Synthesis, Structure, and Magnetic Properties of Linear Trinuclear CuII and NiII Complexes of Porphyrin Analogues Embedded with Binaphthol Units

 ,
,
Abstract
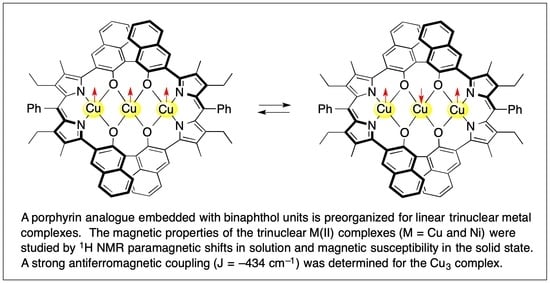
:
1. Introduction
2. Materials and Methods
3. Results and Discussion
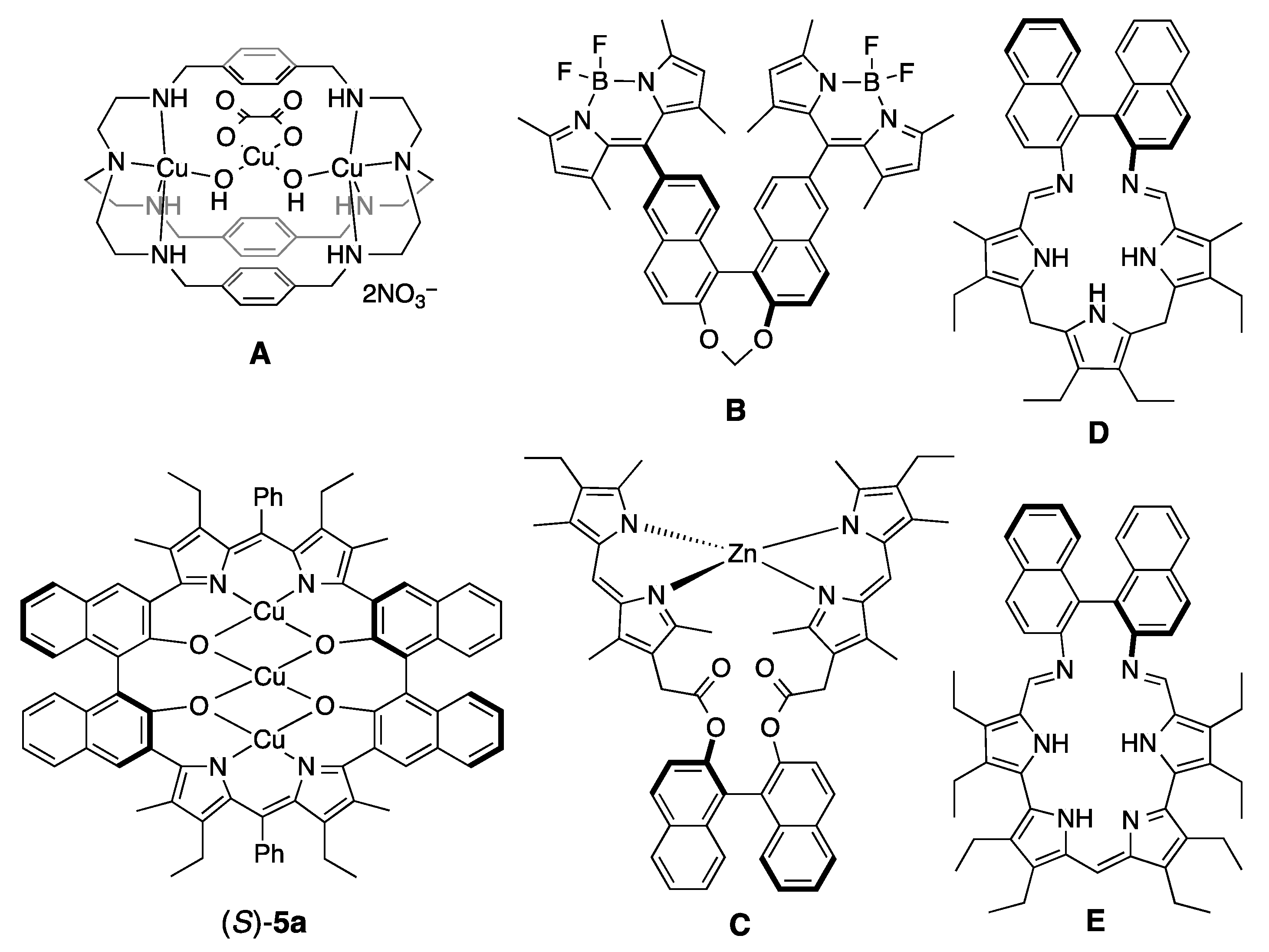
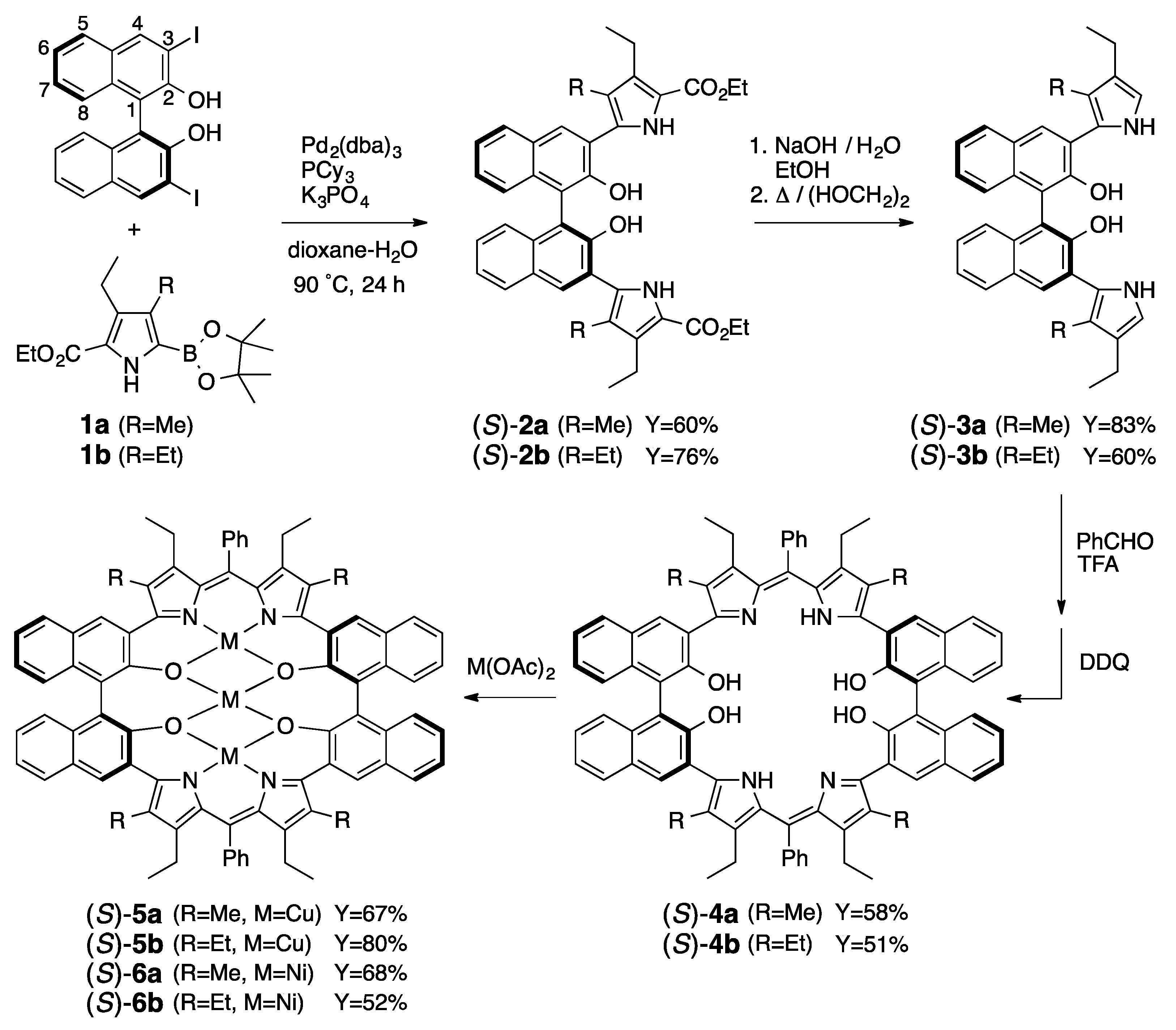
3.1. Synthesis of Porphyrin Analogues
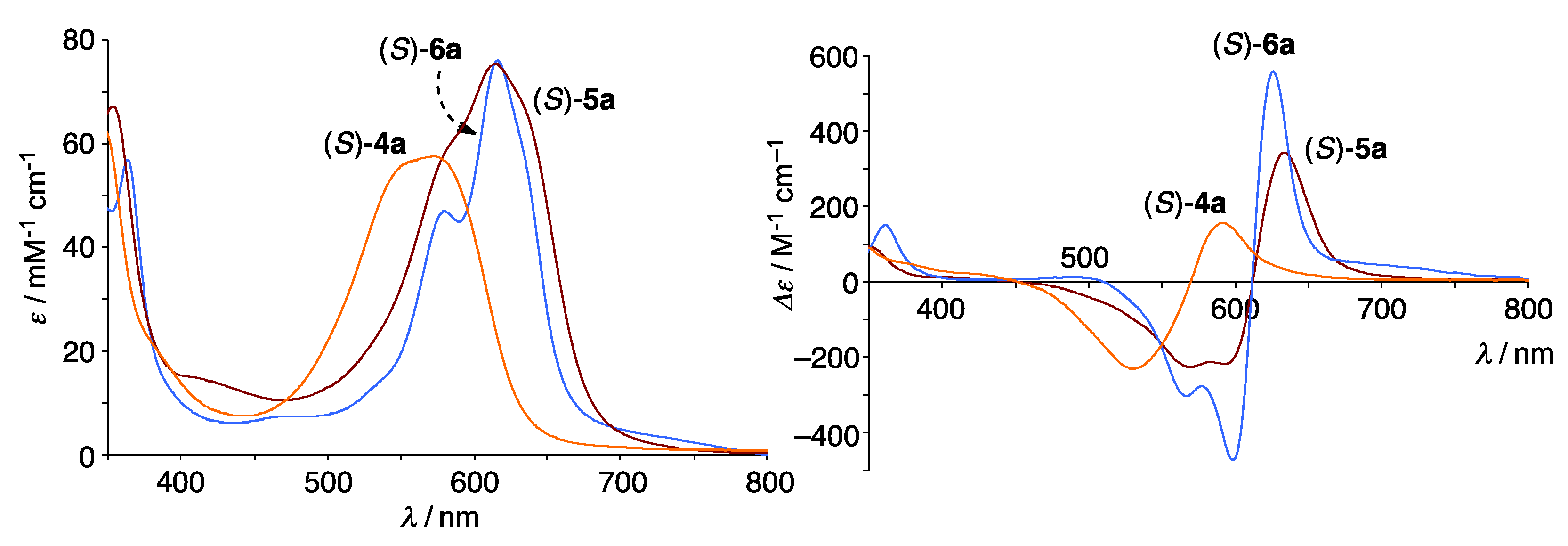
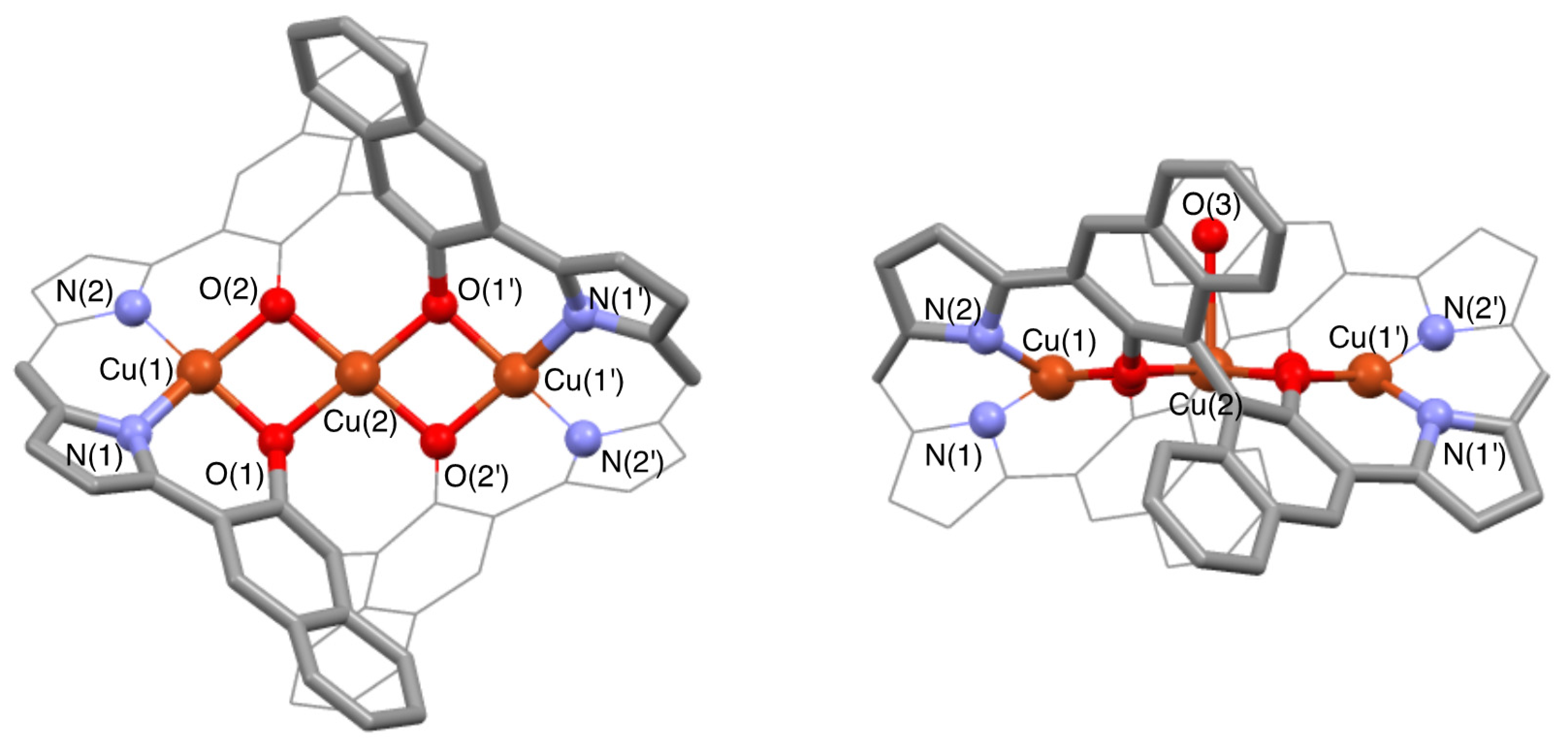
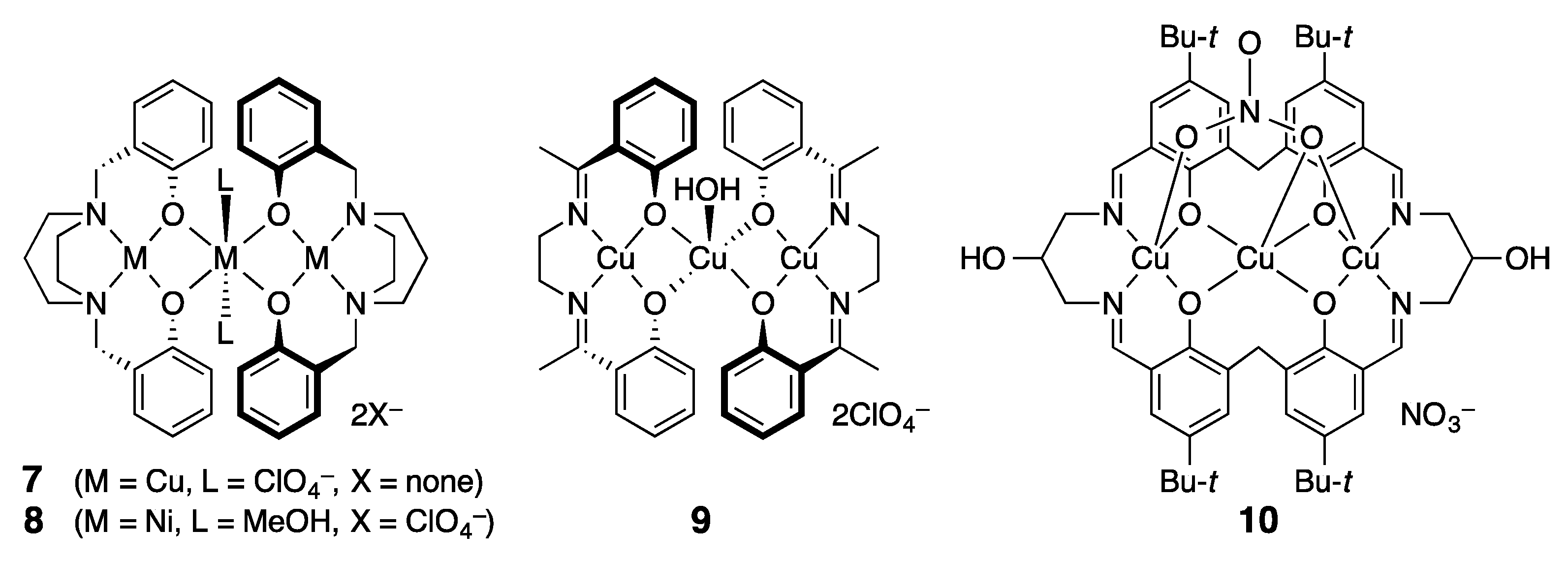
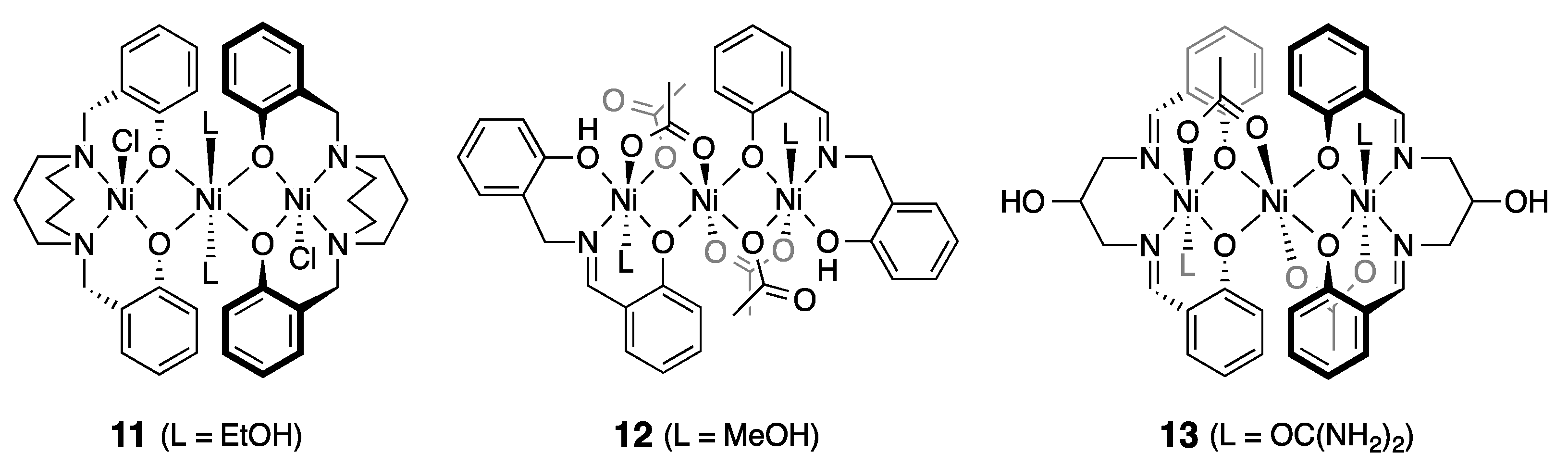
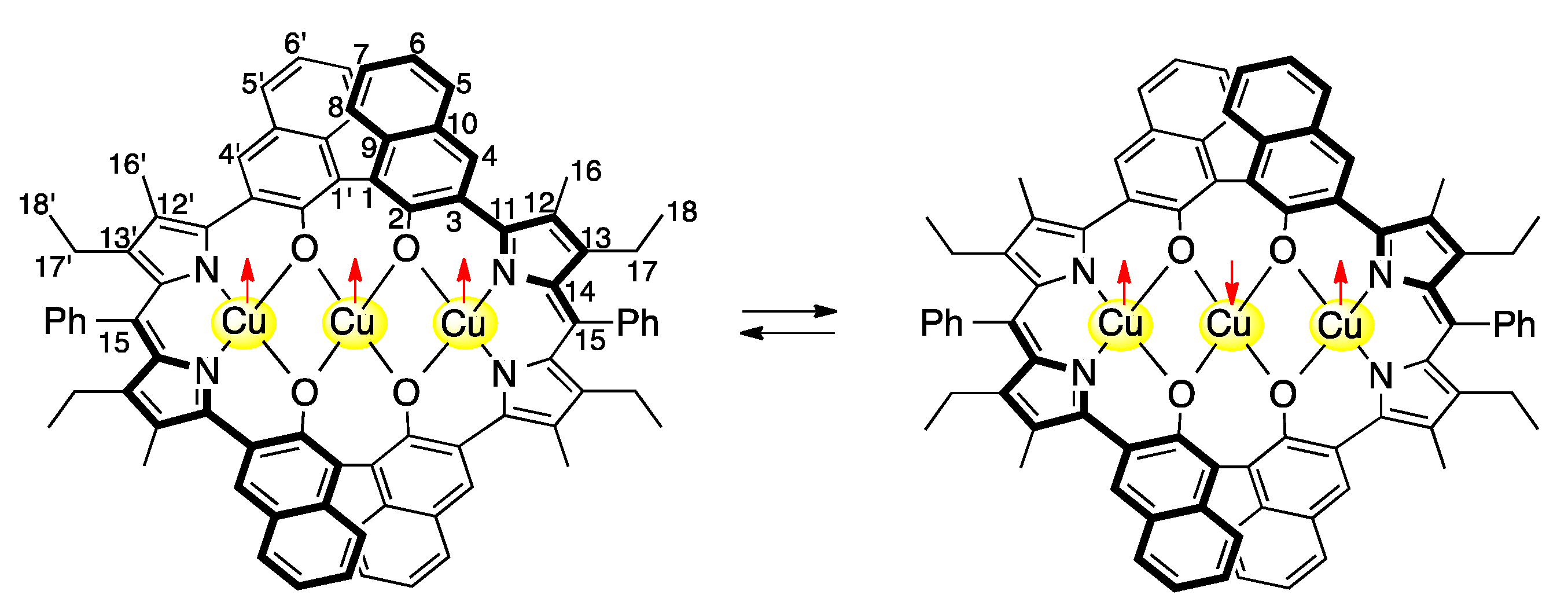
3.2. Structure of Trinuclear Complexes
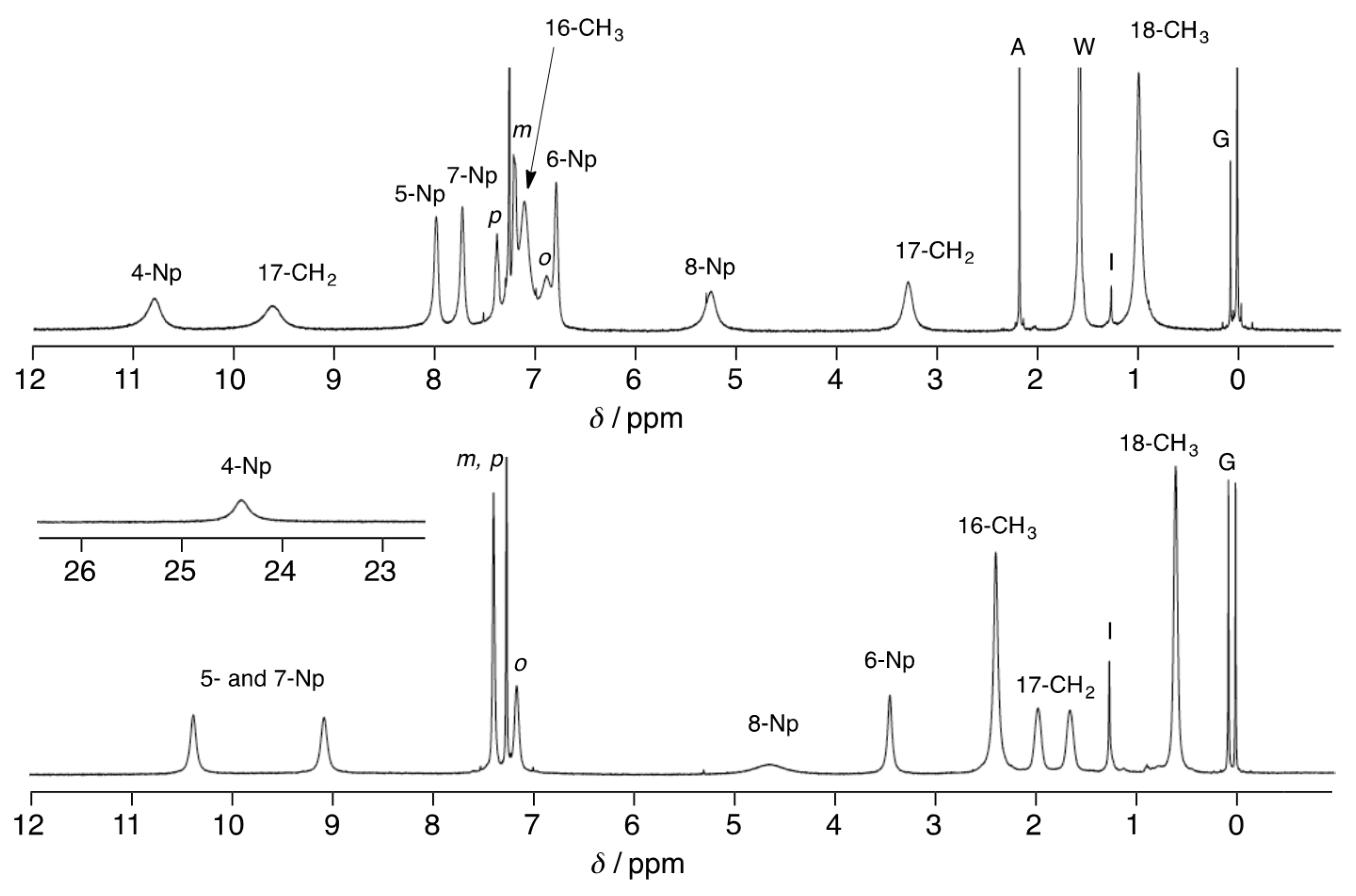
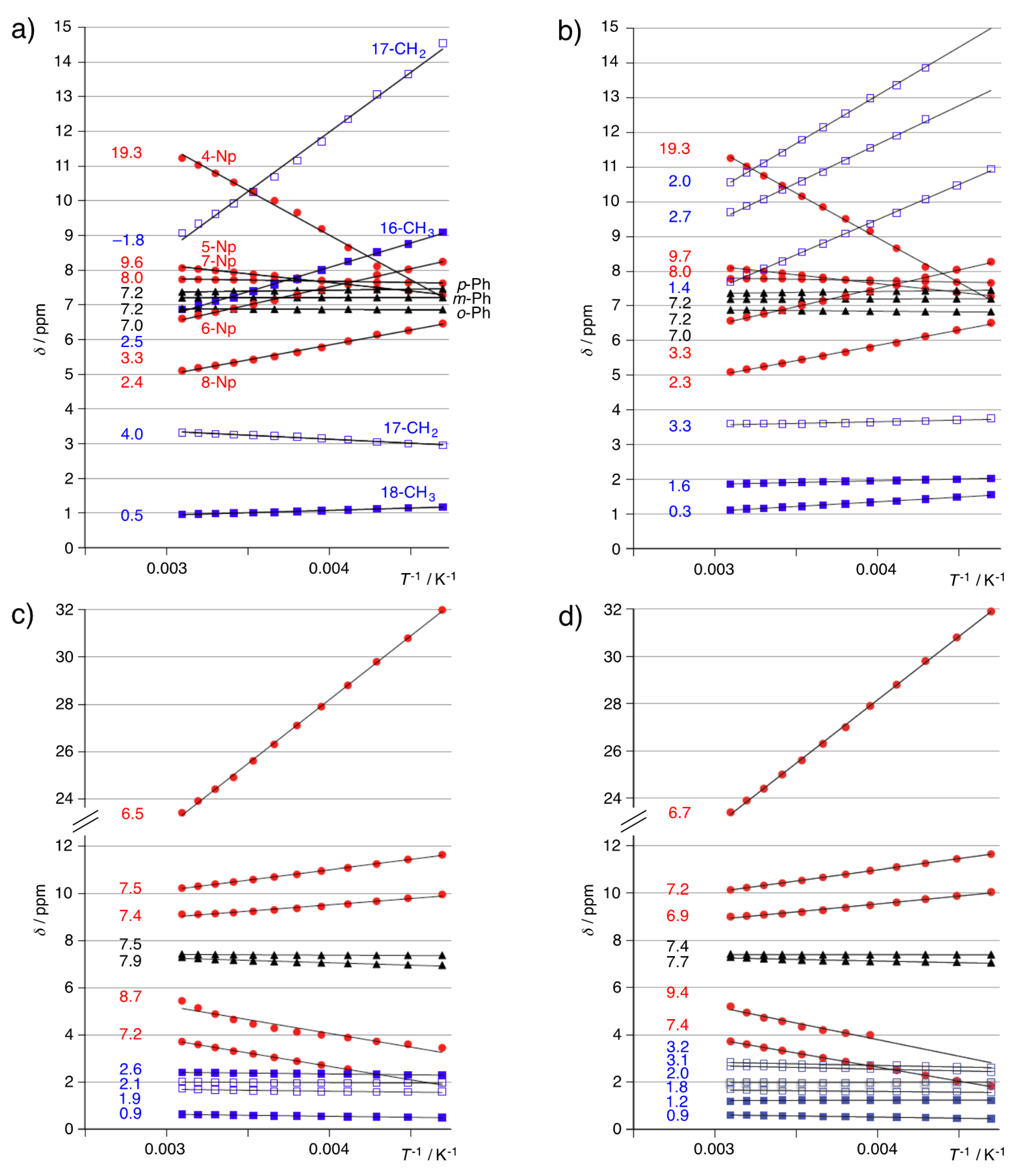
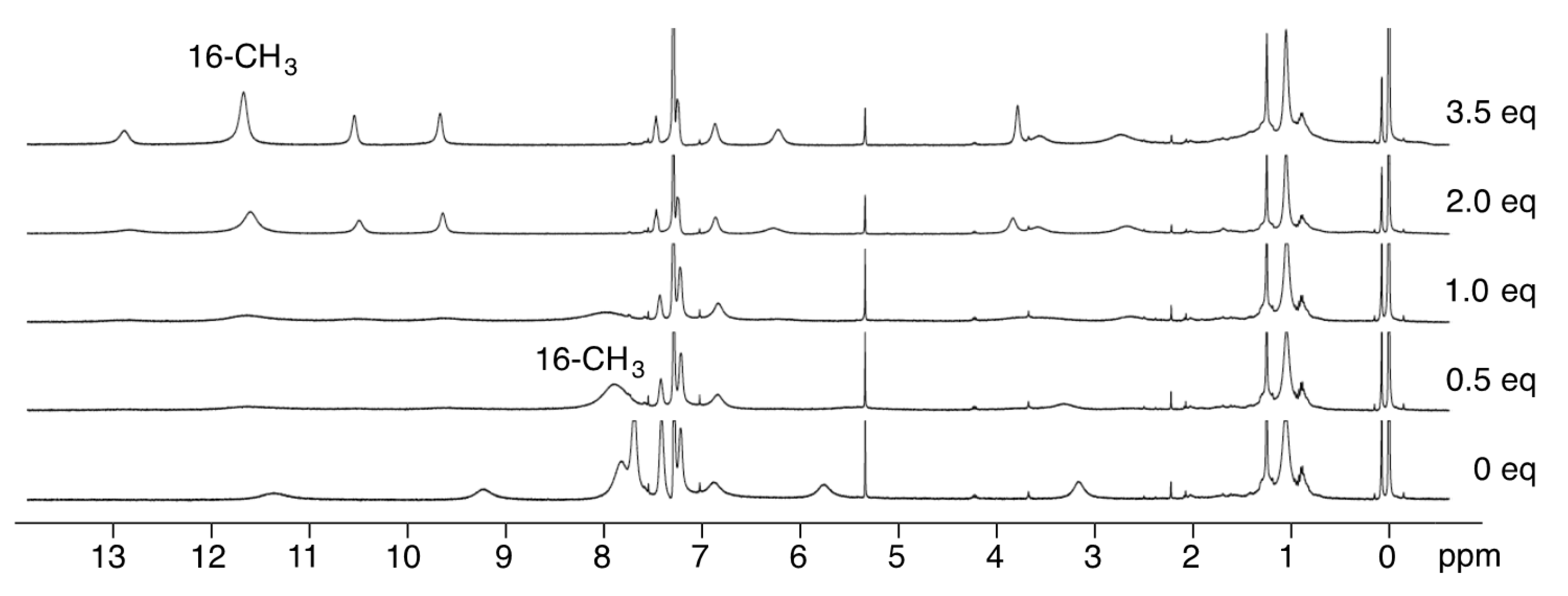
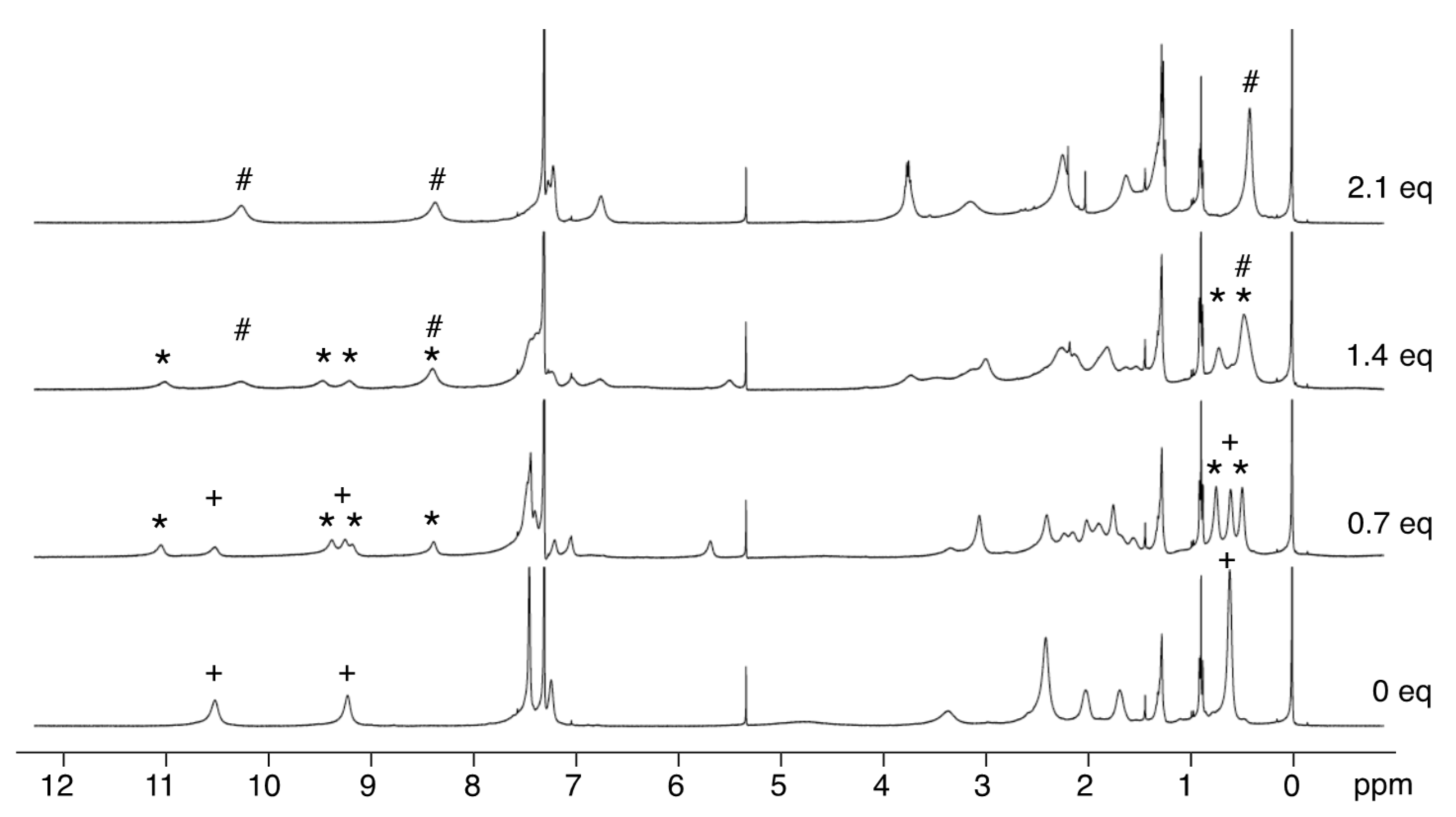
3.3. 1H-NMR Spectra of Paramagnetic Trinuclear Complexes
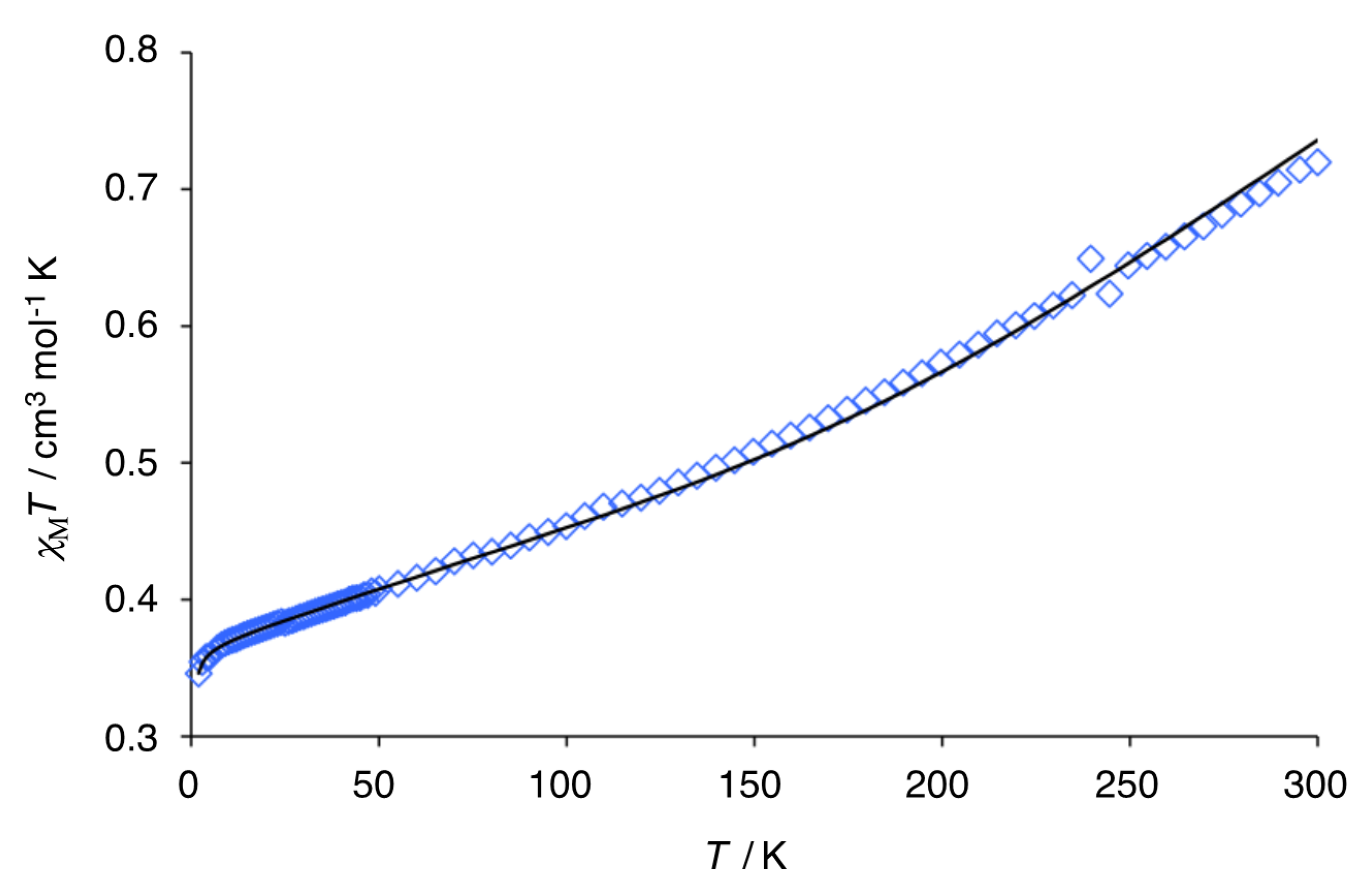
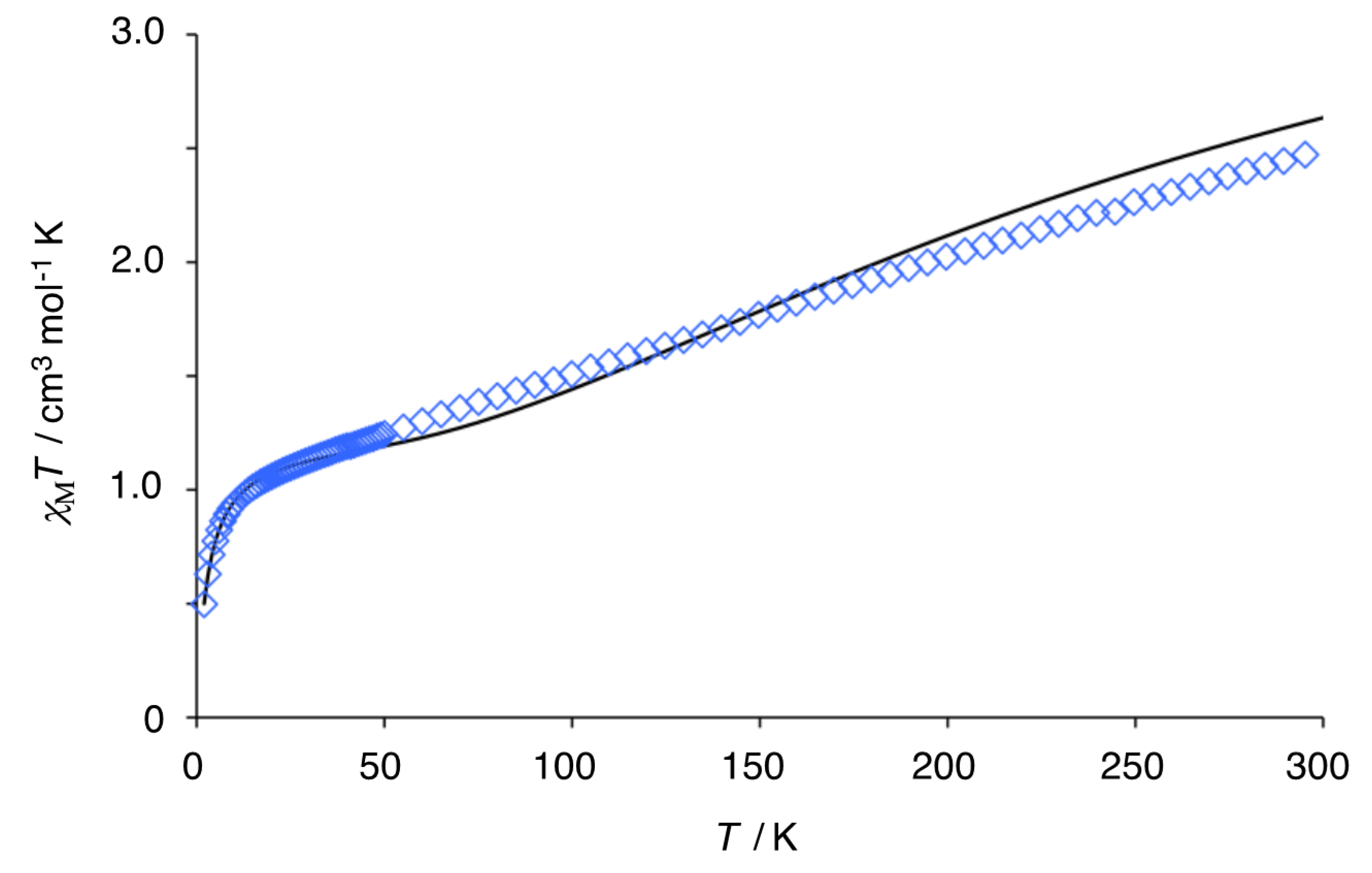
3.4. Magnetic Susceptibility of Trinuclear Complexes
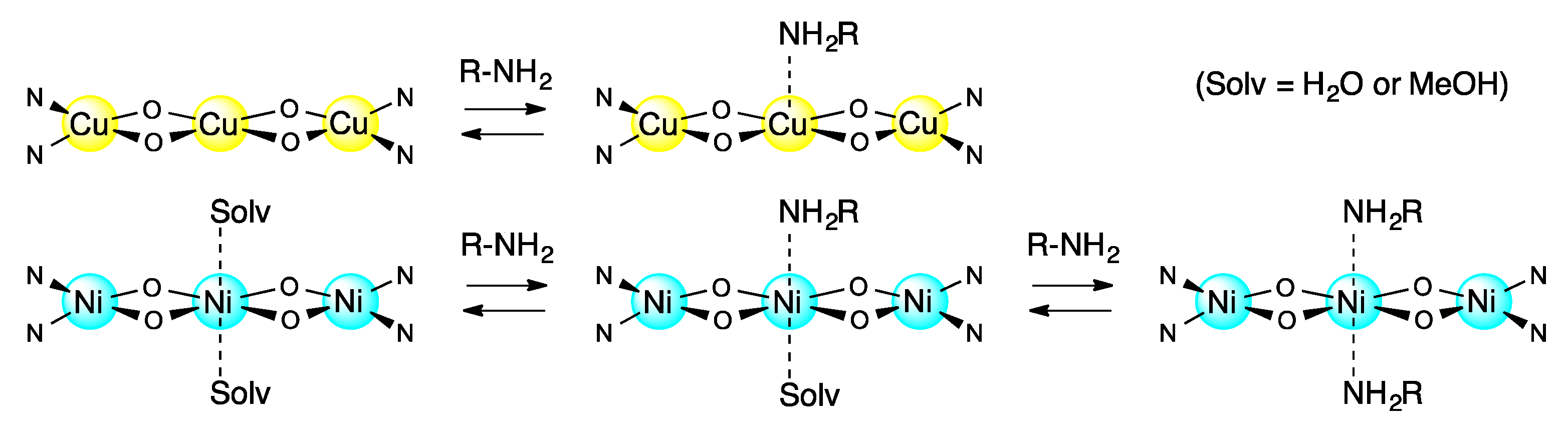
3.5. Reversible Coordination at Apical Sites of the Trinuclear Complexes
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Sessler, J.L.; Tomat, E. Transition-metal Complexes of Expanded Porphyrins. Acc. Chem. Res. 2007, 40, 371–379. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, S.; Osuka, A. Metalation Chemistry of meso-Aryl-Substituted Expanded Porphyrins. Eur. J. Inorg. Chem. 2006, 2006, 1319–1335. [Google Scholar] [CrossRef]
- Vogel, E. Novel Porphyrinoid Macrocycles and Their Metal Complexes. J. Heterocycl. Chem. 1996, 33, 1461–1487. [Google Scholar] [CrossRef]
- Weghorn, S.J.; Sessler, J.L.; Lynch, V.; Baumann, T.F.; Sibert, J.W. Bis[(μ-chloro)copper(II)] Amethyrin: A Bimetallic Copper(II) Complex of an Expanded Porphyrin. Inorg. Chem. 1996, 35, 1089–1090. [Google Scholar] [CrossRef]
- Shimizu, S.; Anand, V.G.; Taniguchi, R.; Furukawa, K.; Kato, T.; Yokoyama, T.; Osuka, A. Biscopper Complexes of meso-Alkyl-Substituted Hexaphyrin: Gable Structures and Varying Antiferromagnetic Coupling. J. Am. Chem. Soc. 2004, 126, 12280–12281. [Google Scholar] [CrossRef]
- Frensch, L.K.; Proepper, K.; John, M.; Demeshko, S.; Brueckner, C.; Meyer, F. Siamese-Twin Porphyrins: A Pyrazole-Based Expanded Porphyrin Providing a Bimetallic Cavity. Angew. Chem. 2011, 123, 1456–1460, Angew. Chem. Int. Ed.2011, 50, 1420–1424. [Google Scholar] [CrossRef]
