PubMed]
- McCombs, J.R.; Owen, S.C. Antibody drug conjugates: Design and selection of linker, payload and conjugation chemistry. AAPS J. 2015, 17, 339–351. [Google Scholar] [CrossRef] [PubMed]
- Del Carpini, J.; Duriga, N.; Kumar, S. Application of octet and doe for rapid screening of assay reagents and assay parameters for total antibody ligand binding assay for Antibody-drug conjugates. In Proceedings of the AAPS National Biotechnology Conference, San Diego, CA, USA, 8–10 June 2014. [Google Scholar]
- Jaramillo, M.L.; Meury, L.; Jolicoeur, N.; Banville, M.; Tao, L.; McCourt, M.O.C. Assays for the selection and functional characterization of antibody-drug conjugates at the National Research Council of Canada. In Proceedings of the 107th Annual Meeting of the American Association for Cancer Research, 16–20 April 2016. [Google Scholar]
- Bornstein, G.G. Antibody Drug Conjugates: Preclinical Considerations. AAPS J. 2015, 17, 525–534. [Google Scholar] [CrossRef] [PubMed]
- Riedl, T.; van Boxtel, E.; Bosch, M.; Parren, P.W.; Gerritsen, A.F. High-Throughput Screening for Internalizing Antibodies by Homogeneous Fluorescence Imaging of a pH-Activated Probe. J. Biomol. Screen. 2016, 21, 12–23. [Google Scholar] [CrossRef] [PubMed]
- Hock, B.M.; Thudium, K.E.; Carrasco-Triguero, M.; Schwabe, N.F. Immunogenicity of Antibody Drug Conjugates: Bioanalytical Methods and Monitoring Strategy for a Novel Therapeutic Modality. AAPS J. 2015, 17, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Shankar, G.; Arkin, S.; Cocea, L.; Devanarayan, V.; Kirshner, S.; Kromminga, A.; Quarmby, V.; Richards, S.; Schneider, C.K.; Subramanyam, M.; et al. Assessment and reporting of the clinical immunogenicity of therapeutic proteins and peptidesharmonized terminology and tactical recommendations. AAPS J. 2014, 16, 658–673. [Google Scholar] [CrossRef] [PubMed]
- Shankar, G.; Devanarayan, V.; Amaravadi, L.; Barrett, Y.C.; Bowsher, R.; Finco-Kent, D.; Fiscella, M.; Gorovits, B.; Kirschner, S.; Moxness, M.; et al. Recommendations for the validation of immunoassays used for detection of host antibodies against biotechnology products. J. Pharm. Biomed. Anal. 2008, 48, 1267–1281. [Google Scholar] [CrossRef] [PubMed]
- Kamath, A.V.; Iyer, S. Preclinical Pharmacokinetic Considerations for the Development of Antibody Drug Conjugates. Pharm. Res. 2015, 32, 3470–3479. [Google Scholar]
- Agarwal, P.; Bertozzi, C.R. Site-specific antibody-drug conjugates: The nexus of bioorthogonal chemistry, protein engineering, and drug development. Bioconjug. Chem. 2015, 26, 176–192. [Google Scholar] [CrossRef] [PubMed]
- Acchione, M.; Kwon, H.; Jochheim, C.M.; Atkins, W.M. Impact of linker and conjugation chemistry on antigen binding, Fc receptor binding and thermal stability of model antibody-drug conjugates. mAbs. 2012, 4, 362–372. [Google Scholar] [CrossRef] [PubMed]
- Hamblett, K.J.; Senter, P.D.; Chace, D.F.; Sun, M.M.; Lenox, J.; Cerveny, C.G.; Kissler, K.M.; Bernhardt, S.X.; Kopcha, A.K.; Zabinski, R.F.; et al. Effects of drug loading on the antitumor activity of a monoclonal antibody drug conjugate. Clin. Cancer Res. 2004, 10, 7063–7070. [Google Scholar] [CrossRef] [PubMed]
