
Synthesis of C3-Symmetric Cinchona-Based Organocatalysts and Their Applications in Asymmetric Michael and Friedel–Crafts Reactions

 , , ,
, , ,  ,
,  ,
,  ,
, 
Abstract
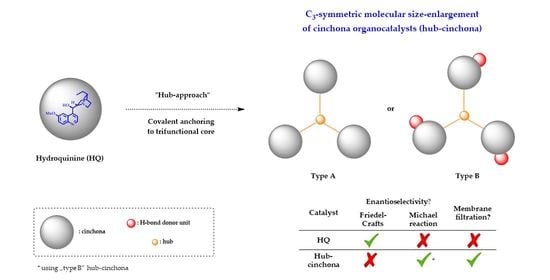
:
1. Introduction
2. Results
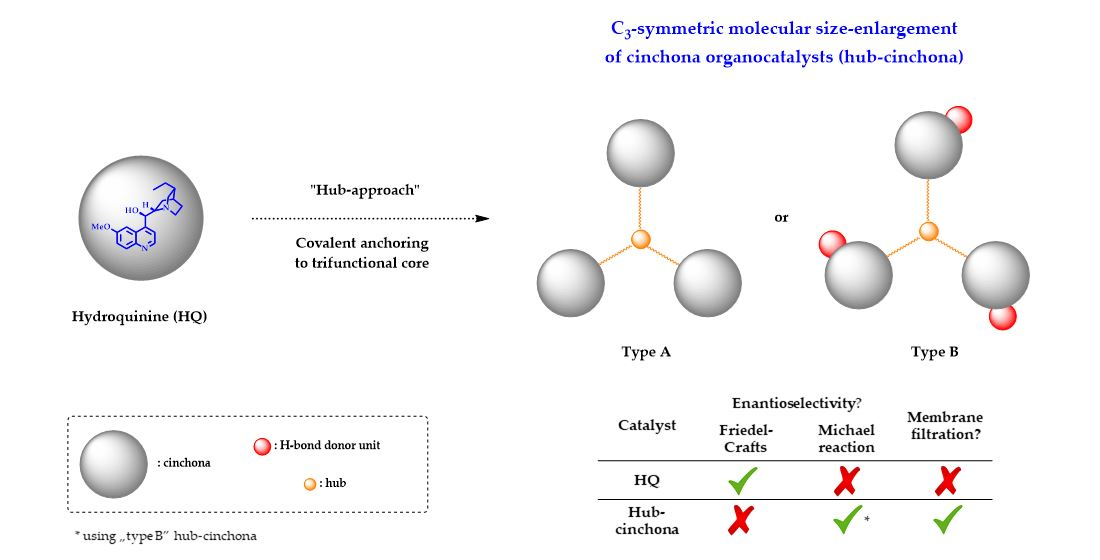
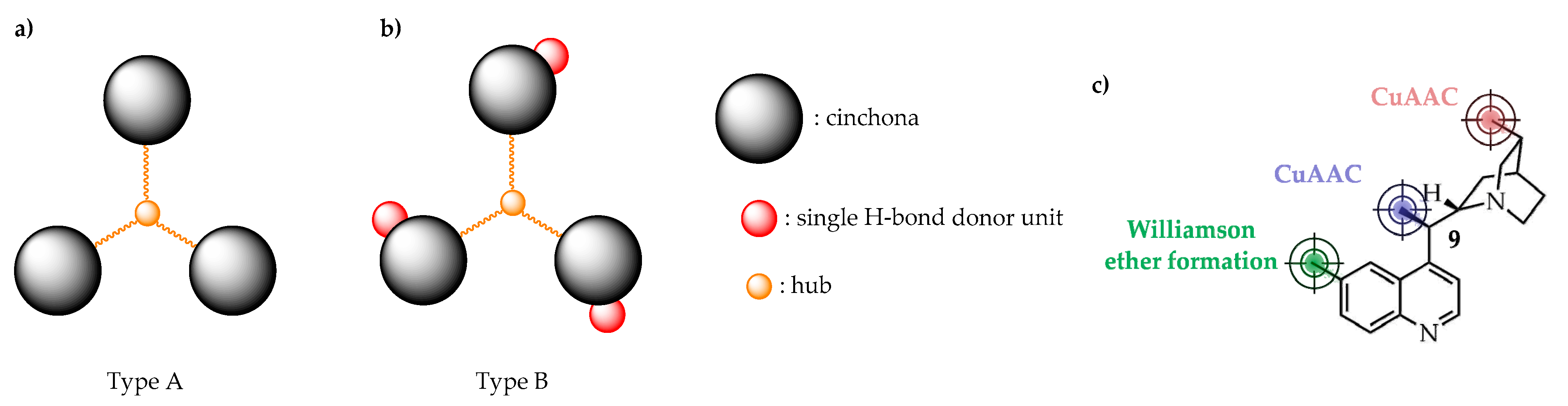
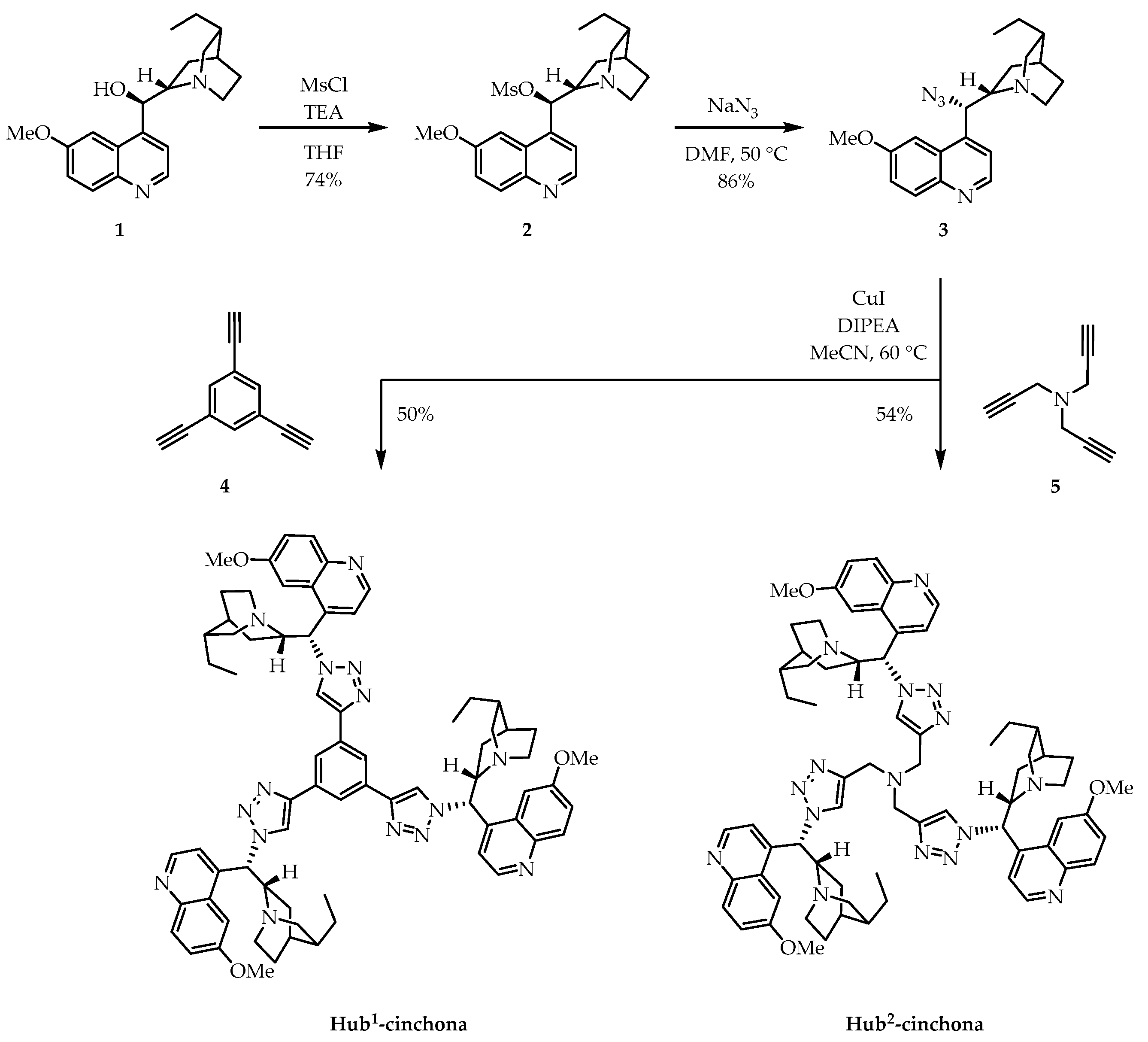
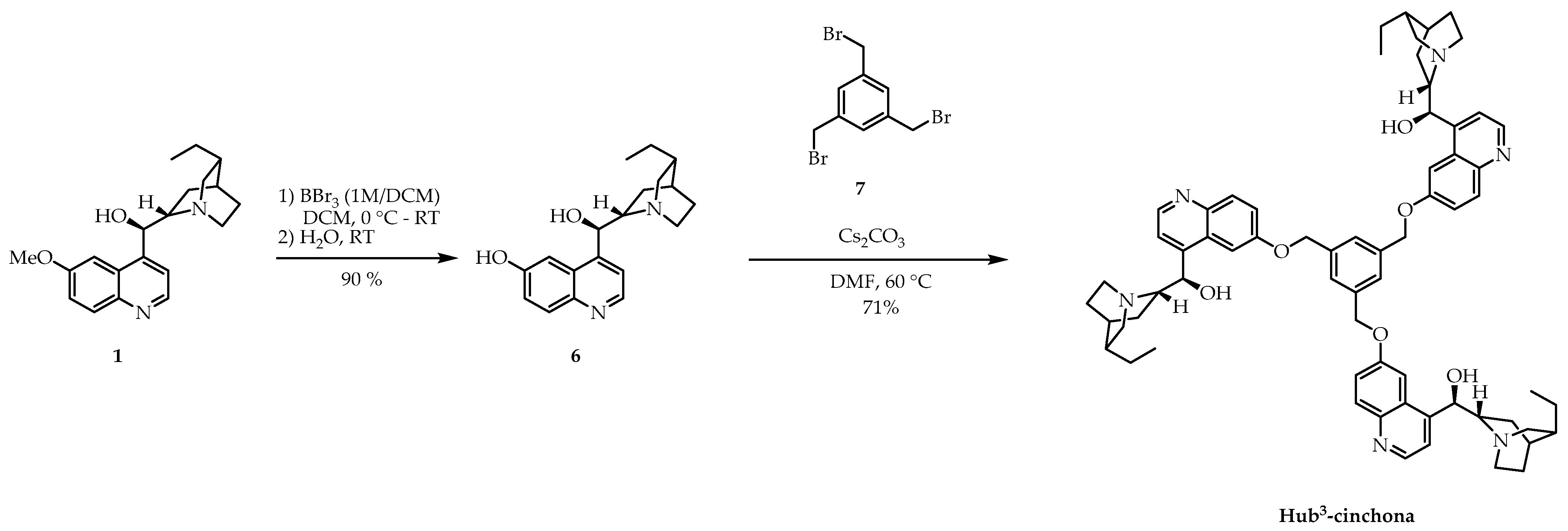
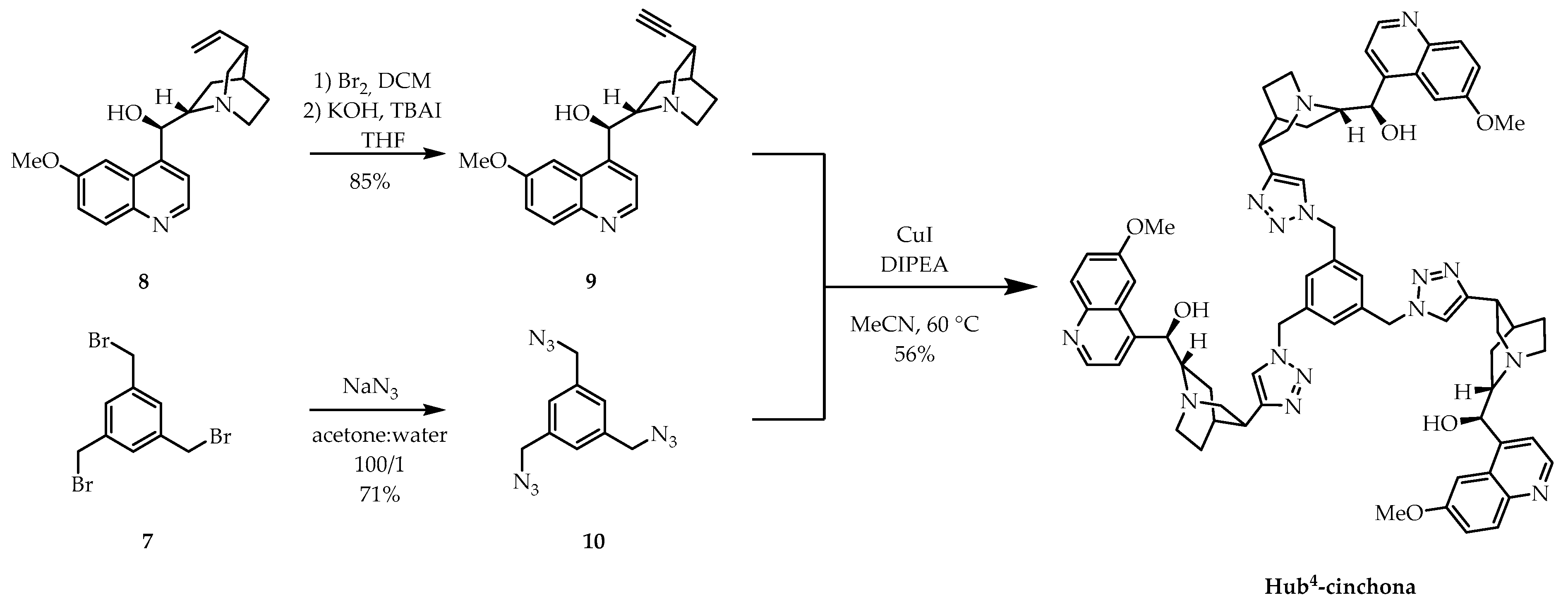
2.1. Synthesis of New C3-Symmetric Hub-Cinchona Catalysts
2.2. Application of Hub-Cinchona Catalysts in Hydroxyalkylation of Indole and Michael Addition Reactions
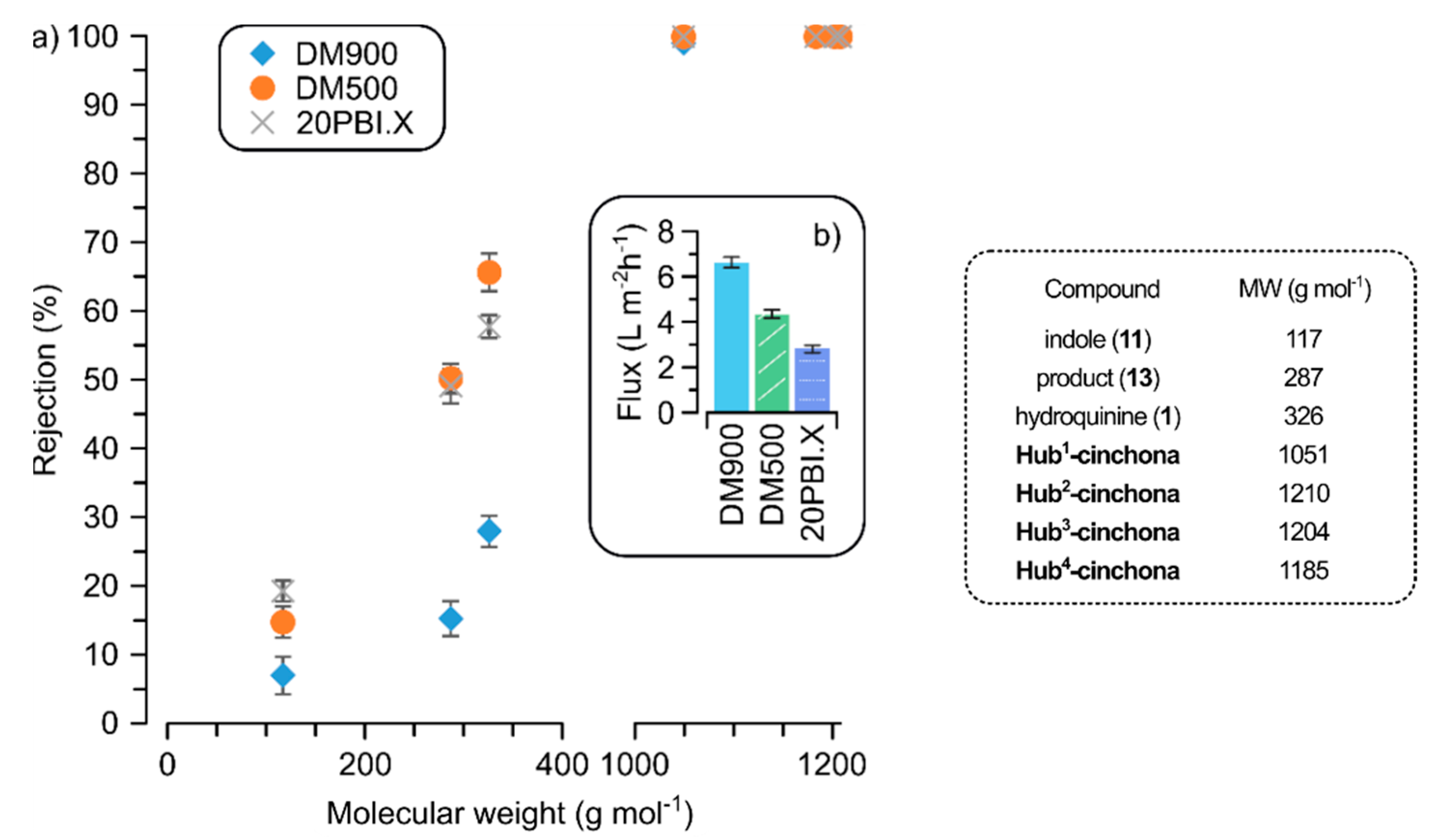
2.3. Membrane Rejection of Hub-Cinchona Organocatalysts
3. Materials and Methods
3.1. General Information
3.2. Preparation of Compounds
3.3. Nanofiltration
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kamer, P.; Vogt, D.; Thybaut, J.W. (Eds.) Contemporary Catalysis: Science, Technology, and Applications, Gld. ed.; The Royal Society of Chemistry: London, UK, 2017. [Google Scholar]
- Jacobsen, E.N.; Pfaltz, A.; Yamamoto, H. Comprehensive Asymmetric Catalysis I–III (Comprehensive Overviews in Chemistry), 1st ed.; Springer: Berlin/Heidelberg, Germany, 1999. [Google Scholar]
- Moberg, C. C3 Symmetry in Asymmetric Catalysis and Chiral Recognition. Angew. Chem. Int. Ed. 1998, 37, 248–268. [Google Scholar] [CrossRef]
- Moberg, C. The Role of Symmetry in Asymmetric Catalysis. Isr. J. Chem. 2012, 52, 653–662. [Google Scholar] [CrossRef]
- Gade, L.H.; Bellemin-Laponnaz, S. Exploiting Threefold Symmetry in Asymmetric Catalysis: The Case of Tris(oxazolinyl)ethanes (“Trisox”). Chem. A Eur. J. 2008, 14, 4142–4152. [Google Scholar] [CrossRef]
- Gibson, S.E.; Castaldi, M.P. Applications of chiral C3-symmetric molecules. Chem. Commun. 2006, 37, 3045–3062. [Google Scholar] [CrossRef]
- Rodríguez, L.-I.; Roth, T.; Fillol, J.L.; Wadepohl, H.; Gade, L.H. The More Gold-The More Enantioselective: Cyclohydroaminations of γ-Allenyl Sulfonamides with Mono-, Bis-, and Trisphospholane Gold(I) Catalysts. Chem. A Eur. J. 2012, 18, 3721–3728. [Google Scholar] [CrossRef]
- Yamanaka, M.; Nakagawa, T.; Aoyama, R.; Nakamura, T. Synthesis and estimation of gelation ability of C3-symmetry tris-urea compounds. Tetrahedron 2008, 64, 11558–11567. [Google Scholar] [CrossRef]