- Givaja, G.; Volpe, M.; Leeland, J.W.; Edwards, M.A.; Young, T.K.; Darby, S.B.; Reid, S.D.; Blake, A.J.; Wilson, C.; Wolowska, J.E.; et al. Design and Synthesis of Binucleating Macrocyclic Clefts Derived from Schiff-Base Calixpyrroles. Chem. Eur. J. 2007, 13, 3707–3723. [Google Scholar] [CrossRef]
- Veauthier, J.M.; Tomat, E.; Lynch, V.M.; Sessler, J.L.; Mirsaidov, U.; Markert, J.T. Calix[4]pyrrole Schiff Base Macrocycles: Novel Binucleating Ligands for Cu(I) and Cu(II). Inorg. Chem. 2005, 44, 6736–6743. [Google Scholar] [CrossRef]
- Volpe, M.; Hartnett, H.; Leeland, J.W.; Wills, K.; Ogunshun, M.; Duncombe, B.J.; Wilson, C.; Blake, A.J.; McMaster, J.; Love, J.B. Binuclear Cobalt Complexes of Schiff-Base Calixpyrroles and Their Roles in the Catalytic Reduction of Dioxygen. Inorg. Chem. 2009, 48, 5195–5207. [Google Scholar] [CrossRef] [Green Version]
- Askarizadeh, E.; Yaghoob, S.B.; Boghaei, D.M.; Slawin, A.M.Z.; Love, J.B. Tailoring Dicobalt Pacman Complexes of Schiff-base Calixpyrroles towards Dioxygen Reduction Catalysis. Chem. Commun. 2010, 46, 710–712. [Google Scholar] [CrossRef] [Green Version]
- Kamimura, Y.; Shimizu, S.; Osuka, A. [40] Nonaphyrin(1.1.1.1.1.1.1.1.1) and Its Heterometallic Complexes with Palladium–Carbon Bonds. Chem. Eur. J. 2007, 13, 1620–1628. [Google Scholar] [CrossRef] [PubMed]
- Inoue, M.; Ikeda, C.; Kawata, Y.; Venkatraman, S.; Furukawa, K.; Osuka, A. Synthesis of Calix [3]dipyrrins by a Modified Lindsey Protocol. Angew. Chem. 2007, 119, 2356–2359, Angew. Chem. Int. Ed.2007, 46, 2306–2309. [Google Scholar] [CrossRef]
- Yoneda, T.; Sung, Y.M.; Lim, J.M.; Kim, D.; Osuka, A. PdII Complexes of [44]- and [46] Decaphyrins: The Largest Hückel Aromatic and Antiaromatic, and Möbius Aromatic Macrocycles. Angew. Chem. Int. Ed. 2014, 53, 13169–13173. [Google Scholar] [CrossRef]
- Soya, T.; Naoda, K.; Osuka, A. NiII Metallations of [40]- and [42] Nonaphyrins(1.1.1.1.1.1.1.1.1): The Largest Doubly Twisted Hückel Antiaromatic Molecule. Chem. Asian J. 2015, 10, 231–238. [Google Scholar] [CrossRef] [PubMed]
- Setsune, J.; Toda, M.; Yoshida, T. Synthesis and Dynamic Structure of Multinuclear Rh Complexes of Porphyrinoids. Chem. Commun. 2008, 1425–1428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Setsune, J.; Toda, M.; Yoshida, T.; Imamura, K.; Watanabe, K. The Synthesis and Dynamic Structures of Multinuclear Complexes of Large Porphyrinoids Expanded by Phenylene and Thienylene Spacers. Chem. Eur. J. 2015, 21, 12715–12727. [Google Scholar] [CrossRef]
