
Novel Coordination Mode in the Potassium Mefenamate Trihydrate Polymeric Structure


Abstract
:1. Introduction
2. Experimental and Calculation Methods
2.1. Crystal Preparation
2.2. X-ray Diffraction
2.3. Computational Details
3. Results and Discussion
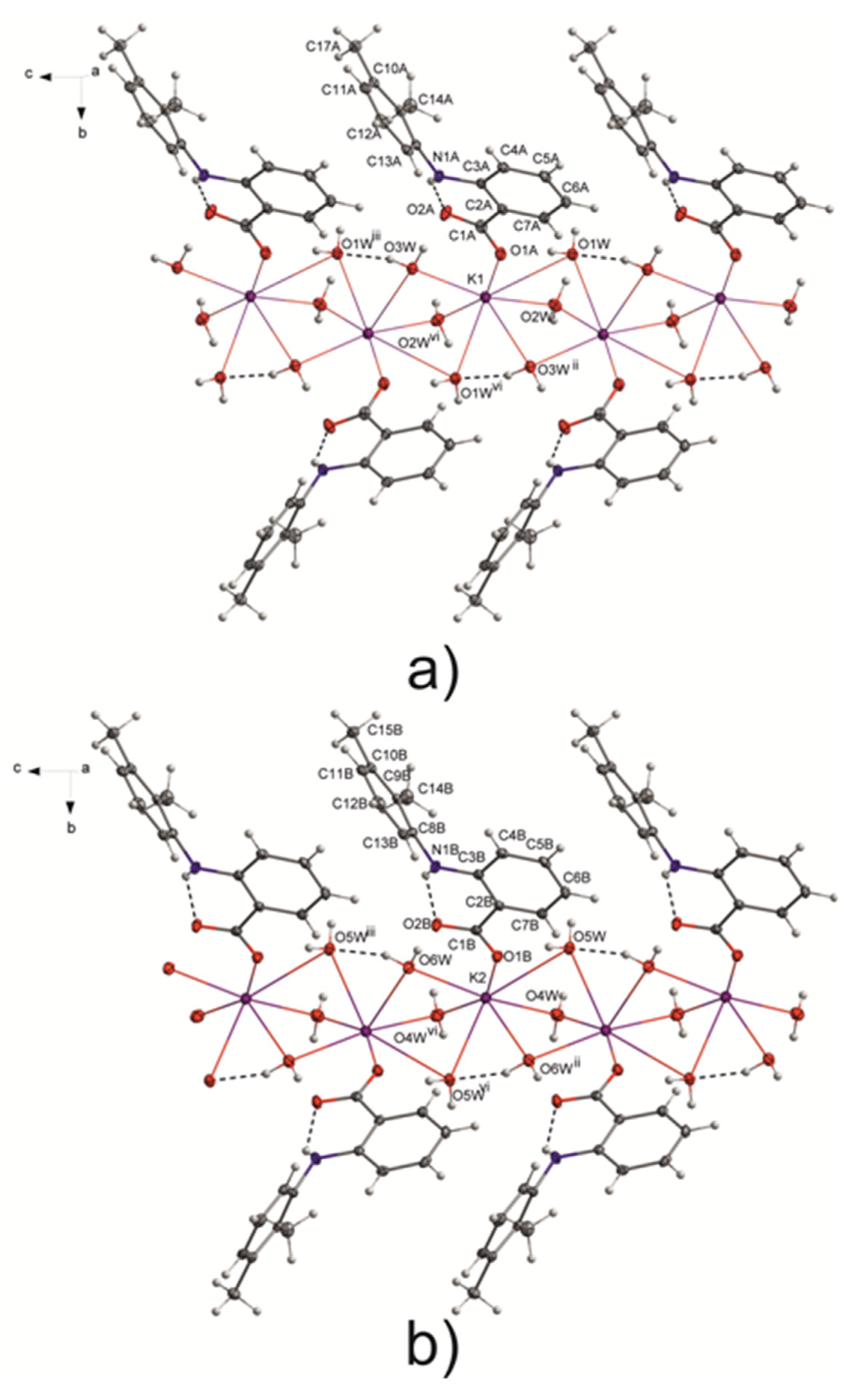
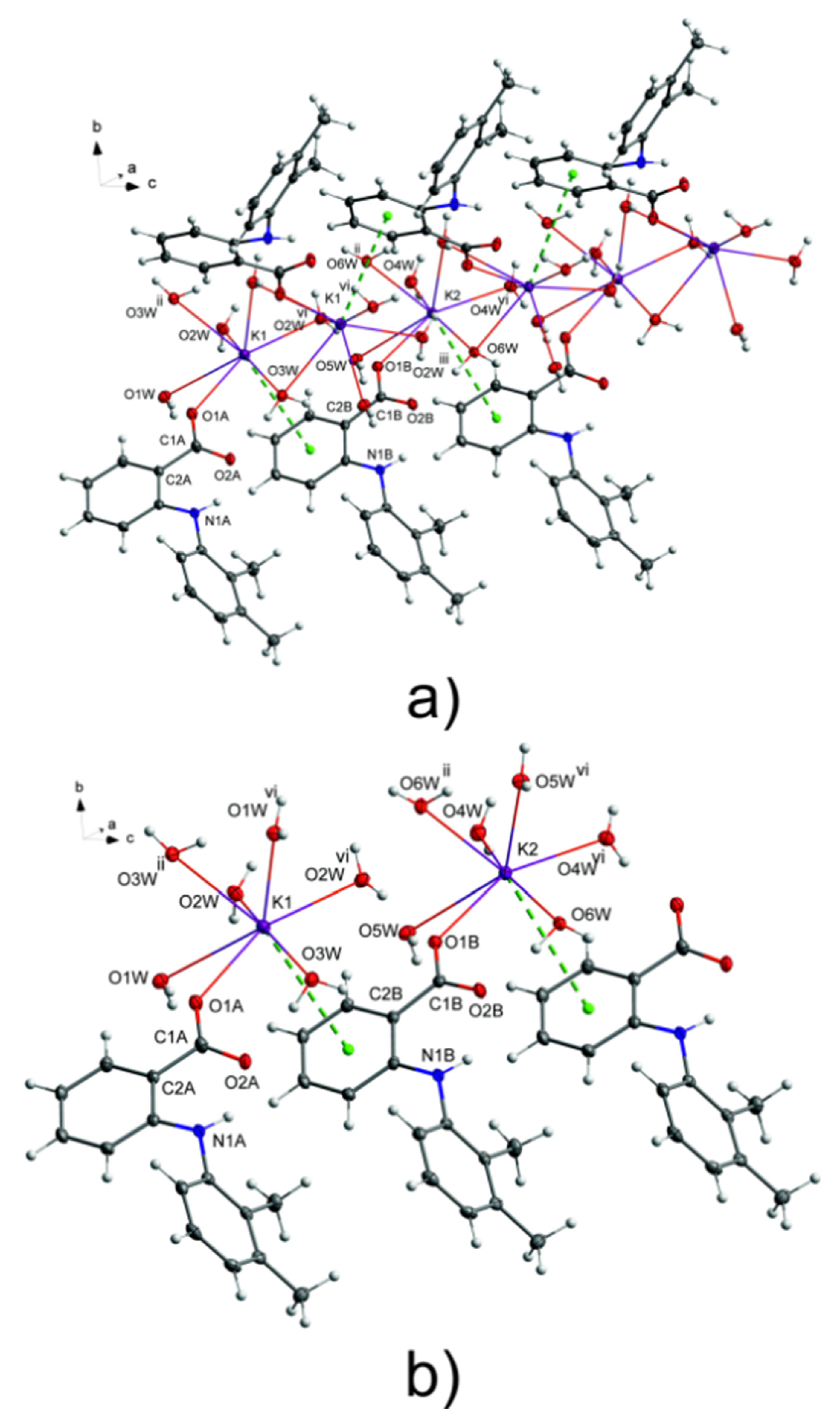
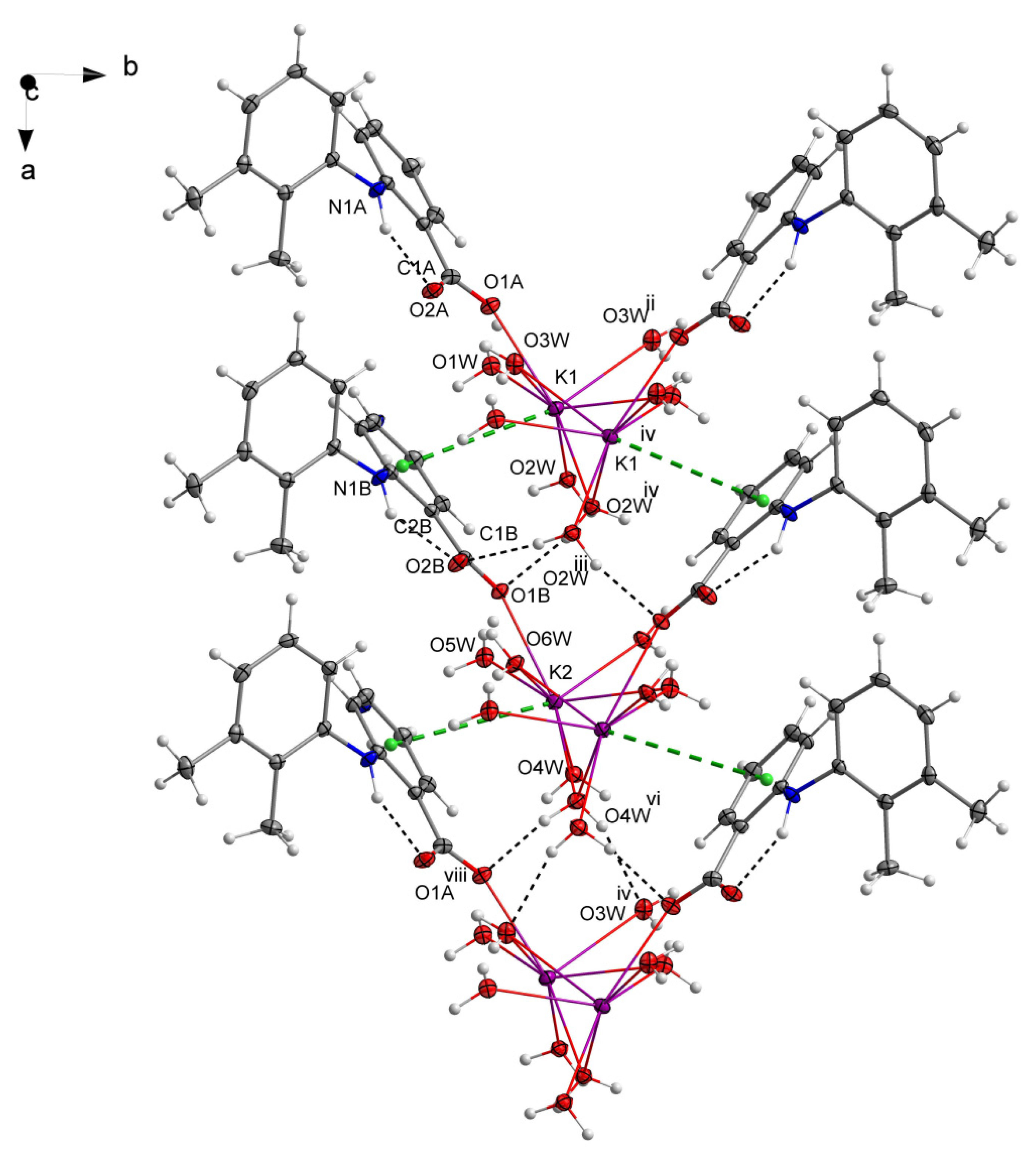
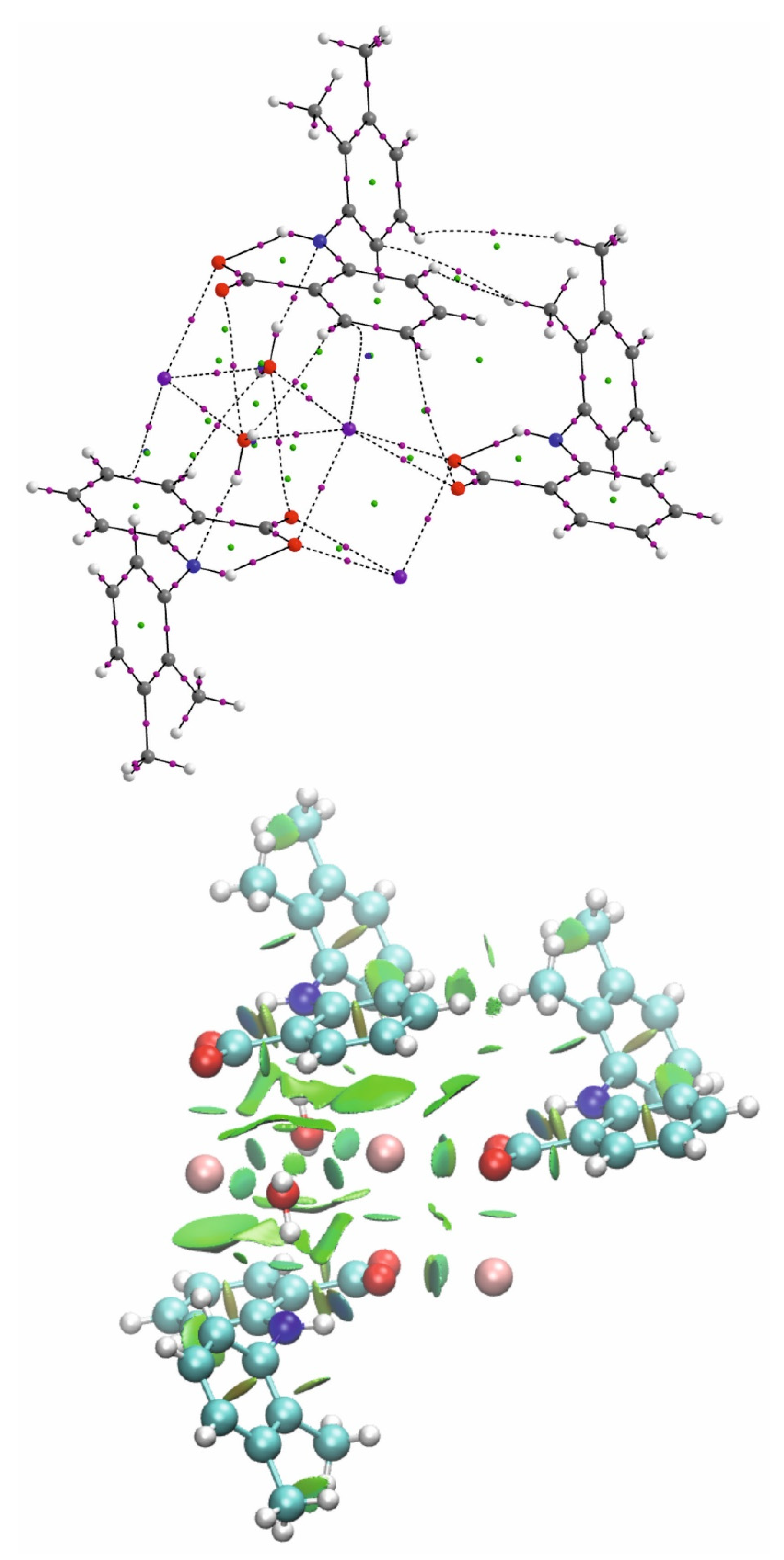
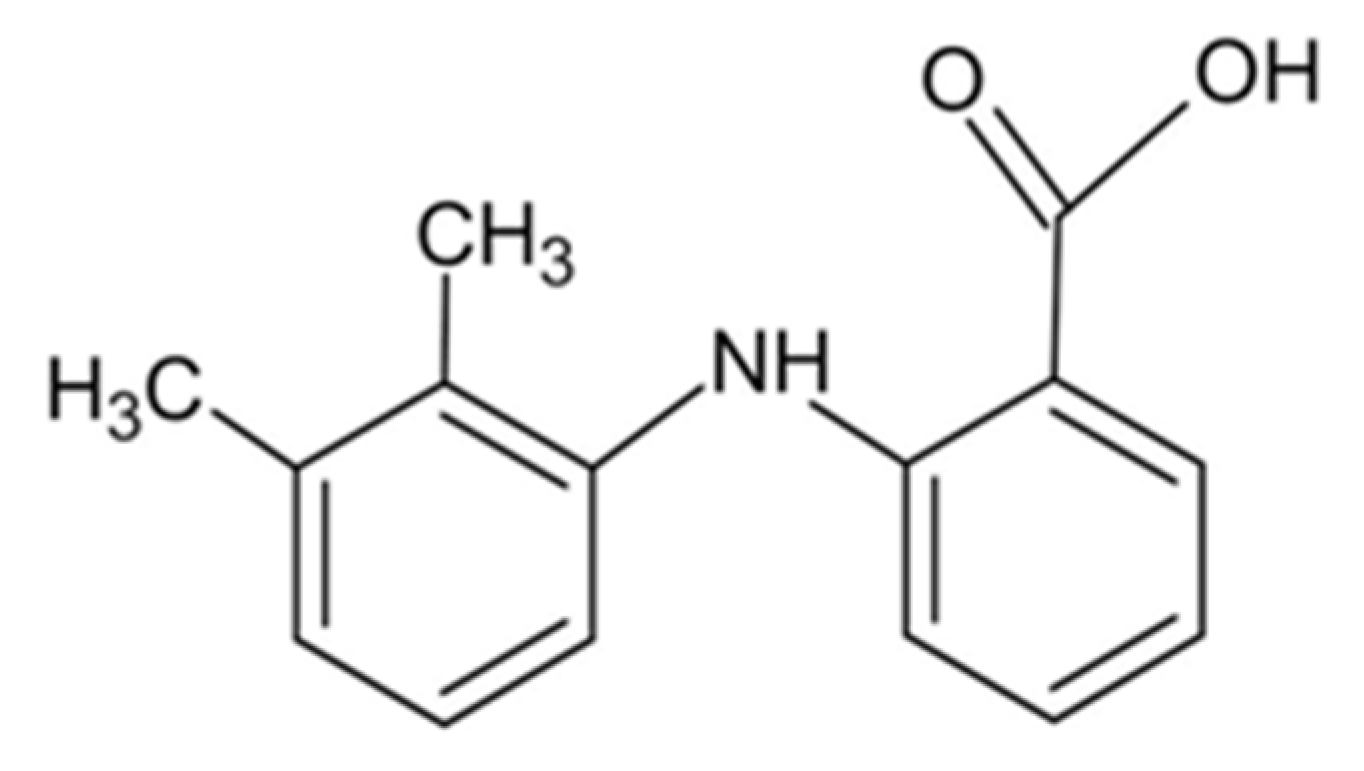
3.1. Structure of Potassium Mefenamate—Water (1/3)
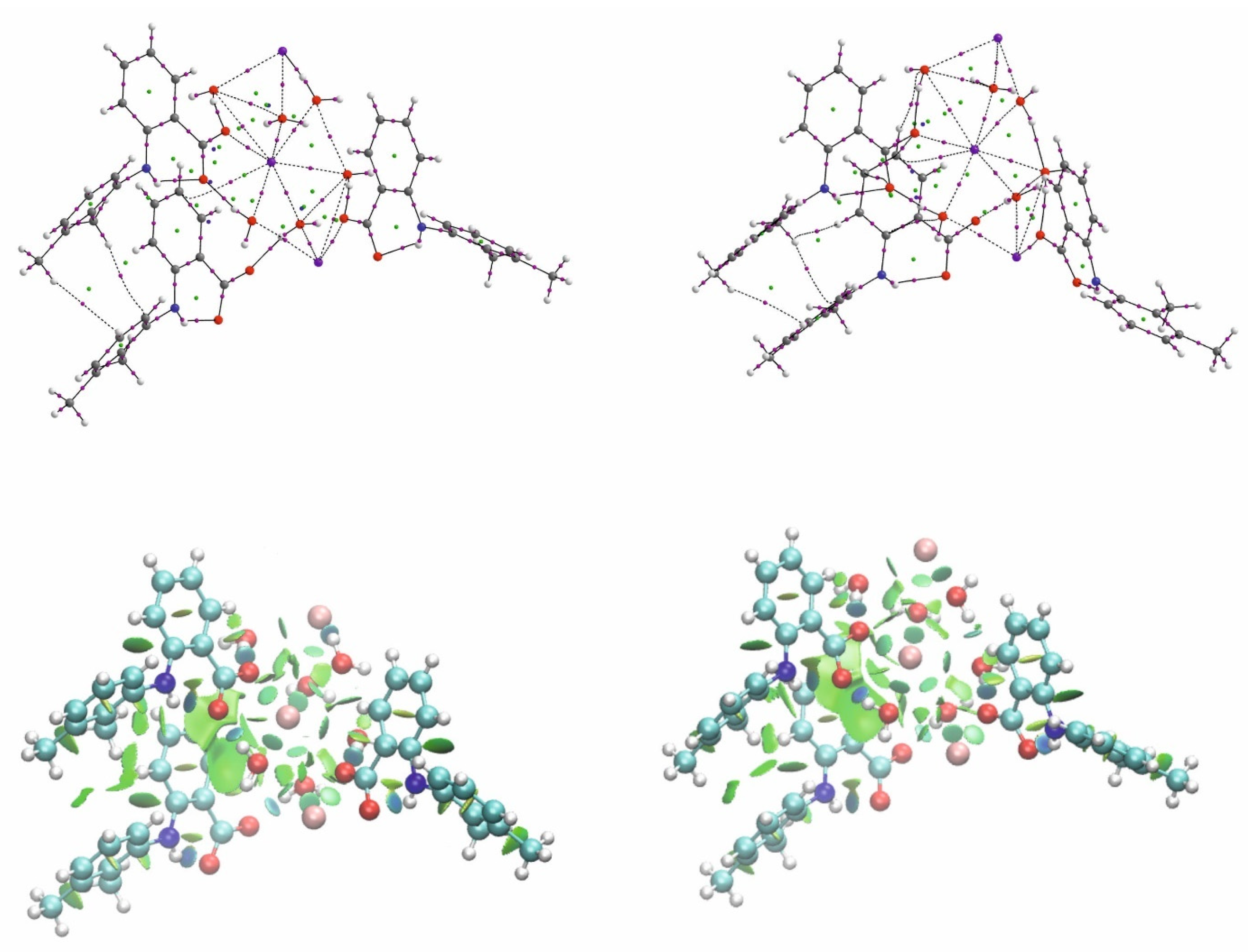
3.2. Analysis of the K—O Bonds
4. Conclusions
- For both known potassium mefenamates, the formation of coordination polymer takes place with the participation of water molecules and the strength of interaction with all oxygen atoms surrounding the cation is similar.