- Lyon, R.P.; Bovee, T.D.; Doronina, S.O.; Burke, P.J.; Hunter, J.H.; Neff-LaFord, H.D.; Jonas, M.; Anderson, M.E.; Setter, J.R.; Senter, P.D. Reducing hydrophobicity of homogeneous antibody-drug conjugates improves pharmacokinetics and therapeutic index. Nat. Biotechnol. 2015, 33, 733–735. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.I.; Decker, S.; Zaharevitz, D.; Rubinstein, L.V.; Venditti, J.M.; Schepartz, S.; Kalyandrug, S.; Christian, M.; Arbuck, S.; Hollingshead, M.; et al. Relationships between drug activity in NCI preclinical in vitro and in vivo models and early clinical trials. Br. J. Cancer 2001, 84, 1424–1431. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Amphlett, G.; Blättler, W.A.; Lambert, J.M.; Zhang, W.E.I. Structural characterization of the maytansinoid−monoclonal antibody immunoconjugate, huN901−DM1, by mass spectrometry. Protein Sci. 2005, 14, 2436–2446. [Google Scholar] [CrossRef] [PubMed]
- Senter, P.D. Potent antibody drug conjugates for cancer therapy. Curr. Opin. Chem. Biol. 2009, 13, 235–244. [Google Scholar] [CrossRef] [PubMed]
- Drake, P.M.; Albers, A.E.; Baker, J.; Banas, S.; Barfield, R.M.; Bhat, A.S.; de Hart, G.W.; Garofalo, A.W.; Holder, P.; Jones, L.C.; et al. Aldehyde tag coupled with HIPS chemistry enables the production of ADCs conjugated site-specifically to different antibody regions with distinct in vivo efficacy and PK outcomes. Bioconjug. Chem. 2014, 25, 1331–1341. [Google Scholar] [CrossRef] [PubMed]
- Hong, E.E.; Chari, R. Linker design for antibody-drug conjugates. In Antibody-Drug Conjugates; The 21st Century Magic Bullets for Cancer; Springer: New York, NY, USA, 2015. [Google Scholar]
- Shepard, H.M.; Phillips, G.L.; Thanos, C.D.; Feldmann, M. Developments in therapy with monoclonal antibodies and related proteins. Clin. Med. 2017, 17, 220–232. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Yang, Q.; Patel, S.; Lei, Y.; McAleer, L.; Singleton, J.; Soltis, D.; Wang, B. Characterization of human tissue factor (TF)–specific monoclonal antibodies prepared using a rapid immunization protocol. Hybridoma 2005, 24, 78–85. [Google Scholar] [CrossRef] [PubMed]
- Carrasco-Triguero, M.; Yi, J.H.; Dere, R.; Qiu, Z.J.; Lei, C.; Li, Y.; Mahood, C.; Wang, B.; Leipold, D.; Poon, K.A.; et al. Immunogenicity assays for antibody-drug conjugates: Case study with ado-trastuzumab emtansine. Bioanalysis 2013, 5, 1007–1023. [Google Scholar] [CrossRef] [PubMed]
- Brentuximab Vedotin: Summary of Product Characteristics, EMEA 2016. Available online: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/002455/WC500135055.pdf (accessed on 12 February 2018).
- Wang, B.; Berger, M.; Masters, G.; Albone, E.; Yang, Q.; Sheedy, J.; Kirksey, Y.; Grimm, L.; Wang, B.; Singleton, J.; et al. Radiotherapy of Human Xenograft NSCLC Tumors in Nude Mice with a 90Y-Labeled Anti-Tissue Factor Antibody. Cancer Biother. Radiopharm. 2005, 20, 300–309. [Google Scholar] [CrossRef] [PubMed]
- Lykke, J.; Nielsen, H.J. The role of tissue factor in colorectal cancer. Eur. J. Surg. Oncol. 2003, 29, 417–422. [Google Scholar] [CrossRef]
- Akashi, T.; Furuya, Y.; Ohta, S.; Fuse, H. Tissue factor expression and prognosis in patients with metastatic prostate cancer. Urology 2003, 62, 1078–1082. [Google Scholar] [CrossRef]
- Ohta, S.; Wada, H.; Nakazaki, T.; Maeda, Y.; Nobori, T.; Shiku, H.; Nakamura, S.; Nagakawa, O.; Furuya, Y.; Fuse, H. Expression of tissue factor is associated with clinical features and angiogenesis in prostate cancer. Anticancer Res. 2002, 22, 2991–2996. [Google Scholar] [PubMed]