- Setsune, J.; Omae, S. Homohelical Porphyrin Analogue Embedded with Binaphthol Units. Chem. Lett. 2012, 41, 168–169. [Google Scholar] [CrossRef]
- Ferguson, G.; Langrick, C.R.; Parker, D.; Matthes, K.E. A Linear Trinuclear Macrocyclic Copper(II) Complexes. J. Chem. Soc. Chem. Commun. 1985, 22, 1609–1610. [Google Scholar] [CrossRef]
- Cronin, L.; Walton, P.H. Synthesis and Single Crystal X-ray Structure of a Novel Trinuclear Copper(II) Methoxide Complex. Inorg. Chim. Acta 1998, 269, 241–245. [Google Scholar] [CrossRef]
- Ruf, M.; Pierpont, C.G. Methoxide Coordination at the Pocket of [CuIITpCum.Me] and a Simple Model for the Center of Galactose Oxidase. Angew. Chem. 1998, 110, 1830–1832, Angew. Chem. Int. Ed.1998, 37, 1736–1739. [Google Scholar] [CrossRef]
- Shakya, R.; Jozwiuk, A.; Powell, D.R.; Houser, R.P. Synthesis and Characterization of Polynuclear Copper(II) Complexes with Pyridylbis(phenol) Ligands. Inorg. Chem. 2009, 48, 4083–4088. [Google Scholar] [CrossRef] [PubMed]
- Barta, C.A.; Bayly, S.R.; Read, P.W.; Patrick, B.O.; Thompson, R.C.; Orvig, C. Molecular Architectures for Trimetallic d/f/d Complexes: Magnetic Studies of a LnCu2 Core. Inorg. Chem. 2008, 47, 2294–2302. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.-F.; van Albada, G.A.; Tang, J.; Mutikainen, I.; Turpeinen, U.; Massera, C.; Roubeau, O.; Costa, J.S.; Gamez, P.; Reedijk, J. Controlled Copper-Mediated Chlorination of Phenol Rings under Mild Conditions. Inorg. Chem. 2007, 46, 4944–4950. [Google Scholar] [CrossRef] [PubMed]
- Thakurta, S.; Chakraborty, J.; Rosair, G.; Tercero, J.; El Fallah, M.S.; Garribba, E.; Mitra, S. Synthesis of Two New Linear Trinuclear CuII Complexes: Mechanism of Magnetic Coupling through Hybrid B3LYP Functional and CShM Studies. Inorg. Chem. 2008, 47, 6227–6235. [Google Scholar] [CrossRef]
- Blake, A.J.; Brechin, E.K.; Codron, A.; Gould, R.O.; Grant, C.M.; Parsons, S.; Rawson, J.M.; Winpenny, R.E.P. New Polynuclear Nickel Complexes with a Variety of Pyridonate and Carboxylate Ligands. J. Chem. Soc. Chem. Commun. 1995, 19, 1983–1985. [Google Scholar] [CrossRef]
- Kavlakoglu, E.; Elmali, A.; Elerman, Y.; Werner, R.; Svoboda, I.; Fuess, H. Crystal Structure and Magnetic Properties of a Linear Trinuclear Ni(II) Complex. Z. Naturforsch. B Chem. Sci. 2001, 56, 43–48. [Google Scholar] [CrossRef]
- Banerjee, S.; Drew, M.G.B.; Lu, C.-Z.; Tercero, J.; Diaz, C.; Ghosh, A. Dinuclear Complexes of MII Thiocyanate (M = Ni and Cu) Containing a Tridentate Schiff-Base Ligand: Synthesis, Structural Diversity and Magnetic Properties. Eur. J. Inorg. Chem. 2005, 12, 2376–2383. [Google Scholar] [CrossRef]