- Crişan, C.V.; Soran, A.; Bende, A.; Hӑdade, N.D.; Terec, A.; Grosu, I. Synthesis, Structure and Supramolecular Properties of a Novel C3 Cryptand with Pyridine Units in the Bridges. Molecules 2020, 25, 3789. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.-H.; Liu, X.-T.; Space, B.; Chang, Z.; Bu, X.-H. Metal-organic materials with triazine-based ligands: From structures to properties and applications. Co-ord. Chem. Rev. 2021, 427, 213518. [Google Scholar] [CrossRef]
- García, A.; Insuasty, B.; Herranz, M.A.; Martínez-Álvarez, R.; Martín, N. New Building Block forC3Symmetry Molecules: Synthesis ofs-Triazine-Based Redox Active Chromophores. Org. Lett. 2009, 11, 5398–5401. [Google Scholar] [CrossRef]
- Burns, B.; King, N.P.; Tye, H.; Studley, J.R.; Gamble, M.; Wills, M. Chiral phosphinamides: New catalysts for the asymmetric reduction of ketones by borane. J. Chem. Soc. Perkin Trans. 1998, 1, 1027–1038. [Google Scholar] [CrossRef]
- Du, D.-M.; Fang, T.; Xu, J.; Zhang, S.-W. Structurally Well-Defined, RecoverableC3-Symmetric Tris(β-hydroxy phosphoramide)-Catalyzed Enantioselective Borane Reduction of Ketones. Org. Lett. 2006, 8, 1327–1330. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, L.; Pan, Y.; Han, J.; Wu, H.; Teng, M.; Li, Z. Novel Tripod l-Prolinamide Catalysts Based on Tribenzyl- and Triphenyl-phosphine Oxide for the Direct Aldol Reaction. Synlett 2009, 2009, 933–936. [Google Scholar] [CrossRef]
- Moorthy, J.N.; Saha, S. C3-Symmetric Proline-Functionalized Organocatalysts: Enantioselective Michael Addition Reactions. Eur. J. Org. Chem. 2010, 2010, 6359–6365. [Google Scholar] [CrossRef]
- Murai, K.; Fukushima, S.; Hayashi, S.; Takahara, Y.; Fujioka, H. C3-Symmetric Chiral Trisimidazoline: Design and Application to Organocatalyst. Org. Lett. 2010, 12, 964–966. [Google Scholar] [CrossRef]
- Murai, K.; Fukushima, S.; Nakamura, A.; Shimura, M.; Fujioka, H. C3-Symmetric chiral trisimidazoline: The role of a third imidazoline and its application to the nitro Michael reaction and the α-amination of β-ketoesters. Tetrahedron 2011, 67, 4862–4868. [Google Scholar] [CrossRef]
