Push or Pull for a Better Selectivity? A Study on the Electronic Effects of Substituents of the Pyridine Ring on the Enantiomeric Recognition of Chiral Pyridino-18-Crown-6 Ethers

 , , ,
, , ,  , , ,
, , ,  , and
, and 
Abstract
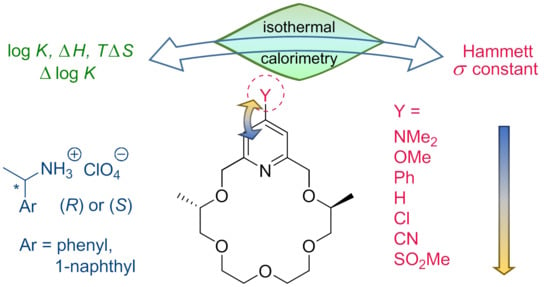
:
1. Introduction
2. Results and Discussion
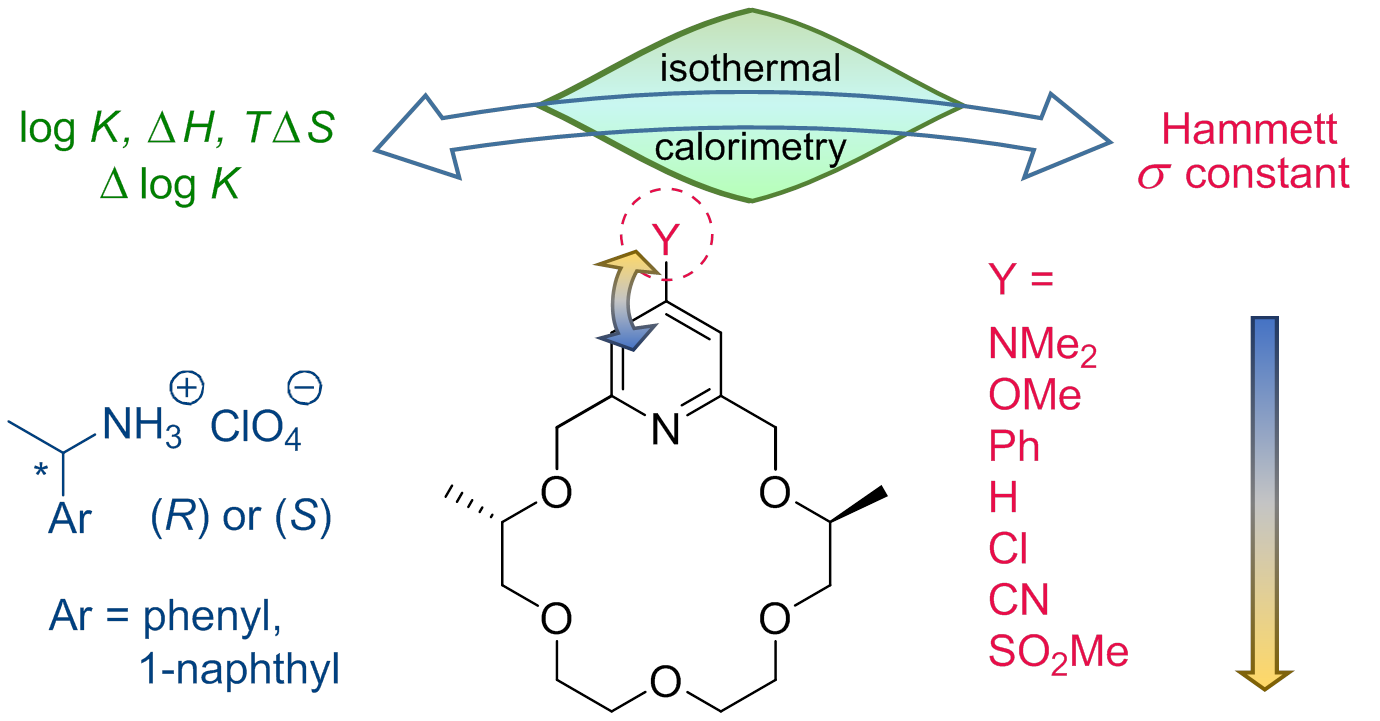
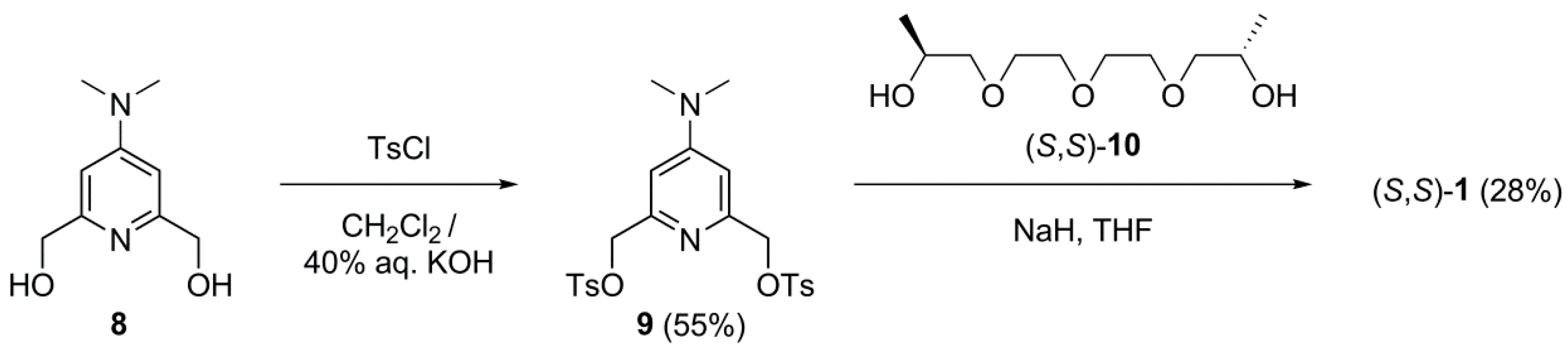
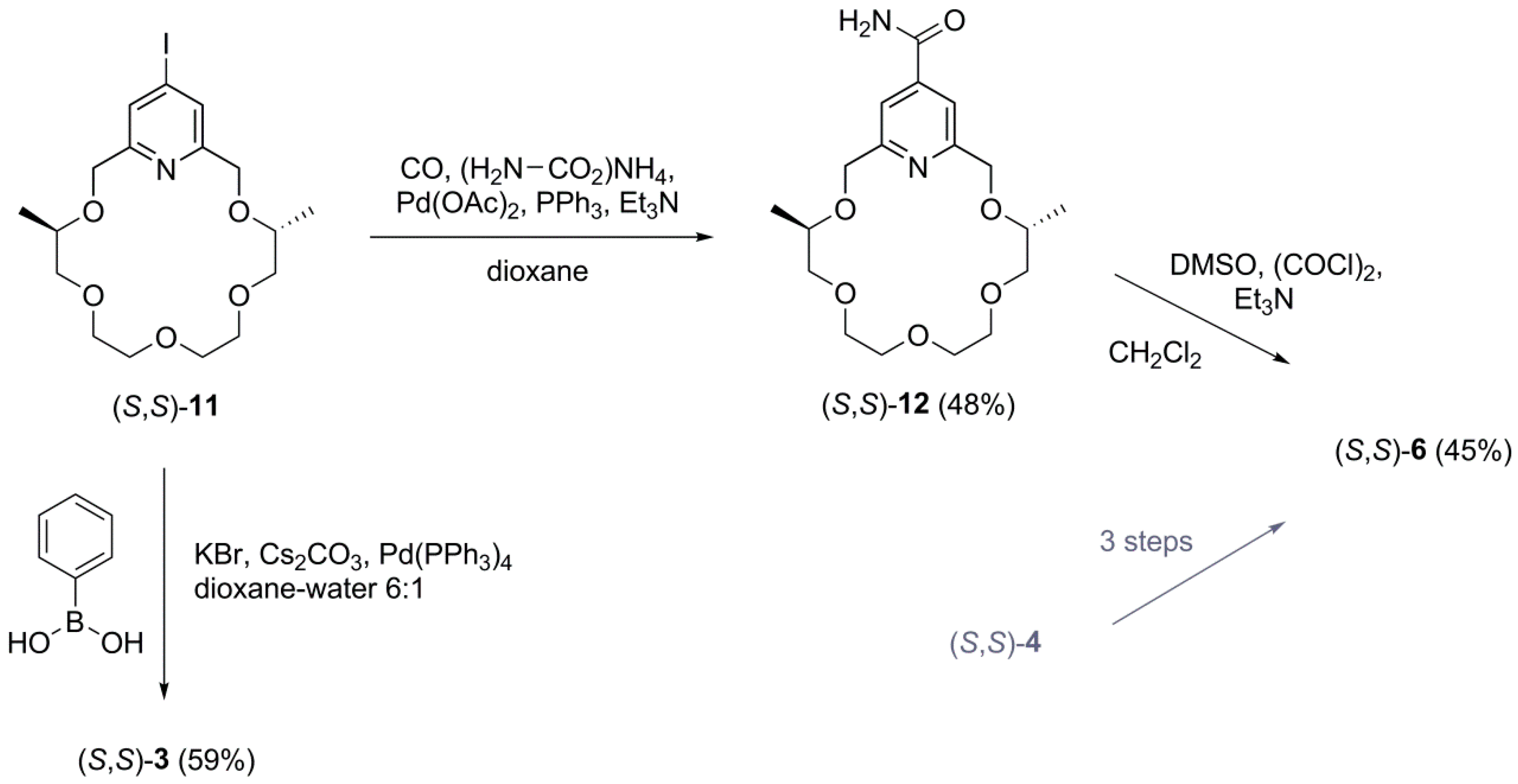
2.1. Synthesis
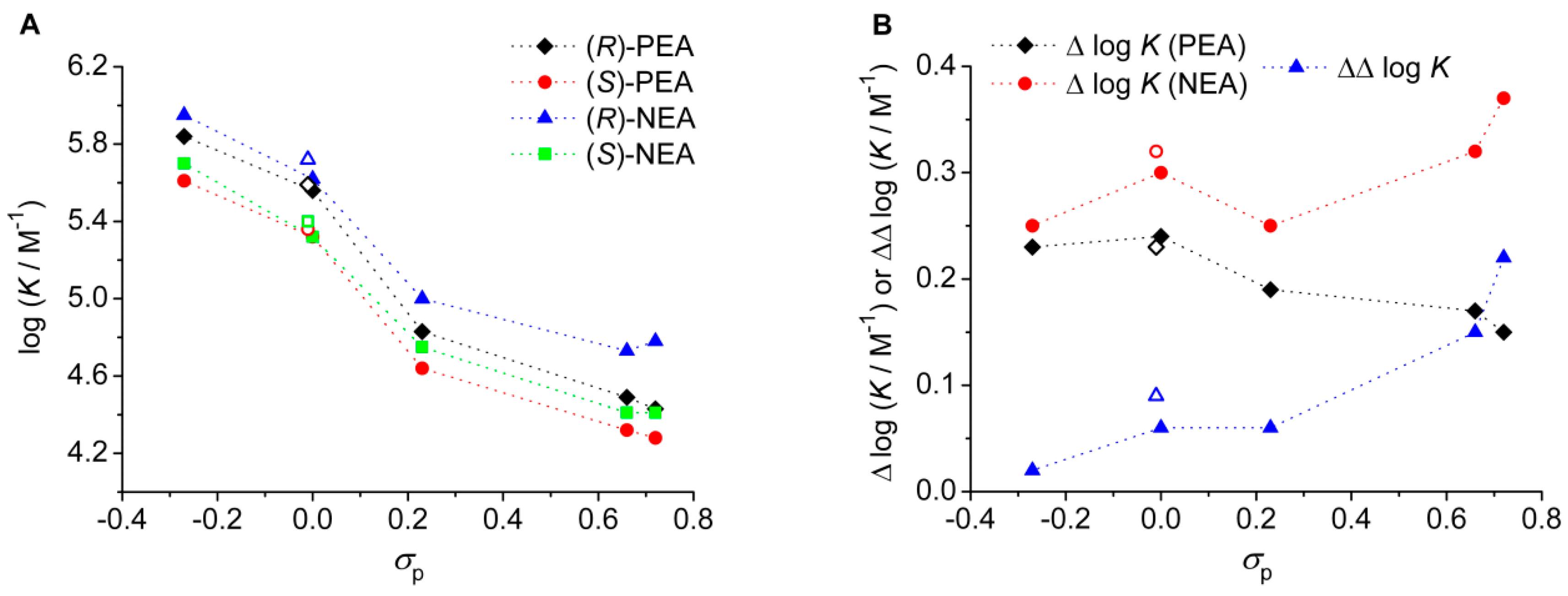
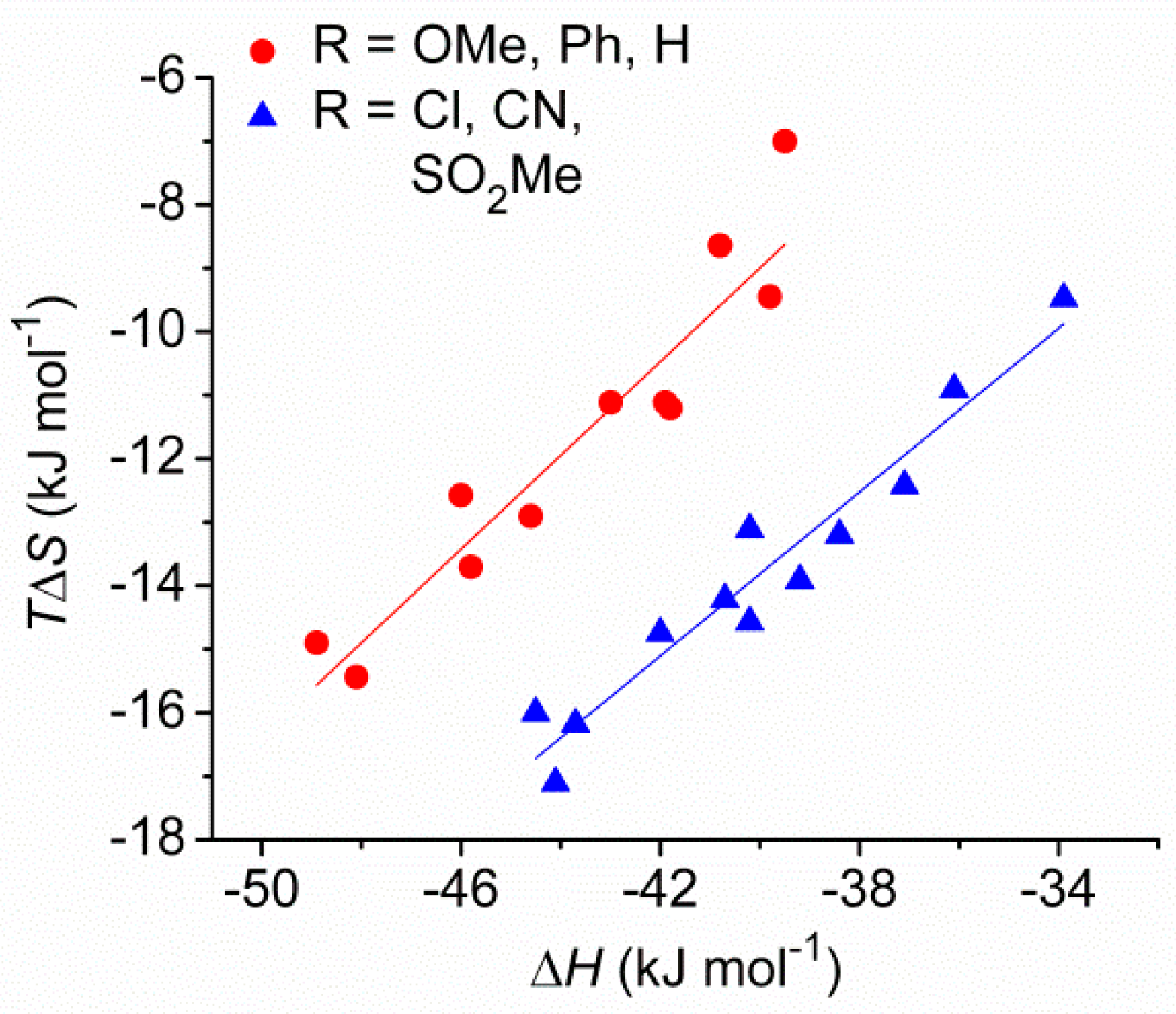
2.2. Enantiomeric Recognition Studies
3. Materials and Methods
3.1. General Information
3.2. Preparation of Compounds (S,S)-1, (S,S)-3, (S,S)-6, (S,S)-7, 9, and (S,S)-12
