Assessing the Molecular Specificity and Orientation Sensitivity of Infrared, Raman, and Vibrational Sum-Frequency Spectra


Abstract
:1. Introduction
2. Background
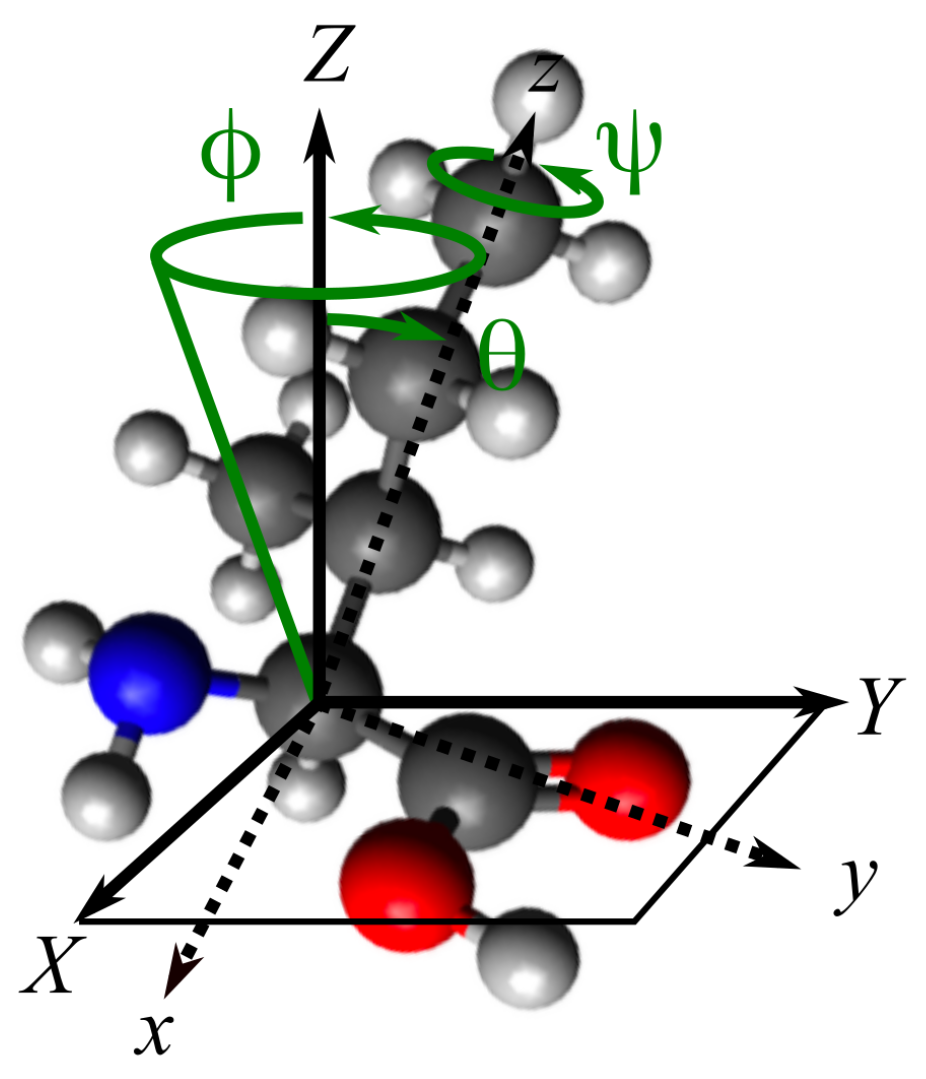
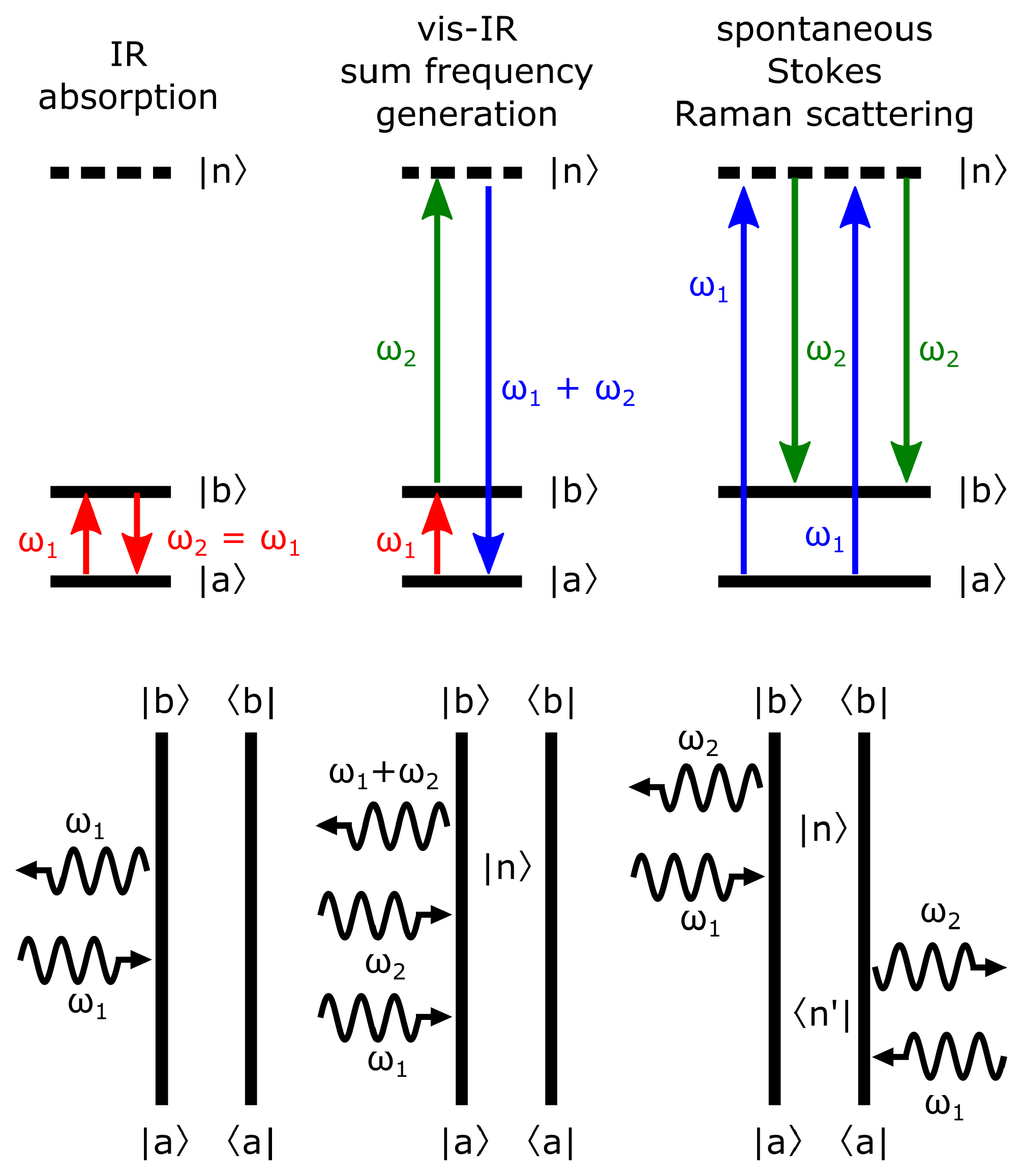
2.1. Molecular and Ensemble Response Functions
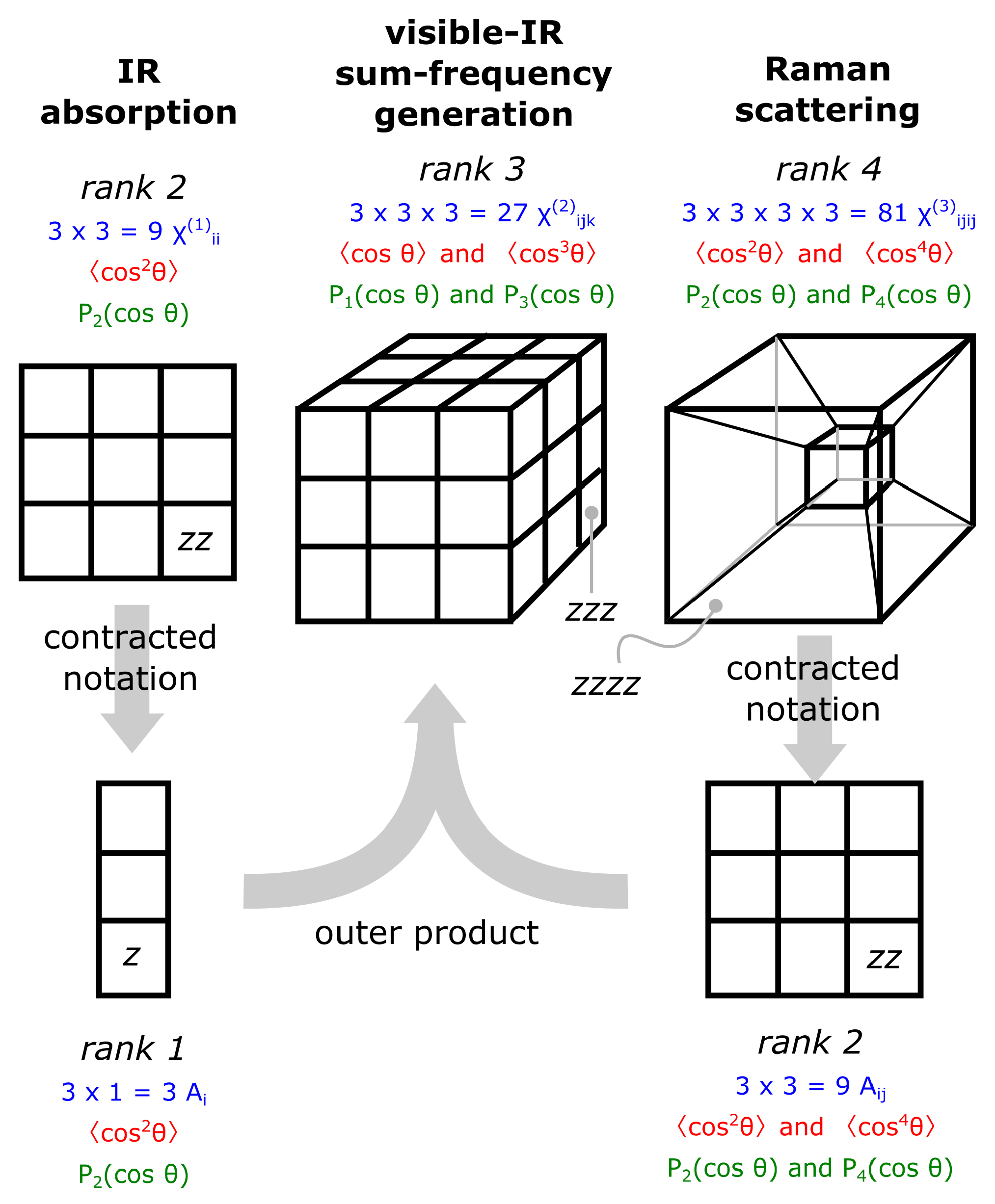
2.2. Accessing Elements of the Response Functions Using Polarized Light
3. Methods
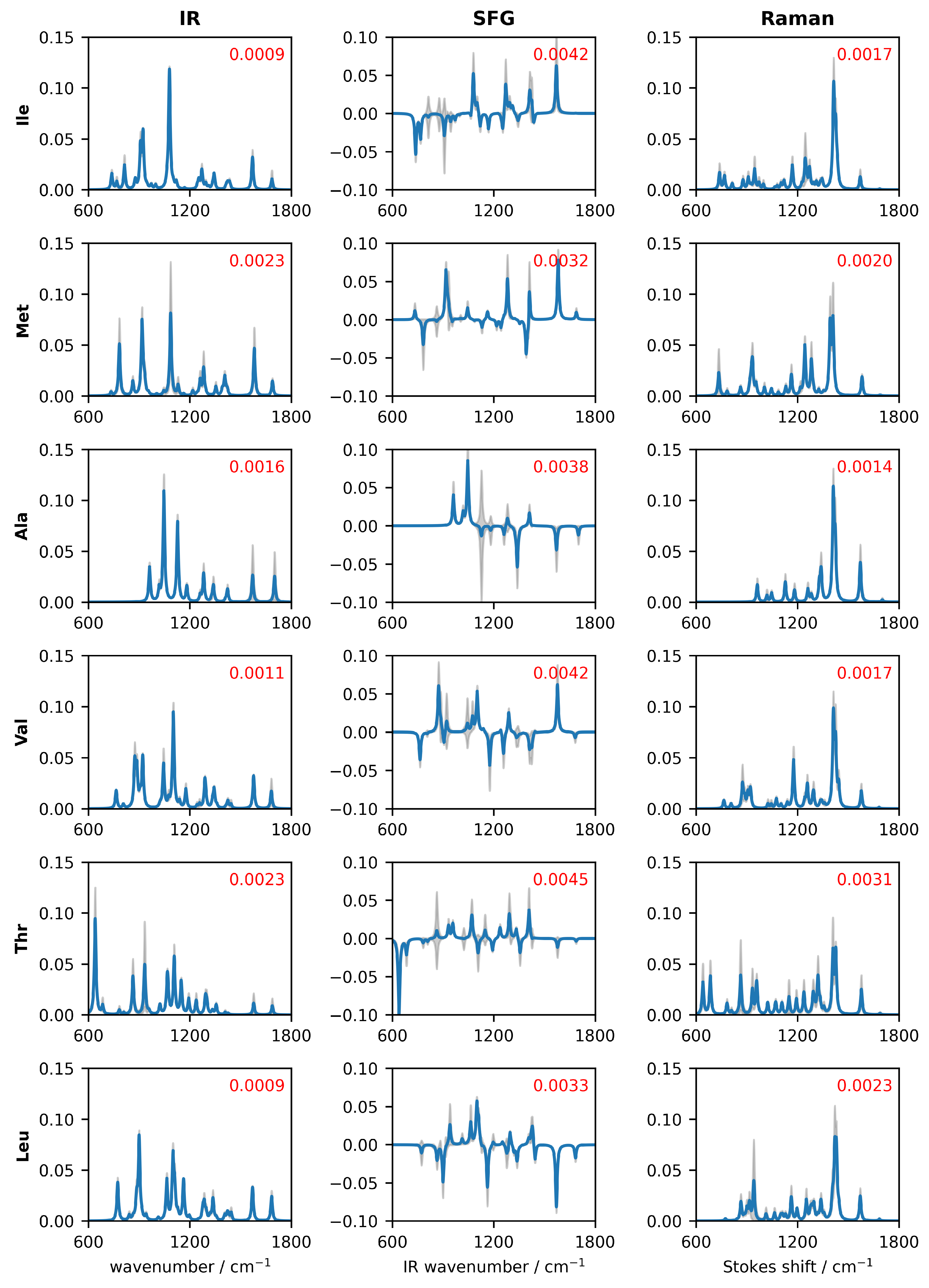
3.1. Generation of the Candidate Spectra
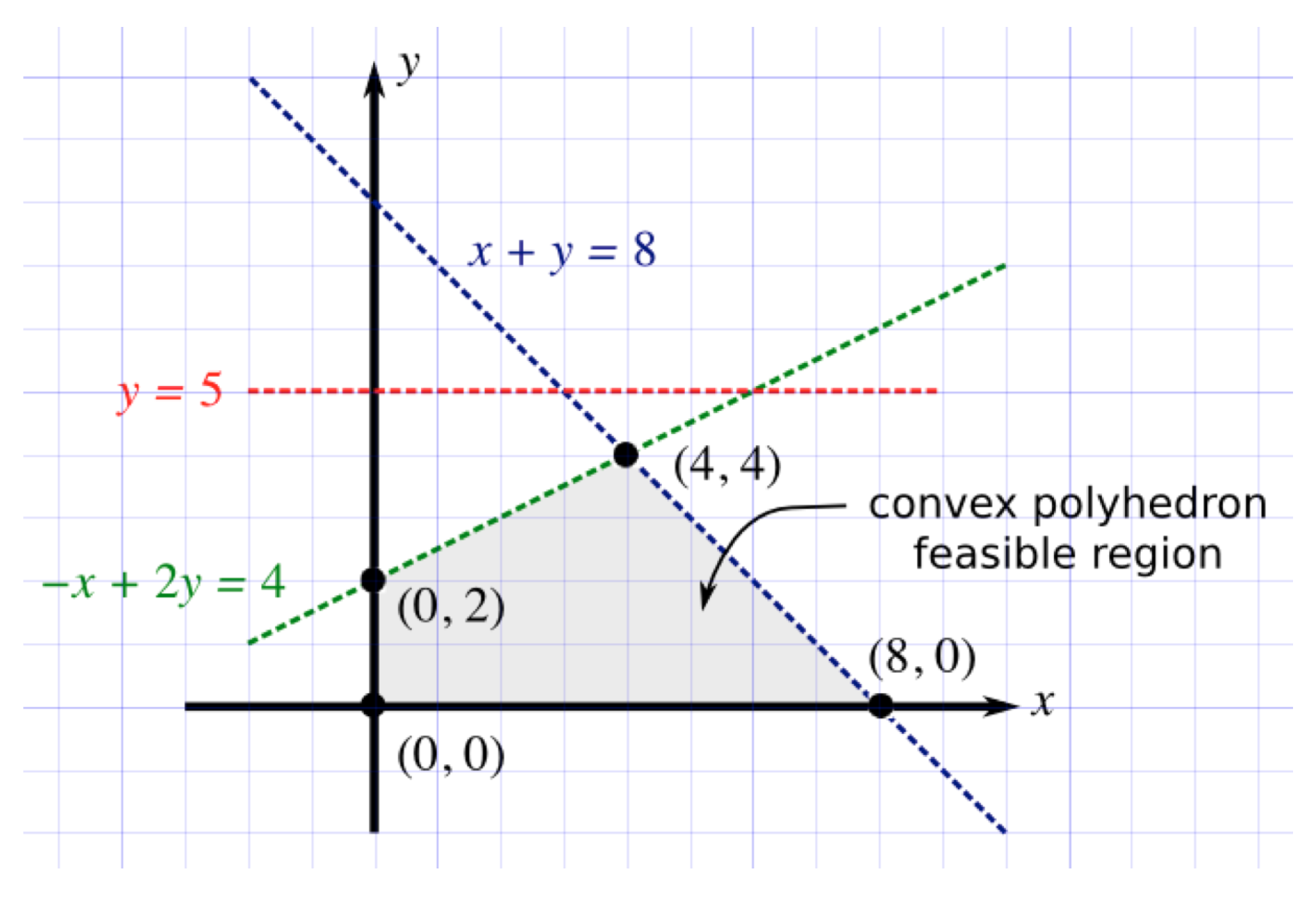
3.2. Linear Programming
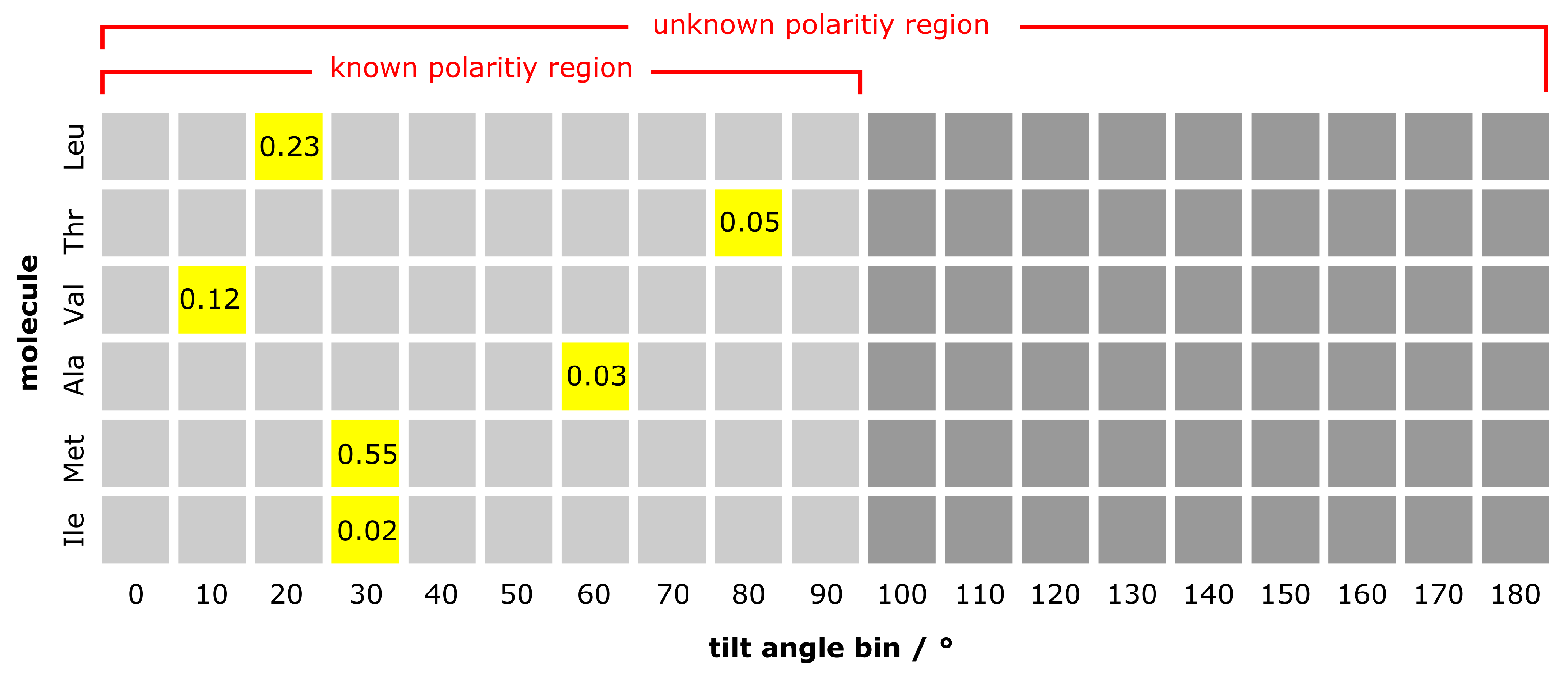
3.3. Construction of Test Cases
4. Results and Discussion
4.1. Known Scaling Factors
4.2. Arbitrary Scaling Factors
4.3. Exploring the Origins of Orientation Sensitivity
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| DCM | direction cosine matrix |
| IR | infrared |
| LP | linear programming |
| SFG | sum-frequency generation |
References
- Ward, I.M. Determination of Molecular Orientation by Spectroscopic Techniques. Adv. Poly. Sci. 1985, 66, 81–115. [Google Scholar]
- McHale, J.L. Molecular Spectroscopy; Pearson Education: New York, NY, USA, 2008. [Google Scholar]
- Kliger, D.S.; Lewis, J.W.; Randal, C.E. Polarized Light in Optics and Spectroscopy; Academic Press, Inc.: San Diego, CA, USA, 1990. [Google Scholar]
- Pelletier, I.; Laurin, I.; Buffeteau, T.; Pézolet, M. Determination of Molecular Orientation in Biaxially Oriented Ultrathin Films. J. Phys. Chem. B 2004, 108, 7162–7169. [Google Scholar] [CrossRef]
- Umemura, J.; Kamata, T.; Kawai, T.; Takenaka, T. Quantitative Evaluation of Molecular Orientation in Thin Langmuir–Blodgett Films by FT-IR Transmission and Reflection-Absorption Spectroscopy. J. Phys. Chem. 1990, 94, 62–67. [Google Scholar] [CrossRef]
- Brunner, H.; Mayer, U.; Hoffmann, M. External Reflection Infrared Spectroscopy of Aniostropic Adsorbate Layers on Dielectric Substrates. Appl. Spectrosc. 1997, 51, 209–217. [Google Scholar] [CrossRef]