- Murai, K.; Nakamura, A.; Matsushita, T.; Shimura, M.; Fujioka, H. C3-Symmetric Trisimidazoline-Catalyzed Enantioselective Bromolactonization of Internal Alkenoic Acids. Chem. A Eur. J. 2012, 18, 8448–8453. [Google Scholar] [CrossRef]
- Takizawa, S.; Sako, M.; Abozeid, M.A.; Kishi, K.; Wathsala, H.D.P.; Hirata, S.; Murai, K.; Fujioka, H.; Sasai, H. Enantio- and Diastereoselective Betti/aza-Michael Sequence: Single Operated Preparation of Chiral 1,3-Disubstituted Isoindolines. Org. Lett. 2017, 19, 5426–5429. [Google Scholar] [CrossRef]
- Boratyński, P.J. Dimeric Cinchona alkaloids. Mol. Divers. 2015, 19, 385–422. [Google Scholar] [CrossRef] [Green Version]
- Park, H.-G.; Jeong, B.-S.; Yoo, M.-S.; Park, M.-K.; Huh, H.; Jew, S.-S. Trimeric Cinchona alkaloid phase-transfer catalyst: α,α′,α′′-tris[O(9)-allylcinchonidinium]mesitylene tribromide. Tetrahedron Lett. 2001, 42, 4645–4648. [Google Scholar] [CrossRef]
- Siva, A.; Murugan, E. A New Trimeric Cinchona Alkaloid as a Chiral Phase-Transfer Catalyst for the Synthesis of Asymmetric α-Amino Acids. Synthesis 2005, 2005, 2927–2933. [Google Scholar] [CrossRef]
- Siva, A.; Murugan, E. New trimeric Cinchona alkaloid-based quaternary ammonium salts as efficient chiral phase transfer catalysts for enantioselective synthesis of α-amino acids. J. Mol. Catal. A Chem. 2006, 248, 1–9. [Google Scholar] [CrossRef]
- Siva, A.; Jayaraman, S.; Kumaraguru, D.; Arockiam, J.B.; Paulpandian, S.; Rajendiran, B. Highly Enantioselective Asymmetric Michael Addition Reactions with New Chiral Multisite Phase-Transfer Catalysts. Synlett 2014, 25, 1685–1691. [Google Scholar] [CrossRef]
- Beneto, A.J.; Sivamani, J.; AshokKumar, V.; Duraimurugan, K.; Balasaravanan, R.; Siva, A. Highly enantioselective Michael addition reactions with new trimeric chiral phase transfer catalysts. New J. Chem. 2015, 39, 3098–3104. [Google Scholar] [CrossRef]
- Károlyi, B.I.; Bősze, S.; Orbán, E.; Sohár, P.; Drahos, L.; Gál, E.; Csámpai, A. Acylated mono-, bis- and tris- Cinchona-Based Amines Containing Ferrocene or Organic Residues: Synthesis, Structure and in Vitro Antitumor Activity on Selected Human Cancer Cell Lines. Molecules 2012, 17, 2316–2329. [Google Scholar] [CrossRef] [Green Version]