- The weak dispersive interaction may be responsible for the formation of a complex between the central ion and the ligand and the cooperative weak interactions are the driving force building polymeric structures.
- The existence of an interaction between the cation and the ligand is confirmed by the presence of a reaction path, even if the interaction is very weak and not very stable.
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Rothan, H.A.; Bahrani, H.; Mohamed, Z.; Teoh, T.C.; Shankar, E.M.; Rahman, N.A.; Yusof, R. Mefenamic acid in combination with ribavirin shows significant effects in reducing chikungunya virus infection in vitro and in vivo. Antivir. Res. 2016, 127, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Lago, E.M.; Silva, M.P.; Queiroz, T.G.; Mazloum, S.F.; Rodrigues, V.C.; Carnaúba, P.U.; Pinto, P.L.; Rocha, J.A.; Ferreira, L.L.G.; Andricopulo, A.D.; et al. Phenotypic screening of nonsteroidal anti-inflammatory drugs identified mefenamic acid as a drug for the treatment of schistosomiasis. EbioMedicine 2019, 43, 370–379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cimolai, N. The potential and promise of mefenamic acid. Expert Rev. Clin. Pharm. 2013, 6, 289–305. [Google Scholar] [CrossRef] [PubMed]
- Bani-Jaber, A.; Hamdan, I.; Al-Khalidi, B. Sodium Mefenamate as a Solution for the Formulation and Dissolution Problems of Mefenamic Acid. Chem. Pharm. Bull. 2007, 55, 1136–1140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kafarska, K.; Wolf, W.M. Novel silver complexes with popular non-steroidal anti-inflammatory drugs. Acta Innov. 2016, 21, 51–59. [Google Scholar]
- Topacli, A.; Ide, S. Molecular structures of metal complexes with mefenamic Acid. J. Pharm. Biomed. Anal. 1999, 21, 975–982. [Google Scholar] [CrossRef]
- Kruszynski, R.; Trzesowska-Kruszynska, A.; Majewski, P.; Łukaszewicz, E.; Majewska, K.; Sierański, T.; Lewiński, B. Structure and properties of the sodium, potassium and calcium salts of 2-(2,3-dimethylphenyl)aminobenzoic acid. J. Mol. Struct. 2010, 970, 79–89. [Google Scholar] [CrossRef]
