A Polar Sulfamide Spacer Significantly Enhances the Manufacturability, Stability, and Therapeutic Index of Antibody–Drug Conjugates

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
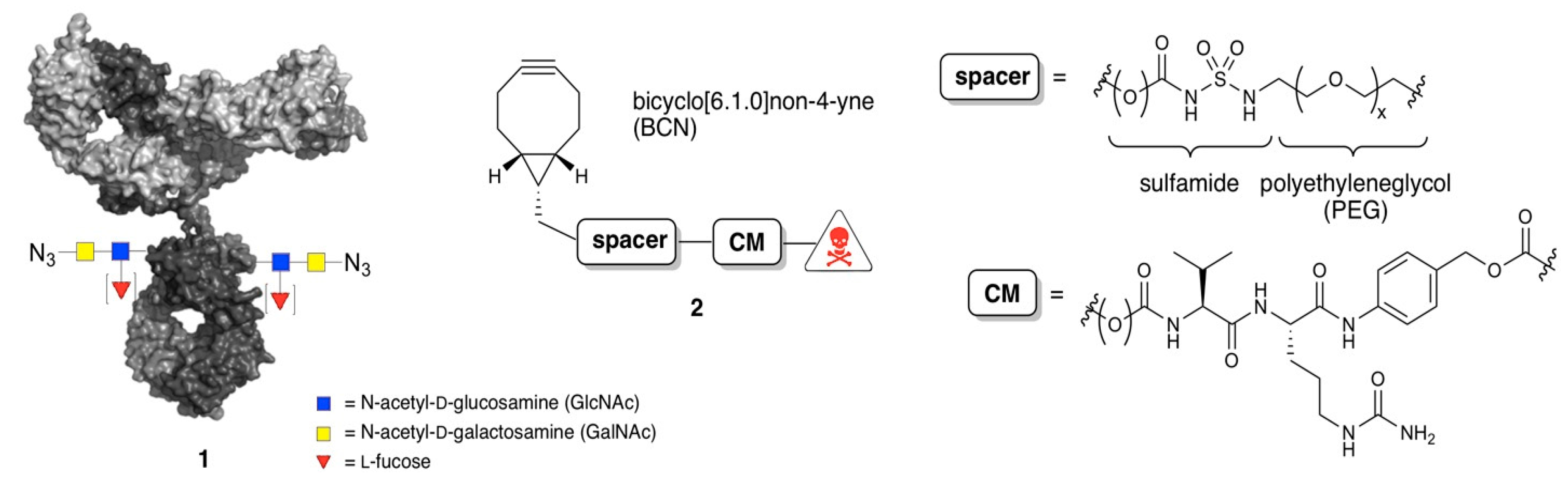
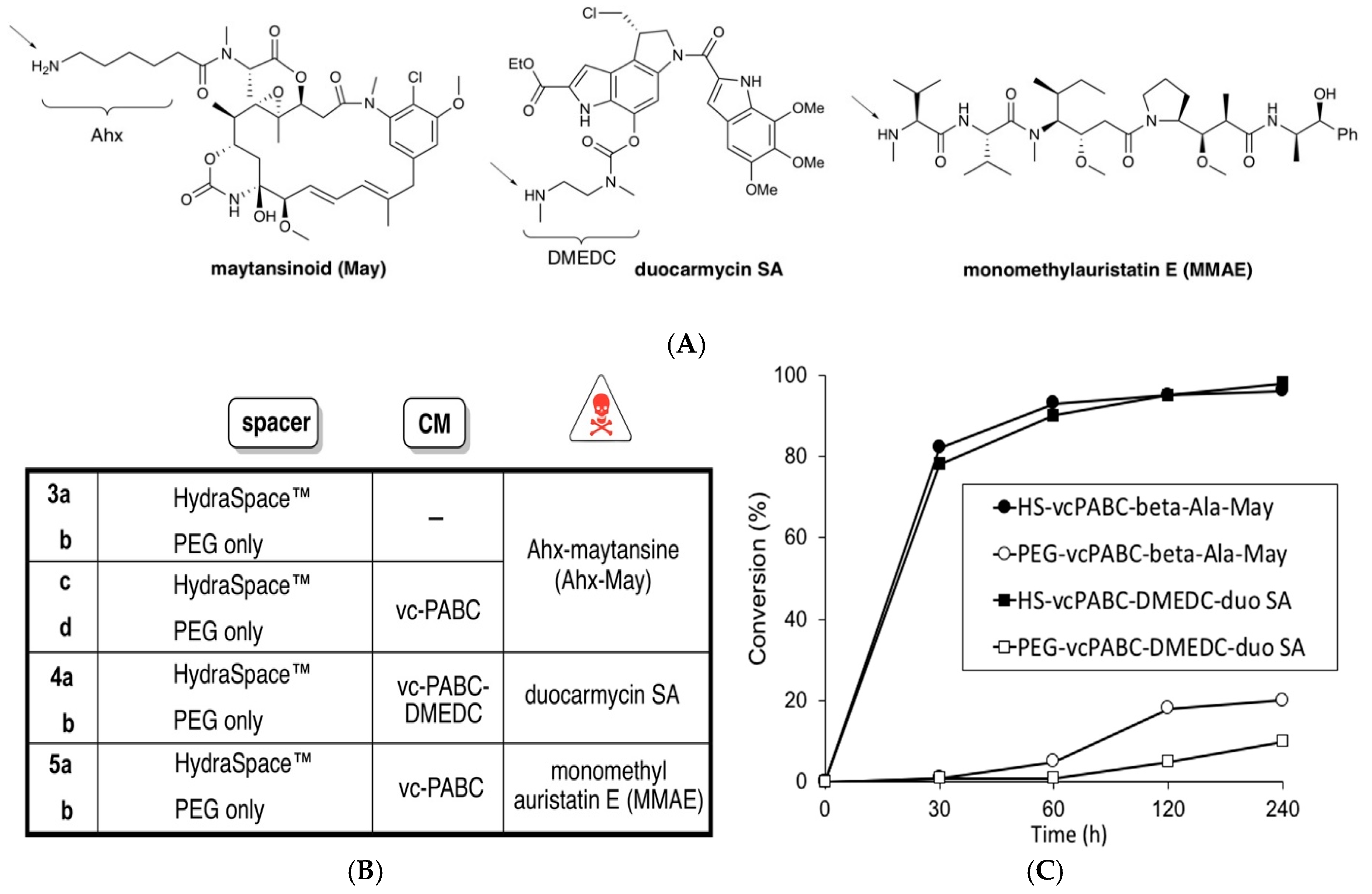
2.1. Synthesis of BCN-Spacer-Payload Constructs
2.2. Remodeling and Conjugation Procedure for Conversion of Antibodies into ADCs
3. Results
3.1. Studies of Conjugation Rates
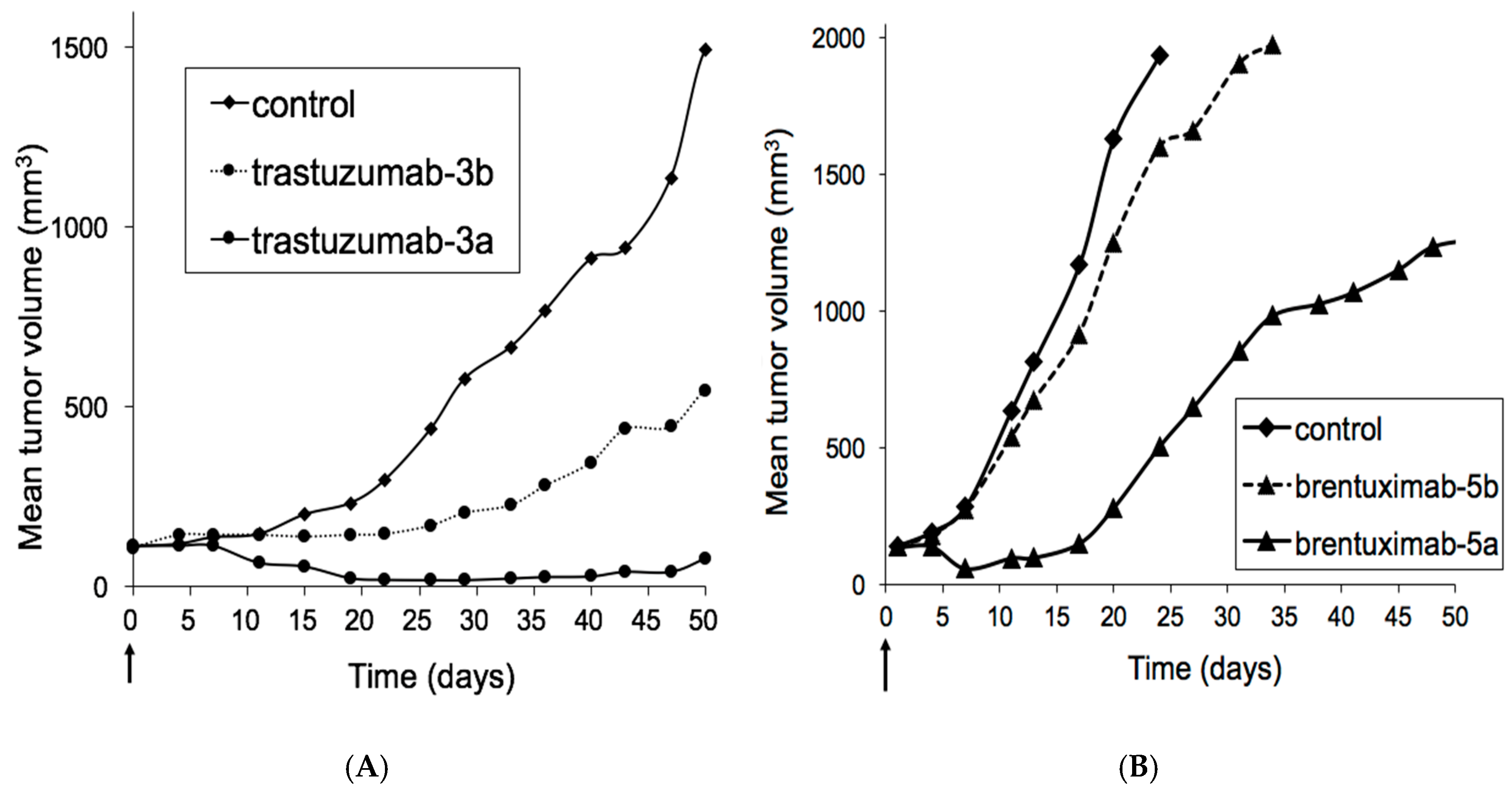
3.2. Xenograft Studies with HER2-Overexpressing Models HBCx-13B or T226
3.3. Xenograft Studies with Karpas-299 Model
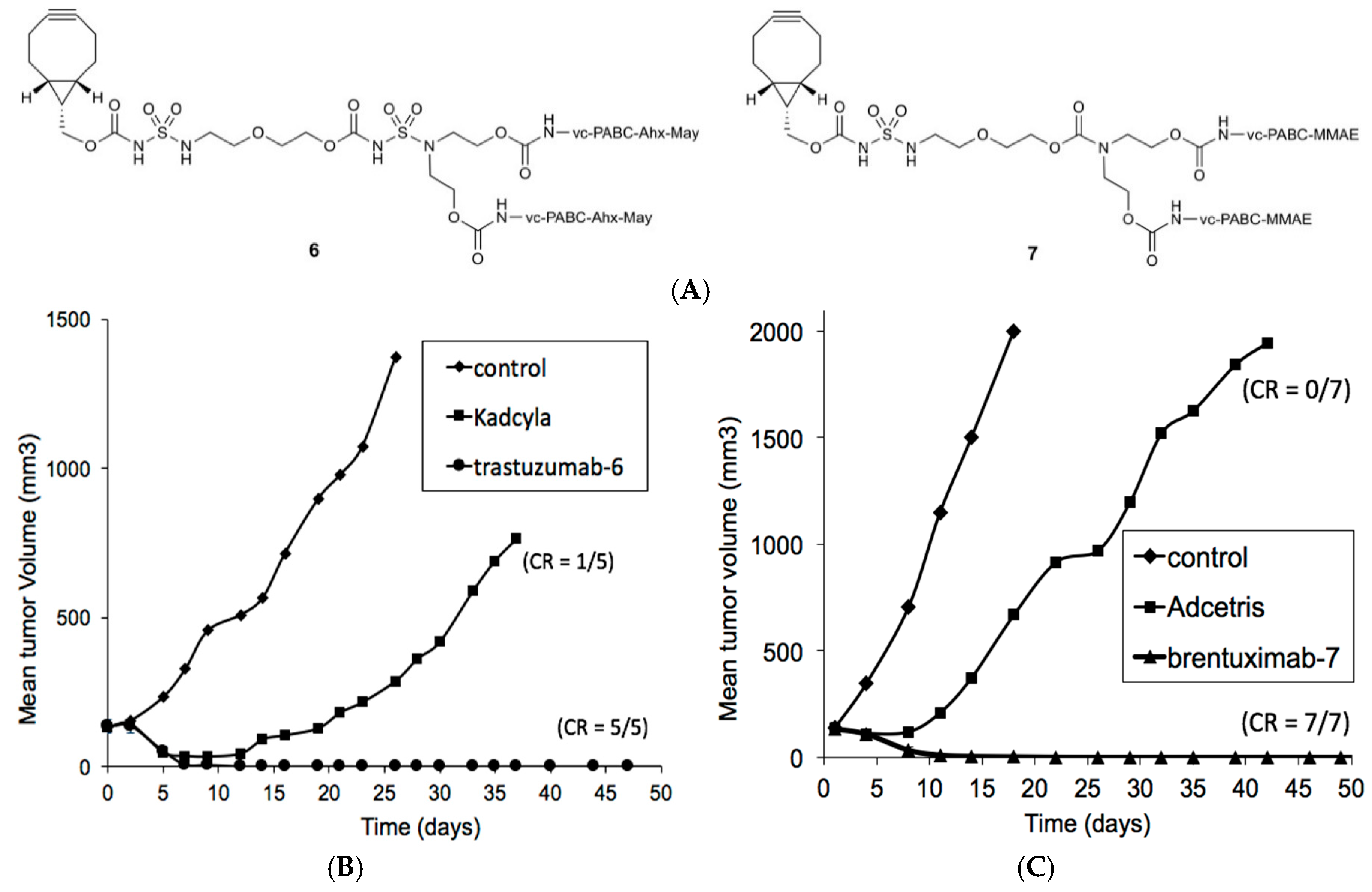
3.4. Tolerability Studies of Trastuzumab-6 and Kadcyla®
3.5. Tolerability Studies of Brentuximab-7 and Adcetris®
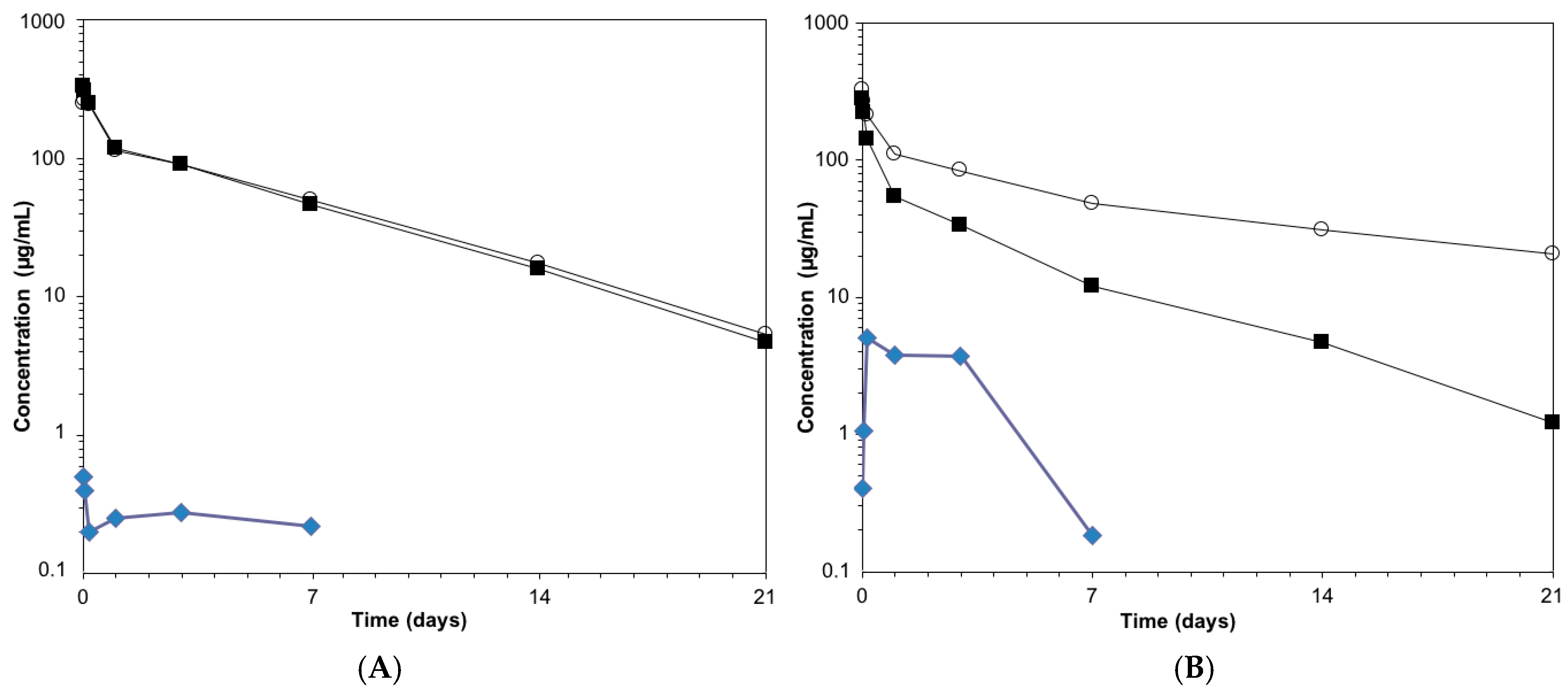
3.6. Pharmacokinetics Studies of Anti-HER2 ADCs
4. Discussion
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Chari, R.V.J.; Miller, M.L.; Widdison, W.C. Antibody-drug conjugates, an emerging concept in cancer therapy. Angew. Chem. Int. Ed. 2014, 53, 3796–3827. [Google Scholar] [CrossRef] [PubMed]
- Hong, E.E.; Chari, R.V.J. Linker design for antibody–drug conjugates. In Antibody–Drug Conjugates; Wang, J., Shen, W.-C., Zaro, J.L., Eds.; AAPS Advances in the Pharmaceutical Sciences Series 17; Springer: Cham, Switzerland, 2015; pp. 49–76. [Google Scholar]
- Jain, N.; Smith, S.W.; Ghone, S.; Tomczuk, B. Current ADC linker chemistry. Pharm. Res. 2015, 32, 3526–3540. [Google Scholar] [CrossRef] [PubMed]
- Beck, A.; Goetsch, L.; Dumontet, C.; Corvaïa, N. Strategies and challenges for the next-generation ofantibody–drug conjugates. Nat. Rev. Drug Discov. 2017, 16, 315–337. [Google Scholar] [CrossRef] [PubMed]