- Min, C.; Han, X.; Liao, Z.; Wu, X.; Zhou, H.-B.; Dong, C. C3-Symmetrical Cinchonine-Squaramide as New Highly Efficient, and Recyclable Organocatalyst for Enantioselective Michael Addition. Adv. Synth. Catal. 2011, 353, 2715–2720. [Google Scholar] [CrossRef]
- Han, X.; Liu, B.; Zhou, H.-B.; Dong, C. Enhanced efficiency of recyclable C3-symmetric cinchonine-squaramides in the asymmetric Friedel–Crafts reaction of indoles with alkyl trifluoropyruvate. Tetrahedron Asymmetry 2012, 23, 1332–1337. [Google Scholar] [CrossRef]
- Han, X.; Dong, C.; Zhou, H.-B. C3-Symmetric Cinchonine-Squaramide-Catalyzed Asymmetric Chlorolactonization of Styrene-Type Carboxylic Acids with 1,3-Dichloro-5,5-dimethylhydantoin: An Efficient Method to Chiral Isochroman-1-ones. Adv. Synth. Catal. 2014, 356, 1275–1280. [Google Scholar] [CrossRef]
- Lv, W.; Guo, C.; Dong, Z.; Tang, S.; Liu, B.; Dong, C. C3-Symmetric cinchonine-squaramide as a recyclable efficient organocatalyst for tandem Michael addition–cyclisation of malononitrile and nitrovinylphenols. Tetrahedron Asymmetry 2016, 27, 670–674. [Google Scholar] [CrossRef]
- Le Phuong, H.A.; Blanford, C.F.; Szekely, G. Reporting the unreported: The reliability and comparability of the literature on organic solvent nanofiltration. Green Chem. 2020, 22, 3397–3409. [Google Scholar] [CrossRef]
- Hu, J.; Kim, C.; Halasz, P.; Kim, J.F.; Kim, J.; Szekely, G. Artificial intelligence for performance prediction of organic solvent nanofiltration membranes. J. Membr. Sci. 2021, 619, 118513. [Google Scholar] [CrossRef]
- Le Phuong, H.A.; Cseri, L.; Whitehead, G.F.S.; Garforth, A.; Budd, P.; Szekely, G. Environmentally benign and diastereoselective synthesis of 2,4,5-trisubstituted-2-imidazolines. RSC Adv. 2017, 7, 53278–53289. [Google Scholar] [CrossRef] [Green Version]
- Dong, R.; Liu, R.; Gaffney, P.R.J.; Schaepertoens, M.; Marchetti, P.; Williams, C.M.; Chen, R.; Livingston, A.G. Sequence-defined multifunctional polyethers via liquid-phase synthesis with molecular sieving. Nat. Chem. 2019, 11, 136–145. [Google Scholar] [CrossRef] [PubMed]
- Székely, G.; Schaepertoens, M.; Gaffney, P.R.J.; Livingston, A.G. Beyond PEG2000: Synthesis and Functionalisation of Monodisperse PEGylated Homostars and Clickable Bivalent Polyethyleneglycols. Chem. A Eur. J. 2014, 20, 10038–10051. [Google Scholar] [CrossRef] [PubMed]