- Krawczyk, M.S.; Majerz, I. The Na—O bond in sodium fenamate. Acta Cryst. 2019, B75, 766–774. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Cryst. 2015, A71, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Cryst. 2015, C71, 3–8. [Google Scholar] [CrossRef]
- Brandenburg, K. DIAMOND; Crystal Impact GbR: Bonn, Germany, 2014. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision A. 03; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef] [Green Version]
- Keith, T.A. AIMALL; Version 19.10.12; TK Gristmill Software: Overland Park, KS, USA, 2019. [Google Scholar]
- Contreras-García, J.; Johnson, E.R.; Keinan, S.; Chaudret, R.; Piquemal, J.-P.; Beratan, D.N.; Yang, W. NCIPLOT: A Program for Plotting Noncovalent Interaction Regions. J. Chem. Theory Comput. 2011, 7, 625–632. [Google Scholar] [CrossRef] [PubMed]
- Bader, R.F.W. Atoms in Molecules: A Quantum Theory; Oxford University Press: New York, NY, USA, 1990. [Google Scholar]
- Rozas, I.; Alkorta, I.; Elguero, J. Behavior of Ylides Containing N, O, and C Atoms as Hydrogen Bond Acceptors. J. Am. Chem. Soc. 2000, 122, 11154–11161. [Google Scholar] [CrossRef]
- Espinosa, E.; Molins, E.; Lecomte, C. Hydrogen bond strengths revealed by topological analyses of experimentally observed electron densities. Chem. Phys. Lett. 1998, 285, 170–173. [Google Scholar] [CrossRef]
- Espinosa, E.; Alkorta, I.; Rozas, I.; Elguero, J.; Molins, E. About the evaluation of the local kinetic, potential and total energy densities in closed-shell interactions. Chem. Phys. Lett. 2001, 336, 457–461. [Google Scholar] [CrossRef]
- Popelier, P.L.A.; Bader, R.F.W. Effect of Twisting a Polypeptide on Its Geometry and Electron Distribution. J. Phys. Chem. 1994, 98, 4473–4481. [Google Scholar] [CrossRef]