- Chollet, P.A.; Messier, J.; Rosilio, C. Infrared Determination of the Orientation of Molecules in Stearamide Monolayers. J. Chem. Phys. 1976, 64, 1042–1050. [Google Scholar] [CrossRef]
- Debe, M.K. Extracting Physical Structure Information from Thin Film Organic Films with Reflection Absorption Infrared Spectroscopy. J. Appl. Phys. 1984, 55, 3354–3366. [Google Scholar] [CrossRef]
- Dluhy, R.A. Quantitative External Reflection Infrared Spectroscopic Analysis of Insoluble Monolayers Spread at the Air–Water Interface. J. Phys. Chem. 1986, 90, 1373–1379. [Google Scholar] [CrossRef]
- Greenler, R.G. Infrared study of adsorbed molecules on metal surfaces by reflection techniques. J. Chem. Phys. 1966, 44, 310–315. [Google Scholar] [CrossRef]
- Hasegawa, T.; Takeda, S.; Kawaguchi, A.; Umemura, J. Quantitative Analysis of Uniaxial Molecular Orientation in Langmuir-Blodgett Films by Infrared Reflection Spectroscopy. Langmuir 1995, 11, 1236–1243. [Google Scholar] [CrossRef] [Green Version]
- Mendelson, R.; Brauner, J.W.; Gericke, A. External Infrared Absorption Spectrometry of Monolayer Films at the Air-Water Interface. Annu. Rev. Phys. Chem. 1995, 46, 305–333. [Google Scholar] [CrossRef]
- Sourisseau, C. Polarization Measurements in Macro- and Micro-Raman Spectoscopies: Molecular Orientations in Thin Films and Azo-Dye Polymer Systems. Chem. Rev. 2004, 104, 3851–3892. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Young, R.J. Polarised Raman Spectroscopy for the Study of Molecular Orientation Distributions in Polymers. J. Mater. Sci. 2006, 41, 963–991. [Google Scholar] [CrossRef]
- Bower, D.I. Investigation of Molecular Orientation Distributions by Polarized Raman Scattering and Polarized Fluorescence. J. Poly. Sci. 1972, 10, 2135–2153. [Google Scholar] [CrossRef]