- Razali, M.; Didaskalou, C.; Kim, J.F.; Babaei, M.; Drioli, E.; Lee, Y.M.; Szekely, G. Exploring and Exploiting the Effect of Solvent Treatment in Membrane Separations. ACS Appl. Mater. Interfaces 2017, 9, 11279–11289. [Google Scholar] [CrossRef] [Green Version]
- Voros, V.; Drioli, E.; Fonte, C.; Szekely, G. Process Intensification via Continuous and Simultaneous Isolation of Antioxidants: An Upcycling Approach for Olive Leaf Waste. ACS Sustain. Chem. Eng. 2019, 7, 18444–18452. [Google Scholar] [CrossRef]
- Alammar, A.; Park, S.-H.; Williams, C.J.; Derby, B.; Szekely, G. Oil-in-water separation with graphene-based nanocomposite membranes for produced water treatment. J. Membr. Sci. 2020, 603, 118007. [Google Scholar] [CrossRef]
- Keraani, A.; Nasser, G.; Shahane, S.; Renouard, T.; Bruneau, C.; Rabiller-Baudry, M.; Fischmeister, C. Syntheses and characterization of molecular weight enlarged olefin metathesis pre-catalysts. Comptes Rendus Chim. 2017, 20, 717–723. [Google Scholar] [CrossRef]
- Kisszékelyi, P.; Nagy, S.; Fehér, Z.; Huszthy, P.; Kupai, J. Membrane-Supported Recovery of Homogeneous Organocatalysts: A Review. Chemestry 2020, 2, 742–758. [Google Scholar] [CrossRef]
- Siew, W.E.; Ates, C.; Merschaert, A.; Livingston, A.G. Efficient and productive asymmetric Michael addition: Development of a highly enantioselective quinidine-based organocatalyst for homogeneous recycling via nanofiltration. Green Chem. 2013, 15, 663–674. [Google Scholar] [CrossRef]
- Shao, Z.; Zhang, H. Combining transition metal catalysis and organocatalysis: A broad new concept for catalysis. Chem. Soc. Rev. 2009, 38, 2745–2755. [Google Scholar] [CrossRef]
- Zhong, C.; Shi, X. When Organocatalysis Meets Transition-Metal Catalysis. Eur. J. Org. Chem. 2010, 2010, 2999–3025. [Google Scholar] [CrossRef]
- Didaskalou, C.; Kupai, J.; Cseri, L.; Barabas, J.; Vass, E.; Holtzl, T.; Szekely, G. Membrane-Grafted Asymmetric Organocatalyst for an Integrated Synthesis–Separation Platform. ACS Catal. 2018, 8, 7430–7438. [Google Scholar] [CrossRef]
- Kisszekelyi, P.; Alammar, A.; Kupai, J.; Huszthy, P.; Barabas, J.; Holtzl, T.; Szente, L.; Bawn, C.; Adams, R.; Szekely, G. Asymmetric synthesis with cinchona-decorated cyclodextrin in a continuous-flow membrane reactor. J. Catal. 2019, 371, 255–261. [Google Scholar] [CrossRef]