- Minamiya, Y.; Matsuzaki, I.; Sageshima, M.; Saito, H.; Taguchi, K.; Nakagawa, T.; Ogawa, J. Expression of tissue factor mRNA and invasion of blood vessels by tumor cells in non-small cell lung cancer. Surg. Today 2004, 34, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Minamiya, Y.; Matsuzaki, I.; Sageshima, M.; Saito, H.; Taguchi, K.; Nakagawa, T.; Ogawa, J.I. Expression of tissue factor in non-small-cell lung cancers and its relationship to metastasis. Br. J. Cancer 1999, 79, 472–477. [Google Scholar]
- Contrino, J.; Hair, G.; Kreutzer, D.L.; Rickles, F.R. In situ detection of tissue factor in vascular endothelial cells: Correlation with the malignant phenotype of human breast disease. Nat. Med. 1996, 2, 209–215. [Google Scholar] [CrossRef] [PubMed]
- Mueller, B.M.; Reisfeld, R.A.; Edgington, T.S.; Ruf, W. Expression of tissue factor by melanoma cells promotes efficient hematogenous metastasis. Proc. Natl. Acad. Sci. USA 1992, 89, 11832–11836. [Google Scholar] [CrossRef] [PubMed]
- Beck, E.P.; Moldenhauer, A.; Merkle, E.; Kiesewetter, F.; Jager, W.; Wildt, L.; Lang, N. CA 125 production and release by ovarian cancer cells in vitro. Int. J. Biol. Markers 1998, 13, 200. [Google Scholar] [PubMed]
- Leake, J.; Woolas, R.P.; Daniel, J.; Oram, D.H.; Brown, C.L. Immunocytochemical and serological expression of CA 125: A clinicopathological study of 40 malignant ovarian epithelial tumors. Histopathology 1994, 24, 57. [Google Scholar] [CrossRef] [PubMed]
- Marth, C.; Egle, D.; Auer, D.; Rössler, J.; Zeimet, A.G.; Vergote, I.; Daxenbichler, G. Modulation of CA-125 tumor marker shedding in ovarian cancer cells by erlotinib or cetuximab. Gynecol. Oncol. 2007, 105, 716–721. [Google Scholar] [CrossRef] [PubMed]
- Berger, M.A.; Masters, G.R.; Singleton, J.; Scully, M.S.; Grimm, L.G.; Soltis, D.A.; Albone, E.F. Pharmacokinetics, Biodistribution, and Radioimmunotherapy with Monoclonal Antibody 776.1 in a Murine Model of Human Ovarian Cancer. Cancer Biother. Radiopharm 2005, 20, 589–602. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| ADC | Target | Indication | Study Sponsor |
|---|---|---|---|
| Phase III | |||
| Depatuxizumab mafodotin (ABT-414) | Epidermal growth factor receptor (EGFR) |
| Abbvie, North Chicago, IL, USA |
| Mirvetuximab soravtansine | Folate receptor alpha |
| Immunogen Inc., Waltham, MA, USA |
| Polatuzumab vedotin | CD79b |
| Genentech, South San Francisco, CA, USA/Roche, Switzerland |
| Rovalpituzumab tesirine | DLL3 |
| Abbvie, North Chicago, IL, USA |
| Sacituzumab govitecan (IMMU-132) | TROP-2 receptor |
| Immunomedics Inc., Morris Plains, NJ, USA |
| SYD985 | Human epidermal growth factor receptor 2 (HER2) |
| Synthon Biopharmaceuticals, The Netherlands |
| Vadastuximab talirine | CD33 |
| Seattle Genetics, Bothell, WA, USA |
| Phase II | |||
| AGS-16C3F | Ectonucleotide pyrophosphatase /phosphodiesterase family member 3 (ENPP3) |
| Agensys Inc., Santa Monica, CA, USA;Astellas Pharma Inc., Japan |
| Anetumab ravtansine | Mesothelin |
| National Cancer Institute, Rockville, MD, USA |
| BMS-986148 | Mesothelin |
| Bristol-Myers Squibb, New York, NY, USA |
| CDX-014 | TIM-1 |