- Junutula, J.R.; Raab, H.; Clark, S.; Bhakta, S.; Leipold, D.D.; Weir, S.; Chen, Y.; Simpson, M.; Tsai, S.P.; Dennis, M.S.; et al. Site-specific conjugation of a cytotoxic drug to an antibody improves the therapeutic index. Nat. Biotechnol. 2008, 26, 925–932. [Google Scholar] [CrossRef] [PubMed]
- Drake, P.M.; Rabuka, D. An emerging playbook forantibody–drug conjugates: Lessons from the laboratory and clinic suggest a strategy for improving efficacy and safety. Curr. Opin. Chem. Biol. 2015, 28, 174–180. [Google Scholar] [CrossRef] [PubMed]
- Strop, P.; Delaria, K.; Foletti, D.; Witt, J.M.; Hasa-Moreno, A.; Poulsen, K.; Casas, M.G.; Dorywalska, M.; Farias, S.; Pios, A.; et al. Site-specific conjugation improves therapeutic index of antibody drug conjugates with high drug loading. Nat. Biotechnol. 2015, 33, 694–696. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, P.; Bertozzi, C.R. Site-specificantibody–drug conjugates: The nexus of bioorthogonal chemistry, protein engineering, and drug development. Bioconjug. Chem. 2014, 26, 176–192. [Google Scholar] [CrossRef] [PubMed]
- Behrens, C.R.; Liu, B. Methods for site-specific drug conjugation to antibodies. MAbs 2014, 6, 46–53. [Google Scholar] [CrossRef] [PubMed]
- Tian, F.; Lu, Y.; Manibusan, A.; Sellers, A.; Tran, H.; Sun, Y.; Phuong, T.; Barnett, R.; Hehli, B.; Song, F.; et al. A general approach to site-specific antibody drug conjugates. Proc. Natl. Acad. Sci. USA 2014, 111, 1766–1771. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Kumar, S.; Chipley, M.; Marcq, O.; Gupta, D.; Jin, Z.; Tomar, D.S.; Swabowski, C.; Smith, J.; Starkey, J.A.; et al. Characterization and higher-order structure assessment of an interchain cysteine-based ADC: Impact of drug loading and distribution on the mechanism of aggregation. Bioconjug. Chem. 2016, 27, 604–615. [Google Scholar] [CrossRef] [PubMed]
- Boswell, C.A.; Mundo, E.E.; Zhang, C.; Bumbaca, D.; Valle, N.R.; Kozak, K.R.; Fourie, A.; Chuh, J.; Koppada, N.; Saad, O.; et al. Impact of drug conjugation on pharmacokinetics and tissue distribution of anti-STEAP1antibody–drug conjugates in rats. Bioconjug. Chem. 2011, 22, 1994–2004. [Google Scholar] [CrossRef] [PubMed]
- Lin, K.; Tibbits, J. Pharmacokinetic considerations for antibody drug conjugates. Pharm. Res. 2012, 29, 2354–2366. [Google Scholar] [CrossRef] [PubMed]
- Zhao, R.Y.; Wilhelm, S.D.; Audette, C.; Jones, G.; Leece, B.A.; Lazar, A.C.; Goldmacher, V.S.; Singh, R.; Kovtun, Y.; Widdison, W.C.; et al. Synthesis and evaluation of hydrophilic linkers for antibody-maytansinoid conjugates. J. Med. Chem. 2011, 54, 3606–3623. [Google Scholar] [CrossRef] [PubMed]
- Ponte, J.F.; Ab, O.; Lanieri, L.; Lee, J.; Coccia, J.; Bartle, L.M.; Themeles, M.; Zhou, Y.; Pinkas, J.; Ruiz-Soto, R. Mirvetuximab soravtansine (IMGN853), a folate receptor alpha-targetingantibody–drug conjugate, potentiates the activity of standard of care therapeutics in ovarian cancer models. Neoplasia 2016, 18, 775–784. [Google Scholar] [CrossRef] [PubMed]
- ADC Review. PEG Linkers. Journal of Antibody–Drug Conjugates. 2015. Available online: http://adcreview.com/adc-university/adcs-101/stable-linker-technologies/peg-linkers/ (accessed on 5 January 2018).