- Schaepertoens, M.; Didaskalou, C.; Kim, J.F.; Livingston, A.G.; Szekely, G. Solvent recycle with imperfect membranes: A semi-continuous workaround for diafiltration. J. Membr. Sci. 2016, 514, 646–658. [Google Scholar] [CrossRef] [Green Version]
- Zhao, D.; Kim, J.F.; Ignacz, G.; Pogany, P.; Lee, Y.M.; Szekely, G. Bio-Inspired Robust Membranes Nanoengineered from Interpenetrating Polymer Networks of Polybenzimidazole/Polydopamine. ACS Nano 2019, 13, 125–133. [Google Scholar] [CrossRef]
- Ignacz, G.; Fei, F.; Szekely, G. Ion-Stabilized Membranes for Demanding Environments Fabricated from Polybenzimidazole and Its Blends with Polymers of Intrinsic Microporosity. ACS Appl. Nano Mater. 2018, 1, 6349–6356. [Google Scholar] [CrossRef]
- Cassani, C.; Martín-Rapún, R.; Arceo, E.; Bravo, F.; Melchiorre, P. Synthesis of 9-amino(9-deoxy)epi cinchona alkaloids, general chiral organocatalysts for the stereoselective functionalization of carbonyl compounds. Nat. Protoc. 2013, 8, 325–344. [Google Scholar] [CrossRef] [PubMed]
- Skarżewski, J.; Zielińska-Błajet, M.; Kucharska, M. Simple Enantiospecific Synthesis of Sulfides ofCinchonaAlkaloids. Synthesis 2006, 2006, 1176–1182. [Google Scholar] [CrossRef]
- Yaegashi, K.; Mikami, M. Preparation of 9-azido Cinchona Alkaloids, Their 9-amino Derivatives, and Their 9-(substituted thioureido) Derivatives. JP Patent 2010024173 (20100204), 4 February 2010. [Google Scholar]
- Heidelberger, M.; Jacobs, W.A. Syntheses in the cinchona series. I. The simpler cinchona alkaloids and their dihydro derivatives. J. Am. Chem. Soc. 1919, 41, 817–833. [Google Scholar] [CrossRef] [Green Version]
- Brandes, S.; Niess, B.; Bella, M.; Prieto, A.; Overgaard, J.; Jørgensen, K.A. Non-Biaryl Atropisomers in Organocatalysis. Chem. A Eur. J. 2006, 12, 6039–6052. [Google Scholar] [CrossRef]
- Braje, W.M.; Frackenpohl, J.; Schrake, O.; Wartchow, R.; Beil, W.; Hoffmann, H.M.R. Synthesis of 10,11-DidehydroCinchona Alkaloids and Key Derivatives. Helvetica Chim. Acta 2000, 83, 777–792. [Google Scholar] [CrossRef]
- Song, Y.; Kohlmeir, E.K.; Meade, T.J. Synthesis of Multimeric MR Contrast Agents for Cellular Imaging. J. Am. Chem. Soc. 2008, 130, 6662–6663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilbert, E.E.; Voreck, W.E. Evaluation of a new organic azide: Hexakis(azidomethyl)benzene (HAB). Propellants Explos. Pyrotech. 1989, 14, 19–23. [Google Scholar] [CrossRef]