- Popelier, P.L.A. Characterization of a Dihydrogen Bond on the Basis of the Electron Density. J. Phys. Chem. 1998, 102, 1873–1878. [Google Scholar] [CrossRef]
- Johnson, E.R.; Keinan, S.; Mori-Sanchez, P.; Contreras-García, J.; Cohen, A.J.; Yang, W. Revealing Noncovalent Interactions. J. Am. Chem. Soc. 2010, 132, 6498–6506. [Google Scholar] [CrossRef] [Green Version]
- Alonso, M.; Woller, T.; Martin-Martinez, F.J.; Contreras-García, J.; Geerlings, P.; De Proft, F. Understanding the Fundamental Role of π/π, σ/σ, and σ/π Dispersion Interactions in Shaping Carbon-Based Materials. Chem. Eur. J. 2014, 20, 4931–4941. [Google Scholar] [CrossRef]
- Wiberg, K.B.; Bader, R.F.W.; Lau, C.D.H. Theoretical analysis of hydrocarbon properties. 1. Bonds, structures, charge concentrations, and charge relaxations. J. Am. Chem. Soc. 1987, 109, 1001–1012. [Google Scholar] [CrossRef]
- Fradera, X.; Austen, M.A.; Bader, R.F.W. The Lewis Model and Beyond. J. Phys. Chem. A 1999, 103, 304–314. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Krawczyk, M.S.; Majerz, I. Novel Coordination Mode in the Potassium Mefenamate Trihydrate Polymeric Structure. Symmetry 2021, 13, 1761. https://doi.org/10.3390/sym13101761
Krawczyk MS, Majerz I. Novel Coordination Mode in the Potassium Mefenamate Trihydrate Polymeric Structure. Symmetry. 2021; 13(10):1761. https://doi.org/10.3390/sym13101761
Chicago/Turabian StyleKrawczyk, Marta S., and Irena Majerz. 2021. "Novel Coordination Mode in the Potassium Mefenamate Trihydrate Polymeric Structure" Symmetry 13, no. 10: 1761. https://doi.org/10.3390/sym13101761
APA StyleKrawczyk, M. S., & Majerz, I. (2021). Novel Coordination Mode in the Potassium Mefenamate Trihydrate Polymeric Structure. Symmetry, 13(10), 1761. https://doi.org/10.3390/sym13101761