- Lagugné Labarthet, F. Polarized Measurements in Raman Microscopy. Annu. Rep. Prog. Chem. 2007, 103, 326–350. [Google Scholar] [CrossRef]
- Richard-Lacroix, M.; Pellerin, C. Novel Method for Quantifying Molecular Orientation by Polarized Raman Spectroscopy: A Comparative Simulations Study. Appl. Spectrosc. 2013, 67, 409–419. [Google Scholar] [CrossRef] [PubMed]
- Richard-Lacroix, M.; Pellerin, C. Accurate New Method for Molecular Orientation Quantification Using Polarized Raman Spectroscopy. Macromolecules 2013, 46, 5561–5569. [Google Scholar] [CrossRef]
- Tsuboi, M.; Benevides, J.M.; Tomas, G.J., Jr. Raman Tensors and their Application in Structural Studies of Biological Systems. Proc. Jpn. Acad. Ser. B 2009, 85, 83–97. [Google Scholar] [CrossRef]
- Yang, S.; Michielsen, S. Orientation Distribution Functions Obtained via Polarized Raman Spectroscopy of Poly(ethylene terephthalate) Fibers. Macromolecules 2003, 36, 6484–6492. [Google Scholar] [CrossRef]
- Takenaka, T.; Fukuzaki, H. Resonance Raman Spectra of Insoluble Monolayers Spread on a Water Surface. J. Raman Spectrosc. 1979, 8, 151–154. [Google Scholar] [CrossRef]
- Takenaka, T.; Nakanaga, T. Resonance Raman Spectra of Monolayers Adsorbed at the Interface between Carbon Tetrachloride and an Aqueous Solution of a Surfactant and a Dye. J. Phys. Chem. 1976, 80, 475–480. [Google Scholar] [CrossRef]
- Takenaka, T. Effect of Electrolyte on the Molecular Orientation in Monolayers Adsorbed at the Liquid–Liquid Interface: Studies by Resonance Raman Spectra. Chem. Phys. Lett. 1978, 55, 515–518. [Google Scholar] [CrossRef]
- Nakanaga, T.; Takenaka, T. Resonance Raman Spectra of Monolayers of a Surface–Active Dye Adsorbed at the Oil–Water Interface. J. Phys. Chem. 1977, 81, 645–649. [Google Scholar] [CrossRef]
- Morita, A. Theory of Sum Frequency Generation Spectroscopy; Springer: Singapore, 2018. [Google Scholar]