- Sharma, A.K.; Lloret, F.; Mukherjee, R. Phenolate and Acetate (Both μ2-1,1 and μ2-1,3 Mode)-Bridged Face-Shared Trioctahedral Linear NiII3, NiII2MII (M = Mn, Co) Complexes: Ferro-and Antiferromagnetic Coupling. Inorg. Chem. 2007, 46, 5128–5130. [Google Scholar] [CrossRef]
- Beissel, T.; Birkelbach, F.; Bill, E.; Glaser, T.; Kesting, F.; Krebs, C.; Weyhermuller, T.; Wieghardt, K.; Butzlaff, C.; Trautwein, A.X. Exchange and Double-Exchange Phenomena in Linear Homo- and Heterotrinuclear Nickel (II,III,IV) Complexes Containing Six μ2-Phenolato or μ2-Thiophenolato Bridging Ligand. J. Am. Chem. Soc. 1996, 118, 12376–12390. [Google Scholar] [CrossRef]
- Xu, Z.; Thompson, L.K.; Milway, V.A.; Zhao, L.; Kelly, T.; Miller, D.O. Self-Assembled Dinuclear, Trinuclear, Tetranuclear, Pentanuclear, and Octanuclear Ni(II) Complexes of a Series of Polytopic Diazine Based Ligands: Structural and Magnetic Properties. Inorg. Chem. 2003, 42, 2950–2959. [Google Scholar] [CrossRef]
- Fontecha, J.B.; Goetz, S.; McKee, V. Di-, Tri-, and Tetracopper(II) Complexes of a Pseudocalixarene Macrocycle. Angew. Chem. 2002, 114, 4735–4738, Angew. Chem. Int. Ed.2002, 41, 4553–4556. [Google Scholar] [CrossRef]
- Esteves, C.V.; Mateus, P.; Andreé, V.; Bandeira, N.A.G.; Calhorda, M.J.; Ferreira, L.P.; Delgado, R. Di-versus Trinuclear Copper(II) Cryptate for the Uptake of Dicarboxylate Anions. Inorg. Chem. 2016, 55, 7051–7060. [Google Scholar] [CrossRef] [PubMed]
- Izzet, G.; Akdas, H.; Hucher, N.; Giorgi, M.; Prange, T.; Reinaud, O. Supramolecular Assemblies with Calix[6]arenes and Copper Ions: From Dinuclear to Trinuclear Linear Arrangements of Hydroxo-Cu(II) Complexes. Inorg. Chem. 2006, 45, 1069–1077. [Google Scholar] [CrossRef] [PubMed]
- Beer, G.; Niederaalt, C.; Grimme, S.; Daub, J. Redox Switches with Chiroptical Signal Expression Based on Binaphthyl Boron Dipyrromethene Conjugates. Angew. Chem. 2000, 112, 3385–3388, Angew. Chem. Int. Ed.2000, 39, 3252–3255. [Google Scholar] [CrossRef]
- Al-Sheikh-Ali, A.; Benson, R.E.; Blumentritt, S.; Cameron, T.S.; Linden, A.; Wolstenholme, D.; Thompson, A. Asymmetric Synthesis of Mono- and Dinuclear Bis(dipyrrinato) Complexes. J. Org. Chem. 2007, 72, 4947–4952. [Google Scholar]
- Gunst, K.; Seggewies, S.; Breitmaier, E. New Chiral Macrocyclic Di-imines Containing Tetrapyrrole and Tripyrrane Subunits. Synthesis 2001, 12, 1856–1860. [Google Scholar] [CrossRef]