3.2.1. (4S,14S)-19-Dimethylamino-4,14-dimethyl-3,6,9,12,15-pentaoxa-21-azabicyclo [15.3.1]heneicosa-1(21),17,19-triene ((S,S)-1)
3.2.2. (4S,14S)-19-Phenyl-4,14-dimethyl-3,6,9,12,15-pentaoxa-21-azabicyclo[15.3.1]heneicosa-1. (21),17,19-triene ((S,S)-3)
3.2.3. (4S,14S)-4,14-Dimethyl-3,6,9,12,15-pentaoxa-21-azabicyclo[15.3.1]heneicosa-1(21),17,19-triene-19-carbonitrile ((S,S)-6)
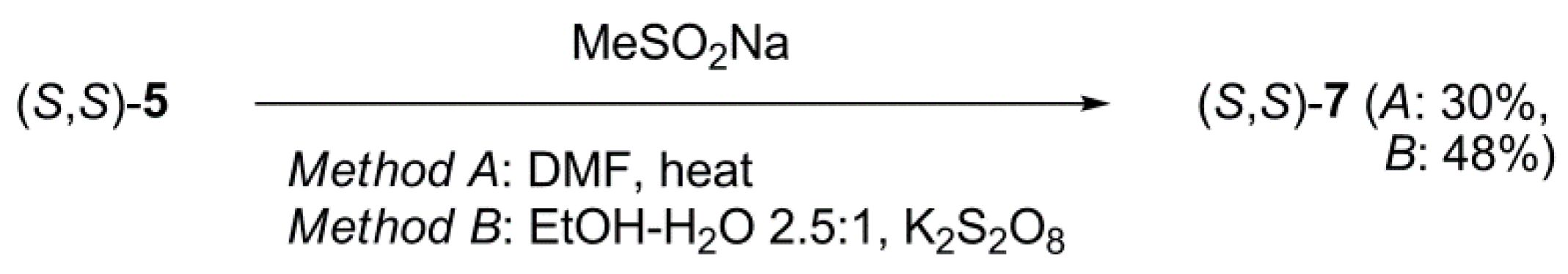
3.2.4. (4S,14S)-19-Methanesulfonyl-4,14-dimethyl-3,6,9,12,15-pentaoxa-21-azabicyclo[15.3.1] heneicosa-1(21),17,19-triene ((S,S)-7)
Method A
Method B
3.2.5. [4-(Dimethylamino)pyridine-2,6-diyl]bis(methylene) bis(4-methylbenzenesulfonate) (9)
3.2.6. (4S,14S)-4,14-Dimethyl-3,6,9,12,15-pentaoxa-21-azabicyclo[15.3.1]heneicosa-1(21),17,19-triene-19-carboxamide ((S,S)-12)