- Hall, S.A.; Jena, K.C.; Covert, P.A.; Roy, S.; Trudeau, T.G.; Hore, D.K. Molecular-Level Surface Structure from Nonlinear Vibrational Spectroscopy Combined with Simulations. J. Phys. Chem. B 2014, 118, 5617–5636. [Google Scholar] [CrossRef] [PubMed]
- Richmond, G.L. Molecular Bonding and Interactions at Aqueous Surfaces as Probed by Vibrational Sum Frequency Spectroscopy. Chem. Rev. 2002, 102, 2693–2724. [Google Scholar] [CrossRef] [PubMed]
- Bain, C.D. Sum-Frequency Vibrational Spectroscopy of the Solid/Liquid Interface. J. Chem. Soc. Faraday Trans. 1995, 91, 1281–1296. [Google Scholar] [CrossRef]
- Lambert, A.G.; Davies, P.B.; Neivandt, D.J. Implementing the Theory of Sum Frequency Generation Vibrational Spectroscopy: A Tutorial Review. Appl. Spectrosc. Rev. 2005, 40, 103–145. [Google Scholar] [CrossRef]
- Vidal, F.; Tadjeddine, A. Sum-Frequency Generation Spectroscopy of Interfaces. Rep. Prog. Phys. 2005, 68, 1095–1127. [Google Scholar] [CrossRef]
- Buck, M.; Himmelhaus, M. Vibrational Spectroscopy of Interfaces by Infrared-Visible Sum Frequency Generation. J. Vac. Sci. Technol. A 2001, 19, 2717–2736. [Google Scholar] [CrossRef]
- Shen, Y.R. Basic Theory of Surface Sum-Frequency Generation. J. Phys. Chem. C 2012, 116, 15505–15509. [Google Scholar] [CrossRef]
- Pezzotti, S.; Serva, A.; Gaigeot, M.P. 2D-HB Network at the Air–Water Interface: A Structural and Dynamical Characterization by Means of ab initio and Classical Molecular Dynamics Simulations. J. Chem. Phys. 2018, 148, 174701. [Google Scholar] [CrossRef]
- Ojha, D.; Kühne, T.D. “On-The-Fly” Calculation of the Vibrational Sum-Frequency Generation Spectrum at the Air-Water Interface. Molecules 2020, 25, 3939. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.R. Surfaces Probed by Nonlinear Optics. Surf. Sci. 1994, 299–300, 551–562. [Google Scholar] [CrossRef]
- Boyd, R.W. Nonlinear Optics, 2nd ed.; Academic Press: San Diego, CA, USA, 2003. [Google Scholar]