- Miyaji, H.; Hong, S.-J.; Jeong, S.-D.; Yoon, D.-W.; Na, H.-K.; Hong, J.; Ham, S.; Sessler, J.L.; Lee, C.-H. A Binol-Strapped Calix[4]pyrrole as a Model Chirogenic Receptor for the Enantioselective Recognition of Carboxylate Anions. Angew. Chem. 2007, 119, 2560–2563, Angew. Chem. Int. Ed.2007, 46, 2508–2511. [Google Scholar] [CrossRef]
- Setsune, J.; Toda, M.; Watanabe, K.; Panda, P.K.; Yoshida, T. Synthesis of Bis(pyrrol-2-yl)arenes by Pd-Catalyzed Cross Coupling. Tetrahedron Lett. 2006, 47, 7541–7544. [Google Scholar]
- Setsune, J.; Watanabe, K. Cryptand-like Porphyrinoid Assembled with Three Dipyrrylpyridine Chains: Synthesis, Structure, and Homotropic Positive Allosteric Binding of Carboxylic Acids. J. Am. Chem. Soc. 2008, 130, 2404–2405. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXTL 5.10 for Windows NT: Structure Determination Software Programs; Bruker Analytical X-ray Systems, Inc.: Madison, WI, USA, 1997. [Google Scholar]
- Bain, G.A.; Bery, J.E. Diamagnetic Corrections and Pascal’s Constants. J. Chem. Educ. 2008, 85, 532. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Wu, T.R.; Shen, L.; Chong, J.M. Asymmetric Allylboration of Aldehyde and Ketones Using 3,3′-Disubstitutedbinaphthol-Modified Boronates. Org. Lett. 2004, 6, 2701–2704. [Google Scholar] [CrossRef] [PubMed]
- Suresh, P.; Srimurugan, S.; Babu, B.; Pati, H.N. Synthesis of Some Acetylene-Tethered Chiral and Achiral Dialdehydes. Acta Chim. Slov. 2008, 55, 453–457. [Google Scholar]
- Kudo, N.; Perseghini, M.; Fu, G.C. A Versatile Method for Suzuki Cross-Coupling Reactions of Nitrogen Heterocycles. Angew. Chem. 2006, 118, 1304–1306, Angew. Chem. Int. Ed.2006, 45, 1282–1284. [Google Scholar] [CrossRef]
- Fu, G.C. The Development of Versatile Methods for Palladium-Catalyzed Coupling Reactions of Aryl Electrophiles through the Use of P(t-Bu)3 and PCy3 as Ligands. Acc. Chem. Res. 2008, 41, 1555–1564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rothemund, P.A. New Porphyrin Synthesis. The Synthesis of Porphin. J. Am. Chem. Soc. 1936, 58, 625–627. [Google Scholar] [CrossRef]
- Song, Y.; Gamez, P.; Roubeau, O.; Lutz, M.; Spek, A.L.; Reedijk, J. Structural and Magnetic Characterization of a Linear Trinuclear Copper Complex Formed through Ligand Sharing. Eur. J. Inorg. Chem. 2003, 16, 2924–2928. [Google Scholar] [CrossRef]