- Kovtun, Y.V.; Audette, C.A.; Mayo, M.F.; Jones, G.E.; Doherty, H.; Maloney, E.K.; Erickson, H.K.; Sun, X.; Wilhelm, S.; Ab, O.; et al. Antibody-maytansinoid conjugates designed to bypass multidrug resistance. Cancer Res. 2010, 70, 2528–2537. [Google Scholar] [CrossRef] [PubMed]
- Lyon, R.P.; Bovee, T.D.; Doronina, S.O.; Burke, P.J.; Hunter, J.H.; Neff-LaFord, H.D.; Jonas, M.; Anderson, M.E.; Setter, J.R.; Senter, P.D.; et al. Reducing hydrophobicity of homogeneousantibody–drug conjugates improves pharmacokinetics and therapeutic index. Nat. Biotechnol. 2015, 33, 733–736. [Google Scholar] [CrossRef] [PubMed]
- Burke, P.J.; Hamilton, J.Z.; Jeffrey, S.C.; Hunter, J.H.; Doronina, S.O.; Okeley, N.M.; Miyamoto, J.B.; Anderson, M.E.; Stone, I.J.; Ulrich, M.L.; et al. Optimization of a PEGylated glucuronide-monomethylauristatin E linker forantibody–drug conjugates. Mol. Cancer Ther. 2017, 16, 116–123. [Google Scholar] [CrossRef] [PubMed]
- Kern, J.C.; Cancilla, M.; Dooney, D.; Kwasnjuk, K.; Zhang, R.; Beaumont, M.; Figueroa, I.; Hsieh, S.; Liang, L.; Tomazela, D.; et al. Discovery of pyrophosphate diesters as tunable, soluble, and bioorthogonal linkers for site-specificantibody–drug conjugates. J. Am. Chem. Soc. 2016, 138, 1430–1445. [Google Scholar] [CrossRef] [PubMed]
- Rodwell, J.D.; Alvarez, V.L.; Lee, C.; Lopes, A.D.; Goers, J.W.F.; King, H.D.; Powsner, H.J.; McKearn, T.J. Site-specific covalent modification of monoclonal antibodies: In vitro and in vivo evaluations. Proc. Natl. Acad. Sci. USA 1986, 83, 2632–2636. [Google Scholar] [CrossRef] [PubMed]