- Superfine, R.; Huang, J.Y.; Shen, Y.R. Phase Measurement For Surface Infrared-Visible Sum-Frequency Generation. Opt. Lett. 1990, 15, 1276–1278. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.R.; Ostroverkhov, V. Sum-frequency Vibrational Spectroscopy on Water Interfaces: Polar Orientation of Water Molecules at Interfaces. Chem. Rev. 2006, 106, 1140–1154. [Google Scholar] [CrossRef] [PubMed]
- Mondal, J.; Nihonyanagi, S.; Yamaguchi, S.; Tahara, T. Structure and Orientation of Water at Charged Lipid Monolayer/Water Interfaces Probed by Heterodyne-Detected Vibrational Sum Frequency Generation Spectroscopy. J. Am. Chem. Soc. 2010, 132, 10656–10657. [Google Scholar] [CrossRef] [PubMed]
- Nihonyanagi, S.; Yamaguchi, S.; Tahara, T. Direct Evidence for Orientational Flip–Flop of Water Molecules at Charged Interfaces: A Heterodyne-Detected Vibrational Sum Frequency Generation Study. J. Chem. Phys. 2009, 130, 204704. [Google Scholar]
- Jena, K.; Hung, K.K.; Schwantje, T.; Hore, D.K. Methyl groups at dielectric and metal surfaces studied by sum-frequency generation in co- and counter-propagating configurations. J. Chem. Phys. 2011, 135, 044704. [Google Scholar] [CrossRef]
- Jena, K.C.; Covert, P.A.; Hore, D.K. Phase Measurement in Non-Degenerate Three-Wave Mixing Spectroscopy. J. Chem. Phys. 2011, 134, 044712. [Google Scholar] [CrossRef]
- Long, D.A. The Raman Effect: A Unified Treatment of The Theory of Raman Scattering by Molecules; John Wiley & Sons: Hoboken, NJ, USA, 2002. [Google Scholar]
- Potma, E.O.; Mukamel, S. Coherent Raman Scattering Microscopy; CRC Press: Boca Raton, FL, USA, 2013; Chapter 1; pp. 3–42. [Google Scholar]
- Mukamel, S. Principles of Nonlinear Optical Spectroscopy; Oxford University Press: New York, NY, USA, 1995. [Google Scholar]