- Song, Y.; Gamez, P.; Roubeau, O.; Mutikainen, I.; Turpeinen, U.; Reedijk, J. Structure and Magnetism of Two New Linear Trinuclear Copper(II) Clusters Obtained from the Tetradentate N2O2 Ligand Bis(2-hydroxybenzyl)-1,3-diaminopropane. Inorg. Chim. Acta 2005, 358, 109–115. [Google Scholar] [CrossRef]
- Song, Y.; van Albada, G.A.; Quesada, M.; Mutikainen, I.; Turpeinen, U.; Reedijk, J. A New Linear Trinuclear Cu(II) Complex [Cu3L2(MeCN)2I2](MeCN)2 with Semi-Coordinated Iodides Formed through Ligand Sharing (H2L = 1,7-Bis(2-hydroxyphenyl)-2,6-diaza-4-hydroxyl-heptane). Inorg. Chem. Commun. 2005, 8, 975–978. [Google Scholar] [CrossRef]
- Bu, W.-H.; Du, M.; Shang, Z.-L.; Zhang, R.-H.; Liao, D.-Z.; Shionoya, M.; Clifford, T. Varying Coordination Modes and Magnetic Properties of Copper(II) Complexes with Diazamesocyclic Ligands by Altering Additional Donor Pendants on 1,5-Diazacyclooctane. Inorg. Chem. 2000, 39, 4190–4199. [Google Scholar] [CrossRef]
- Du, M.; Zhao, X.-J.; Guo, J.-H.; Bu, X.-H.; Ribas, J. Towards the Design of Linear Homo-Trinuclear Metal Complexes Based on a New Phenol-Functionalised Diazamesocyclic Ligand: Structural Analysis and Magnetism. Eur. J. Inorg. Chem. 2005, 2, 294–304. [Google Scholar] [CrossRef]
- Epstein, J.M.; Figgis, B.N.; White, A.H.; Willis, A.C. Crystal Structures of Two Trinuclear Schiff-Based Copper(II) Complexes. J. Chem. Soc. Dalton Trans. 1974, 1954–1961. [Google Scholar] [CrossRef]
- Bu, X.-H.; Du, M.; Zhang, L.; Liao, D.-Z.; Tang, J.-K.; Zhang, R.-H.; Shionoya, M. Novel Nickel(II) Complexes with Diazamesocyclic Ligands Functionalized by Additional Phenol Donor Pendant(s): Synthesis, Characterization, Crystal Structures and Magnetic Properties. J. Chem. Soc. Dalton Trans. 2001, 593–598. [Google Scholar] [CrossRef]
- Wang, Q.-L.; Yang, C.; Qi, L.; Liao, D.-Z.; Yang, G.-M.; Ren, H.-X. A Trinuclear Nickel(II) Complex with Dissimilar Bridges: Synthesis, Crystal Structure, Spectroscopy and Magnetism. J. Mol. Struct. 2008, 892, 88–92. [Google Scholar] [CrossRef]
- Lu, J.-W.; Chen, C.-Y.; Kao, M.-C.; Cheng, C.-M.; Wei, H.-H. Synthesis, Crystal Structure, and Magnetic Properties of μ-Phenoxo/μ-Carboxylato-Bridged Trinuclear Nickel(II) Complexes with Schiff Base, DMF, and Urea Ligands. J. Mol. Struct. 2009, 936, 228–233. [Google Scholar] [CrossRef]
- Mukherjee, P.; Drew, M.G.B.; Gomez-Garcıa, C.J.; Ghosh, A. (Ni2), (Ni3), and (Ni2 + Ni3): A Unique Example of Isolated and Cocrystallized Ni2 and Ni3 Complexes. Inorg. Chem. 2009, 48, 4817–4827. [Google Scholar] [CrossRef]