- Boeggeman, E.; Ramakrishnan, B.; Pasek, M.; Manzoni, M.; Puri, A.; Loomis, K.H.; Waybright, T.J.; Qasba, P.K. Site specific conjugation of fluoroprobes to the remodeled Fc N-glycans of monoclonal antibodies using mutant glycosyltransferases: Application for cell surface antigen detection. Bioconjug. Chem. 2009, 20, 1228–1236. [Google Scholar] [CrossRef] [PubMed]
- Van Geel, R.; Wijdeven, M.A.; Heesbeen, R.; Verkade, J.M.; Wasiel, A.A.; van Berkel, S.S.; van Delft, F.L. Chemoenzymatic conjugation of toxic payloads to the globally conserved N-glycan of native mAbs provides homogeneous and highly efficaciousantibody–drug conjugates. Bioconjug. Chem. 2015, 26, 2233–2242. [Google Scholar] [CrossRef] [PubMed]
- Dommerholt, J.; Schmidt, S.; Temming, R.; Hendriks, L.J.; Rutjes, F.P.; van Hest, J.C.; Lefeber, D.J.; Friedl, P.; van Delft, F.L. Readily accessible bicyclononynes for bioorthogonal labeling and three-dimensional imaging of living cells. Angew. Chem. Int. Ed. 2010, 49, 9422–9425. [Google Scholar] [CrossRef] [PubMed]
- Somu, R.V.; Boshoff, H.; Qiao, C.; Bennett, E.M.; Barry, C.E.; Aldrich, C.C. Rationally designed nucleoside antibiotics that inhibit siderophore biosynthesis of Mycobacterium tuberculosis. J. Med. Chem. 2006, 49, 31–34. [Google Scholar] [CrossRef] [PubMed]
- Adem, Y.T.; Schwarz, K.A.; Duenas, E.; Patapoff, T.W.; Galush, W.J.; Esue, O. Auristatin antibody drug conjugate physical instability and the role of drug payload. Bioconjug. Chem. 2014, 25, 656–664. [Google Scholar] [CrossRef] [PubMed]