- Hung, K.K.; Stege, U.; Hore, D.K. IR Absorption, Raman Scattering, and IR-Vis Sum-Frequency Generation Spectroscopy as Quantitative Probes of Surface Structure. Appl. Spectrosc. Rev. 2015, 50, 351–376. [Google Scholar] [CrossRef]
- Lagugné Labarthet, F.; Buffeteau, T.; Sourisseau, C. Orientation Distribution Functions in Uniaxial Systems Centered Perpendicularly to a Constraint Direction. Appl. Spectrosc. 2000, 54, 699–705. [Google Scholar] [CrossRef]
- Wang, Y.; Cui, Z.F.; Wang, H.F. Experimental Observables and Macroscopic Susceptibility/Microscopic Polarizability Tensors for Third and Fourth-Order Nonlinear Spectroscopy of Ordered Molecular System. Chin. J. Chem. Phys. 2007, 20, 449–460. [Google Scholar] [CrossRef]
- Hall, S.A.; Hickey, A.D.; Hore, D.K. Structure of Phenylalanine Adsorbed on Polystyrene From Nonlinear Vibrational Spectroscopy Measurements and Electronic Structure Calculations. J. Phys. Chem. C 2010, 114, 9748–9757. [Google Scholar] [CrossRef]
- Schmidt, M.W.; Baldridge, K.K.; Boatz, J.A.; Elbert, S.T.; Gordon, M.S.; Jensen, J.H.; Koseki, S.; Matsunaga, N.; Nguyen, K.A.; Su, S.; et al. General atomic and molecular electronic structure system. J. Comput. Chem. 1993, 14, 1347–1363. [Google Scholar] [CrossRef]
- Linder, R.; Nispeal, M.; Häber, T.; Kleinermanns, K. Gas-phase FT-IR spectra of natural amino acids. Chem. Phys. Lett. 2005, 409, 260–264. [Google Scholar] [CrossRef]
- Merrick, J.R.; Moran, D.; Radom, L. An Evaluation of Harmonic Vibrational Frequency Scale Factors. J. Phys. Chem. A 2007, 111, 11683–11700. [Google Scholar] [CrossRef]
- Matousek, J.; Gärtner, B. Understanding and Using Linear Programming; Springer: Heidelberg/Berlin, Germany, 2007. [Google Scholar]
- Dantzig, G.B. Reminiscences about the origins of linear programming. Oper. Res. Lett. 1982, 1, 43–48. [Google Scholar] [CrossRef]
- Karmarkar, N. A new polynomial-time algorithm for linear programming. Combinatorica 1984, 4, 373–395. [Google Scholar] [CrossRef]