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Budhathoki-Uprety, J.; Shah, J.; Korsen, J.A.; Wayne, A.E.; Galassi, T.V.; Cohen, J.R.; Harvey, J.D.; Jena, P.V.; Ramanathan, L.V.; Jaimes, E.A.; et al. Synthetic molecular recognition nanosensor paint for microalbuminuria. Nat. Commun. 2019, 10, 3605. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, L.A.; He, H.; Pham-Huy, C. Chiral drugs: An overview. Int. J. Biomed. Sci. 2006, 2, 85–100. [Google Scholar] [PubMed]
- Núñez, M.C.; García-Rubiño, M.E.; Conejo-García, A.; Cruz-López, O.; Kimatrai, M.; Gallo, M.A.; Espinosa, A.; Campos, J.M. Homochiral drugs: A demanding tendency of the pharmaceutical industry. Curr. Med. Chem. 2009, 16, 2064–2074. [Google Scholar] [CrossRef] [PubMed]
- Trojanowicz, M. Enantioselective electrochemical sensors and biosensors: A mini-review. Electrochem. Commun. 2014, 38, 47–52. [Google Scholar] [CrossRef]
- Pu, L. Simultaneous determination of concentration and enantiomeric composition in fluorescent sensing. Acc. Chem. Res. 2017, 50, 1032–1040. [Google Scholar] [CrossRef]
- Shang, X.; Park, C.H.; Jung, G.Y.; Kwak, S.K.; Oh, J.H. Highly Enantioselective Graphene-Based Chemical Sensors Prepared by Chiral Noncovalent Functionalization. ACS Appl. Mater. Interfaces 2018, 10, 36194–36201. [Google Scholar] [CrossRef]
- Hyun, M.H. Liquid chromatographic enantioseparations on crown ether-based chiral stationary phases. J. Chromatogr. A 2016, 1467, 19–32. [Google Scholar] [CrossRef]