- Zhang, L.; Gao, W.; Wu, Q.; Su, Q.; Zhang, J.; Mu, Y. Synthesis and Characterization of Chiral Trinuclear Cobalt and Nickel Complexes Supported by Binaphthol-derived Bis(salicylaldimine) Ligands. J. Coord. Chem. 2013, 66, 3182–3192. [Google Scholar] [CrossRef]
- Evans, D.F. The Determination of the Paramagnetic Susceptibility of Substances in Solution by Nuclear Magnetic Resonance. J. Chem. Soc. 1959, 2003–2005. [Google Scholar] [CrossRef]
- Schubert, E.M. Utilizing the Evans Method with a Superconducting NMR Spectrometer in the Undergraduate Laboratory. J. Chem. Educ. 1992, 69, 62. [Google Scholar] [CrossRef]
- Grant, D.H. Paramagnetic Susceptibility by NMR: The “Solvent Correction” Reexamined. J. Chem. Educ. 1995, 72, 39–40. [Google Scholar] [CrossRef]
- Bertini, I.; Luchinat, C. NMR of Paramagnetic Substances. Coord. Chem. Rev. 1996, 150, 1–296. [Google Scholar]
- Bren, K.L. NMR Analysis of Spin States and Spin Densities. In Spin States in Biochemistry and Inorganic Chemistry: Influence on Structure and Reactivity; Swart, M., Costas, M., Eds.; Wiley: Chichester, UK, 2016; Volume 16, pp. 409–434. [Google Scholar]
- Kahn, O. Molecular Magnetism; VCH: New York, NY, USA, 1993. [Google Scholar]
- Thompson, L.K.; Mandal, S.K.; Tandon, S.S.; Bridson, J.N.; Park, M.K. Magnetostructural Correlations in Bis(μ2-phenoxide)-Bridged Macrocyclic Dinuclear Copper(II) Complexes. Influence of Electron-Withdrawing Substituents on Exchange Coupling. Inorg. Chem. 1996, 35, 3117–3125. [Google Scholar] [CrossRef] [PubMed]
- Nanda, K.K.; Das, R.; Thompson, L.K.; Venkatsubramanian, K.; Paul, P.; Nag, K. Magneto-Structural Correlations in Macrocyclic Dinickel(II) Complexes: Tuning of Spin Exchange by Varying Stereochemistry and Auxiliary Ligands. Inorg. Chem. 1994, 33, 1188–1193. [Google Scholar] [CrossRef]
- Carbonaro, L.; Isola, M.; La Pegna, P.; Senatore, L.; Marchetti, F. Spectrophotometric Study of the Equilibria between Nickel(II) Schiff-Base Complexes and Alkaline Earth or Nickel(II) Cations in Acetonitrile Solution. Inorg. Chem. 1999, 38, 5519–5525. [Google Scholar] [CrossRef] [PubMed]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| (S)-5a | 7 | 8 | 9 | 10 | |
|---|---|---|---|---|---|
| M(1)–O(1) | 1.877 | 1.927 | 1.873 | 1.877–1.899 | 1.945–1.957 |
| M(1)–O(2) | 1.893 | 1.929 | |||
| M(2)–O(1) | 1.949 | 1.953 | 2.041 | 1.930, 1.949 | 1.918–1.929 |
| M(2)–O(2) | 1.922 | 1.956 | 2.050 | 1.977, 2.065 | |