- Chvatal, V. Linear Programming; W. H. Freeman and Company: New York, NY, USA, 1983. [Google Scholar]
- Roy, R.; Sevick-Muraca, E. Truncated Newton’s Optimization Scheme for Absorption and Fluorescence Optical Tomography: Part I Theory and Formulation. Opt. Exp. 1999, 4, 353–371. [Google Scholar] [CrossRef]
- Partovi-Azar, P.; Kühne, T.D.; Kaghazchi, P. Evidence for the Existence of Li2S2 clusters in Lithium-Sulfur Batteries: ab initio Raman spectroscopy simulation. Phys. Chem. Chem. Phys. 2015, 17, 22009–22014. [Google Scholar] [CrossRef] [Green Version]
- Li, D.H.; Fukushima, M. A Modified BFGS Method and its Global Convergence in Nonconvex Minimization. J. Comput. Appl. Math. 2001, 129, 15–35. [Google Scholar] [CrossRef] [Green Version]
- Cormen, T.H.; Leiserson, C.E.; Livest, R.L.; Stein, C. Introduction to Algorithms; MIT Press: Cambridge, MA, USA; McGraw-Hill: New York, NY, USA, 2001; Chapter The Simplex Algorithm; pp. 790–804. [Google Scholar]
- GNU Linear Programming Kit, Version 4.64. 2017. Available online: http://www.gnu.org/software/glpk/glpk.html (accessed on 19 February 2015).
- Chen, F.; Hung, K.K.; Stege, U.; Hore, D.K. Linear Programming Applied to Polarized Raman Data for Elucidating Molecular Structure at Surfaces. Chemometr. Intell. Lab. 2020, 196, 103898. [Google Scholar] [CrossRef]
- Buffeteau, T.; Lagugné Labarthet, F.; Sourisseau, C.; Kostromine, S.; Bieringer, T. Biaxial orientation induced in a photoaddressable azopolymer thin film as evidenced by polarized UV-visible, infrared, and Raman spectra. Macromolecules 2004, 37, 2880–2889. [Google Scholar] [CrossRef]
- Lagugné-Labarthet, F.; Sourisseau, C.; Schaller, R.D.; Saykally, R.J.; Rochon, P. Chromophore orientations in a nonlinear optical azopolymer diffraction grating: Even and odd order parameters from far-field Raman and near-field second harmonic generation microscopies. J. Phys. Chem. B 2004, 108, 17059–17068. [Google Scholar] [CrossRef]