- Hock, M.B.; Thudium, K.E.; Carrasco-Triguero, M.; Schwabe, N.F. Immunogenicity of antibody drug conjugates: Bioanalytical methods and monitoring strategy for a novel therapeutic modality. AAPS J. 2015, 17, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Poon, K.A.; Flagella, A.; Beyer, J.; Tibbitts, J.; Kaur, S.; Saada, O.; Yi, J.H.; Girish, S.; Dybdal, N.; Reynolds, T. Preclinical safety profile of trastuzumab emtansine (T-DM1): Mechanism of action of its cytotoxic component retained with improved tolerability. Toxicol. Appl. Pharmacol. 2013, 273, 298–313. [Google Scholar] [CrossRef] [PubMed]
- Lhospice, F.; Brégeon, D.; Belmant, C.; Dennler, P.; Chiotellis, A.; Fischer, E.; Gauthier, L.; Boëdec, A.; Rispaud, H.; Savard-Chambard, S.; et al. Site-specific conjugation of monomethyl auristatin E to anti-CD30 antibodies improves their pharmacokinetics and therapeutic index in rodent models. Mol. Pharm. 2015, 12, 1863–1871. [Google Scholar] [CrossRef] [PubMed]
- Lyon, R.P.; Setter, J.R.; Bovee, T.D.; Doronina, S.A.; Hunter, J.A.; Anderson, M.E.; Balasubramanian, C.L.; Duniho, S.M.; Leiske, C.I.; Li, F.; et al. Self-hydrolyzing maleimides improve the stability and pharmacological properties ofantibody–drug conjugates. Nat. Biotechnol. 2014, 32, 1059–1062. [Google Scholar] [CrossRef] [PubMed]
- Tumey, L.N.; Li, F.; Rago, B.; Han, X.; Loganzo, F.; Musto, S.; Graziani, E.I.; Puthenveetil, S.; Casavant, J.; Marquette, K.; et al. Site Selection: A Case Study in the Identification of Optimal Cysteine Engineered Antibody Drug Conjugates. AAPS J. 2017, 19, 1123–1135. [Google Scholar] [CrossRef] [PubMed]