| M(2)–O(apical) | 2.43 | 2.589 | 2.067 | 2.177 | 2.491 |
| M(1)–M(2) | 2.910 | 2.938 | 3.007 | 2.950, 2.975 | 2.843, 2.861 |
| M(1)–O(1)–M(2) | 98.5 | 98.3 | 100.0 | 97.6–101.6 | 94.1–95.3 |
| M(1)–O(2)–M(2) | 100.0 | 98.4 | 100.4 | ||
| O(1)–M(1)–O(2) | 82.1 | 80.5 | 83.8 | 76.8, 76.7 | 77.5–81.4 |
| O(1)–M(2)–O(2) | 79.5 | 81.9 | 75.4 | ||
| M(1)–M(2)–M(1′) | 174.7 | 180 | 180 | 156.2 | 127.8 |
| S = 1/2 | S = 3/2 | |||
|---|---|---|---|---|
| Cu(1), Cu(1′) | 0.5977 | 0.6083 | ||
| Cu(2) | −0.6169 | 0.6366 | ||
| naphthyl-O(1),O(1′) | −0.0018 | −0.0032 | 0.1428 | 0.1453 |
| dipyrrin-N(1),N(1′) | 0.1061 | 0.1051 | 0.1099 | 0.1093 |
| naphthyl-C(4),C(4′) | 0.0061 | 0.0069 | −0.0082 | −0.0092 |
| naphthyl-C(5),C(5′) | 0.0047 | 0.0053 | −0.0052 | −0.0058 |
| naphthyl-C(6),C(6′) | −0.0060 | −0.0067 | 0.0066 | 0.0073 |
| naphthyl-C(7),C(7′) | 0.0043 | 0.0048 | −0.0048 | −0.0053 |
| naphthyl-C(8),C(8′) | −0.0054 | −0.0060 | 0.0063 | 0.0070 |
| pyrrole β-C(12),C(12′) | 0.0034 | 0.0036 | 0.0047 | 0.0053 |
| pyrrole β-C(13),C(13′) | 0.0072 | 0.0072 | 0.0067 | 0.0062 |
| methyl-C(16),C(16′) | 0.0010 | 0.0010 | 0.0009 | 0.0009 |
| methylene-C(17),C(17′) | 0.0004 | 0.0003 | 0.0005 | 0.0004 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Setsune, J.-i.; Omae, S.; Tsujimura, Y.; Mochida, T.; Sakurai, T.; Ohta, H. Synthesis, Structure, and Magnetic Properties of Linear Trinuclear CuII and NiII Complexes of Porphyrin Analogues Embedded with Binaphthol Units. Symmetry 2020, 12, 1610. https://doi.org/10.3390/sym12101610
Setsune J-i, Omae S, Tsujimura Y, Mochida T, Sakurai T, Ohta H. Synthesis, Structure, and Magnetic Properties of Linear Trinuclear CuII and NiII Complexes of Porphyrin Analogues Embedded with Binaphthol Units. Symmetry. 2020; 12(10):1610. https://doi.org/10.3390/sym12101610
Chicago/Turabian StyleSetsune, Jun-ichiro, Shintaro Omae, Yukinori Tsujimura, Tomoyuki Mochida, Takahiro Sakurai, and Hitoshi Ohta. 2020. "Synthesis, Structure, and Magnetic Properties of Linear Trinuclear CuII and NiII Complexes of Porphyrin Analogues Embedded with Binaphthol Units" Symmetry 12, no. 10: 1610. https://doi.org/10.3390/sym12101610
APA StyleSetsune, J. -i., Omae, S., Tsujimura, Y., Mochida, T., Sakurai, T., & Ohta, H. (2020). Synthesis, Structure, and Magnetic Properties of Linear Trinuclear CuII and NiII Complexes of Porphyrin Analogues Embedded with Binaphthol Units. Symmetry, 12(10), 1610. https://doi.org/10.3390/sym12101610