- Rodriguez, V.; Lagugné Labarthet, F.; Sourisseau, C. Orientation Distribution Functions based upon both 〈P1〉, 〈P3〉 Order Parameters and upon the Four 〈P1〉 up to 〈P4〉 Values: Application to an Electrically Poled Nonlinear Optical Azopolymer Film. Appl. Spectrosc. 2005, 59, 322–328. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Spectral Data | Known Scaling Factors | Arbitrary Scaling Factors | ||
|---|---|---|---|---|
| (a) Known Polarity | (b) Unknown Polarity | (c) Known Polarity | (d) Unknown Polarity | |
| IR only | ✗ | ✗ | ✗ | ✗ |
| SFG only | ✓ | ✗ | ✗ | ✗ |
| Raman only | ✓ | ✗ | ✓ | ✗ |
| IR + Raman | (✓) | ✗ | (✓) | ✗ |
| IR + SFG | (✓) | ✓ | ✗ | ✗ |
| SFG + Raman | (✓) | ✓ | (✓) | ✓ |
| IR + SFG + Raman | (✓) | (✓) | (✓) | (✓) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, F.; Gozdzialski, L.; Hung, K.-K.; Stege, U.; Hore, D.K. Assessing the Molecular Specificity and Orientation Sensitivity of Infrared, Raman, and Vibrational Sum-Frequency Spectra. Symmetry 2021, 13, 42. https://doi.org/10.3390/sym13010042
Chen F, Gozdzialski L, Hung K-K, Stege U, Hore DK. Assessing the Molecular Specificity and Orientation Sensitivity of Infrared, Raman, and Vibrational Sum-Frequency Spectra. Symmetry. 2021; 13(1):42. https://doi.org/10.3390/sym13010042
Chicago/Turabian StyleChen, Fei, Lea Gozdzialski, Kuo-Kai Hung, Ulrike Stege, and Dennis K. Hore. 2021. "Assessing the Molecular Specificity and Orientation Sensitivity of Infrared, Raman, and Vibrational Sum-Frequency Spectra" Symmetry 13, no. 1: 42. https://doi.org/10.3390/sym13010042
APA StyleChen, F., Gozdzialski, L., Hung, K. -K., Stege, U., & Hore, D. K. (2021). Assessing the Molecular Specificity and Orientation Sensitivity of Infrared, Raman, and Vibrational Sum-Frequency Spectra. Symmetry, 13(1), 42. https://doi.org/10.3390/sym13010042