- Lyle, M.P.A.; Draper, N.D.; Wilson, P.D. Enantioselective Friedel−Crafts Alkylation Reactions Catalyzed by a Chiral NonracemicC2-Symmetric 2,2‘-Bipyridyl Copper(II) Complex. Org. Lett. 2005, 7, 901–904. [Google Scholar] [CrossRef]
- Török, B.; Abid, M.; London, G.; Esquibel, J.; Török, M.; Mhadgut, S.C.; Yan, P.; Prakash, G.K.S. Highly Enantioselective Organocatalytic Hydroxyalkylation of Indoles with Ethyl Trifluoropyruvate. Angew. Chem. Int. Ed. 2005, 44, 3086–3089. [Google Scholar] [CrossRef]
- Evans, D.A.; Mito, A.S.; Seidel, D. Scope and Mechanism of Enantioselective Michael Additions of 1,3-Dicarbonyl Compounds to Nitroalkenes Catalyzed by Nickel(II)−Diamine Complexes. J. Am. Chem. Soc. 2007, 129, 11583–11592. [Google Scholar] [CrossRef]
- Tan, B.; Zhang, X.; Chua, P.J.; Zhong, G. Recyclable organocatalysis: Highly enantioselective Michael addition of1,3-diaryl-1,3-propanedioneto nitroolefins. Chem. Commun. 2009, 779–781. [Google Scholar] [CrossRef]
- Gonczi, K.; Kudar, V.; Jaszay, Z.; Bombicz, P.; Faigl, F.; Madarász, J. Solid state structural relation and binary melting phase diagram of (S-) and racemic 2-(2-nitro-1-phenylethyl)-1,3-diphenyl-propane-1,3-dione. Thermochim. Acta 2014, 580, 46–52. [Google Scholar] [CrossRef] [Green Version]
- Fei, F.; Le Phuong, H.A.; Blanford, C.F.; Szekely, G. Tailoring the Performance of Organic Solvent Nanofiltration Membranes with Biophenol Coatings. ACS Appl. Polym. Mater. 2019, 1, 452–460. [Google Scholar] [CrossRef]
- Cseri, L.; Szekely, G. Towards cleaner PolarClean: Efficient synthesis and extended applications of the polar aprotic solvent methyl 5-(dimethylamino)-2-methyl-5-oxopentanoate. Green Chem. 2019, 21, 4178–4188. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||
|---|---|---|---|
| Solvent | Reaction Time (h) | Yield (13, %) 2 | Enantiomeric Excess [(S), %] 3 |
| CPME | 24 | 82 | 73 |
| 2-MeTHF | 24 | 70 | 69 |
| MTBE | 24 | 88 | 67 |
| DME | 24 | 81 | 64 |
| toluene | 24 | 98 | 61 |
| EtOAc | 24 | 70 | 59 |
| PolarClean | 24 | 67 | 52 |
| DMC | 24 | 92 | 52 |
| DCM | 24 | 99 | 48 |