- Pillow, T.H.; Tien, J.; Parsons-Reponte, K.L.; Bhakta, S.; Li, H.; Staben, L.R.; Li, G.; Chuh, J.; Fourie-O’Donohue, A.; Darwish, M.; et al. Site-Specific Trastuzumab Maytansinoid Antibody–Drug Conjugates with Improved Therapeutic Activity through Linker and Antibody Engineering. J. Med. Chem. 2014, 57, 7890–7899. [Google Scholar] [CrossRef] [PubMed]
- Li, J.Y.; Perry, S.R.; Muniz-Medina, V.; Wang, X.; Wetzel, L.K.; Rebelatto, M.C.; Hinrichs, M.J.M.; Bezabeh, B.Z.; Fleming, R.L.; Dimasi, N. A Biparatopic HER2-Targeting Antibody–Drug Conjugate Induces Tumor Regression in Primary Models Refractory to or Ineligible for HER2-Targeted Therapy. Cancer Cell 2016, 29, 117–129. [Google Scholar] [CrossRef] [PubMed]
- Durben, K.R.; Nottoli, M.S.; Catron, N.D.; Richwine, L.; Jenkins, G.J. High-Throughput, Multispecies, Parallelized Plasma Stability Assay for the Determination and Characterization of Antibody–Drug Conjugate Aggregation and Drug Release. ACS Omega 2017, 2, 4207–4215. [Google Scholar] [CrossRef]
- Puthenveetil, S.; He, H.; Loganzo, F.; Musto, S.; Teske, J.; Green, M.; Tan, X.; Hosselet, C.; Lucas, J.; Tumey, L.N.; et al. Multivalent peptidic linker enables identification of preferred sites of conjugation for a potent thialanstatin antibody drug conjugate. PLoS ONE 2017, 12, e0178452. [Google Scholar] [CrossRef] [PubMed]
- Maruani, A.; Smith, M.E.B.; Miranda, E.; Chester, K.A.; Chudasama, V.; Caddick, S. A plug-and-play approach to antibody-based therapeutics via a chemoselective dual click strategy. Nat. Commun. 2015, 6, 6645. [Google Scholar] [CrossRef] [PubMed]
- Puthenveetil, S.; Musto, S.; Loganzo, F.; Tumey, L.N.; O’Donell, C.J.; Graziani, E. Development of Solid-Phase Site-Specific Conjugation and Its Application toward Generation of Dual Labeled Antibody and Fab Drug Conjugates. Bioconjug. Chem. 2016, 27, 1030–1039. [Google Scholar] [CrossRef] [PubMed]