| Celldex Therapeutics, Hampton, NJ, USA |
| Coltuximab ravtansine (SAR3419) | CD19 |
| Sanofi, France |
| Denintuzumab mafodotin (SGN-CD19A) | CD19 |
| Seattle Genetics, Bothell, WA, USA |
| DS-8201a | HER2 |
| Daiichi Sankyo Co. Ltd., Japan |
| Enfortumab vedotin (ASG-22CE) | Nectin-4 |
| Astellas Pharma Global Development Inc., Northbrook, IL, USA |
| Glembatumumab vedotin | Glycoprotein NMB |
| Celldex Therapeutics, Hampton, NJ, USA |
| hLL1-DOX | CD74 |
| National Cancer Institute, Rockville, MD, USA |
| HuMax-AXL-ADC | Axl |
| Genmab, Denmark |
| Labetuzumab govitecan | CEACAM5 |
| Immunomedics Inc., Morris Plains, NJ, USA |
| Lorvotuzumab mertansine | CD56 |
| Children’s Oncology Group, Monrovia, CA, USA |
| PSMA ADC | Prostate Specific Membrane Antigen |
| Progenics Pharmaceuticals Inc., Tarrytown, NY, USA |
| RC48-ADC | HER2 |
| RemeGen |
| Sacituzumab govitecan (IMMU 132) | Tumor-associated calcium signal transducer 2 (TROP-2) receptor |
| Immunomedics Inc., Morris Plains, NJ, USA |
| SAR566658 (ACT14884) | CA6 |
| Sanofi, France |
| SGN15 | Lewis-Y antigen |
| Seattle Genetics Inc., Bothell, WA, USA |
| Tisotumab vedotin (HuMax-TF-ADC) | Tissue factor |
| Genmab, Denmark |
| Vadastuximab Talirine (SGN-CD33A; 33A) | CD33 |
| Seattle Genetics Inc., Bothell, WA, USA |
| W0101 | insulin-like growth factor 1 (IGF-1) receptor |
| Pierre Fabre Medicament, France |
| BIAcore Analysis | Coagulation Time | ||||
|---|---|---|---|---|---|
| Anti-TF MAbs | Isotype | ka (1/Ms) | kd (1/s) | KD (M) | Mean ± SD (s) |
| No Ab | N/A | N/A | N/A | N/A | 185.0 ± 8.7 |
| TF158 | N/A | N/A | N/A | N/A | >450 a |
| TF278 | IgG1, λ | 2.9 × 105 | 1.5 × 10−4 | 5.2 × 10−10 | 190.0 ± 17.3 |
| TF392 | IgG1, λ | 2.1 × 105 | 2.3 × 10−4 | 1.1 × 10−9 | 210.0 ± 0.0 |
| TF260 | IgG1, λ | 2.0 × 105 | 2.6 × 10−4 | 1.3 × 10−9 | 185.0 ± 8.7 |
| TF009 | IgG1, κ | 2.0 × 105 | 3.6 × 10−4 | 1.8 × 10−9 | 195.0 ± 15.0 |
| TF277 | IgG1, κ | 4.4 × 105 | 3.1 × 10−3 | 7.1 × 10−9 | 205.0 ± 8.7 |
| TF124 | IgG1, κ | 6.0 × 105 | 1.5 × 10−2 | 2.5 × 10−8 | 200.0 ± 8.7 |
| TF080 | IgG1, κ | 2.9 × 105 | 2.0 × 10−2 | 6.8 × 10−8 | 202.5 ± 10.6 |
| TF126 | IgG1, κ | 1.7 × 106 | 1.8 × 10−1 | 1.0 × 10−7 | ND |
| TF297 | IgG1, κ | 2.9 × 104 | 7.1 × 10−3 | 2.4 × 10−7 | 225 a |
| TF261 | IgG1, λ | 3.4 × 105 | 1.0 × 10−1 | 3.0 × 10−7 | 225 a |
| TF451 | IgG1, κ | 4.0 × 105 | 1.5 × 10−1 | 3.6 × 10−7 | 180 a |
| TF405 | IgG1, κ | 8.3 × 104 | 3.4 × 10−2 | 4.1 × 10−7 | 220.0 ± 34.6 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mehrling, T.; Soltis, D. Challenges in Optimising the Successful Construction of Antibody Drug Conjugates in Cancer Therapy. Antibodies 2018, 7, 11. https://doi.org/10.3390/antib7010011
Mehrling T, Soltis D. Challenges in Optimising the Successful Construction of Antibody Drug Conjugates in Cancer Therapy. Antibodies. 2018; 7(1):11. https://doi.org/10.3390/antib7010011
Chicago/Turabian StyleMehrling, Thomas, and Daniel Soltis. 2018. "Challenges in Optimising the Successful Construction of Antibody Drug Conjugates in Cancer Therapy" Antibodies 7, no. 1: 11. https://doi.org/10.3390/antib7010011
APA StyleMehrling, T., & Soltis, D. (2018). Challenges in Optimising the Successful Construction of Antibody Drug Conjugates in Cancer Therapy. Antibodies, 7(1), 11. https://doi.org/10.3390/antib7010011