| MeCN | 24 | 82 | 36 |
| EtOH | 24 | 98 | 2 |
| CPME | 4 | 87 | 73 |
| CPME | 2 | 85 | 72 |
| CPME | 1 | 87 | 72 |
 | ||
|---|---|---|
| Catalyst | Yield (13, %) 2 | Enantiomeric Excess [(S), %] 3 |
| hydroquinine (1) | 87 | 72 |
| Hub1-cinchona | 78 | 26 |
| Hub2-cinchona | 77 | 2 |
| Hub3-cinchona | 69 | 18 |
| Hub4-cinchona | 72 | 29 |
 | |||
|---|---|---|---|
| Solvent | Enantiomeric Excess (13, %) 2 | Solvent | Enantiomeric Excess (13, %) 2 |
| CPME | 18 (S) | PolarClean | 10 (S) |
| 2-MeTHF | 12 (S) | DMC | 4 (S) |
| MTBE | 12 (S) | DCM | 11 (R) |
| DME | 13 (S) | MeCN | 13 (R) |
| toluene | 4 (R) | EtOH | 0 |
| EtOAc | 2 (S) | ||
 | ||
|---|---|---|
| Catalyst | Yield (16, %) 3 | Enantiomeric Excess (16, %) 2 |
| hydroquinine (1) | 82 | 14 (R) |
| Hub1-cinchona | 10 | 3 (S) |
| Hub2-cinchona | 24 | 1 (S) |
| Hub3-cinchona | 82 | 32 (S) |
| Hub4-cinchona | 78 | 1 (S) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kisszékelyi, P.; Fehér, Z.; Nagy, S.; Bagi, P.; Kozma, P.; Garádi, Z.; Dékány, M.; Huszthy, P.; Mátravölgyi, B.; Kupai, J. Synthesis of C3-Symmetric Cinchona-Based Organocatalysts and Their Applications in Asymmetric Michael and Friedel–Crafts Reactions. Symmetry 2021, 13, 521. https://doi.org/10.3390/sym13030521
Kisszékelyi P, Fehér Z, Nagy S, Bagi P, Kozma P, Garádi Z, Dékány M, Huszthy P, Mátravölgyi B, Kupai J. Synthesis of C3-Symmetric Cinchona-Based Organocatalysts and Their Applications in Asymmetric Michael and Friedel–Crafts Reactions. Symmetry. 2021; 13(3):521. https://doi.org/10.3390/sym13030521
Chicago/Turabian StyleKisszékelyi, Péter, Zsuzsanna Fehér, Sándor Nagy, Péter Bagi, Petra Kozma, Zsófia Garádi, Miklós Dékány, Péter Huszthy, Béla Mátravölgyi, and József Kupai. 2021. "Synthesis of C3-Symmetric Cinchona-Based Organocatalysts and Their Applications in Asymmetric Michael and Friedel–Crafts Reactions" Symmetry 13, no. 3: 521. https://doi.org/10.3390/sym13030521
APA StyleKisszékelyi, P., Fehér, Z., Nagy, S., Bagi, P., Kozma, P., Garádi, Z., Dékány, M., Huszthy, P., Mátravölgyi, B., & Kupai, J. (2021). Synthesis of C3-Symmetric Cinchona-Based Organocatalysts and Their Applications in Asymmetric Michael and Friedel–Crafts Reactions. Symmetry, 13(3), 521. https://doi.org/10.3390/sym13030521