- Levengood, M.R.; Zhang, X.; Hunter, J.H.; Emmerton, K.K.; Miyamoto, J.B.; Lewis, T.S.; Senter, P.D. Orthogonal Cysteine Protection Enables Homogeneous Multi-Drug Antibody–Drug Conjugates. Angew. Chem. Int. Ed. 2017, 56, 733–737. [Google Scholar] [CrossRef] [PubMed]





© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Verkade, J.M.M.; Wijdeven, M.A.; Van Geel, R.; Janssen, B.M.G.; Van Berkel, S.S.; Van Delft, F.L. A Polar Sulfamide Spacer Significantly Enhances the Manufacturability, Stability, and Therapeutic Index of Antibody–Drug Conjugates. Antibodies 2018, 7, 12. https://doi.org/10.3390/antib7010012
Verkade JMM, Wijdeven MA, Van Geel R, Janssen BMG, Van Berkel SS, Van Delft FL. A Polar Sulfamide Spacer Significantly Enhances the Manufacturability, Stability, and Therapeutic Index of Antibody–Drug Conjugates. Antibodies. 2018; 7(1):12. https://doi.org/10.3390/antib7010012
Chicago/Turabian StyleVerkade, Jorge M. M., Marloes A. Wijdeven, Remon Van Geel, Brian M. G. Janssen, Sander S. Van Berkel, and Floris L. Van Delft. 2018. "A Polar Sulfamide Spacer Significantly Enhances the Manufacturability, Stability, and Therapeutic Index of Antibody–Drug Conjugates" Antibodies 7, no. 1: 12. https://doi.org/10.3390/antib7010012
APA StyleVerkade, J. M. M., Wijdeven, M. A., Van Geel, R., Janssen, B. M. G., Van Berkel, S. S., & Van Delft, F. L. (2018). A Polar Sulfamide Spacer Significantly Enhances the Manufacturability, Stability, and Therapeutic Index of Antibody–Drug Conjugates. Antibodies, 7(1), 12. https://doi.org/10.3390/antib7010012