- Cheng, P.; Jiao, S.; Xu, K.; Wang, C. Progress in Enantioselective Recognition Based on Chiral Crown Ether. Chin. J. Org. Chem. 2013, 33, 280–287. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Yin, J.; Yoon, J. Recent advances in development of chiral fluorescent and colorimetric sensors. Chem. Rev. 2014, 114, 4918–4959. [Google Scholar] [CrossRef] [PubMed]
- Móczár, I.; Huszthy, P. Optically active crown ether-based fluorescent sensor molecules: A mini-review. Chirality 2019, 31, 97–109. [Google Scholar] [CrossRef] [PubMed]
- Szemenyei, B.; Móczár, I.; Pál, D.; Kocsis, I.; Baranyai, P.; Huszthy, P. Synthesis and Enantiomeric Recognition Studies of Optically Active Pyridino-Crown Ethers Containing an Anthracene Fluorophore Unit. Chirality 2016, 28, 562–568. [Google Scholar] [CrossRef] [PubMed]
- Pál, D.; Móczár, I.; Szemenyei, B.; Marczona, D.; Kocsis, I.; Prikler, G.; Vezse, P.; Baranyai, P.; Huszthy, P. Pyridino-18-crown-6 ether type chemosensors containing a benzothiazole fluorophore unit: Synthesis and enantiomeric recognition studies. Tetrahedron 2019, 75, 2900–2909. [Google Scholar] [CrossRef]
- Sousa, L.R.; Sogah, G.D.Y.; Hoffman, D.H.; Cram, D.J. Host–guest complexation. 12. Total optical resolution of amine and amino ester salts by chromatography. J. Am. Chem. Soc. 1978, 100, 4569–4576. [Google Scholar] [CrossRef]
- Bradshaw, J.S.; Huszthy, P.; Wang, T.M.; Zhu, C.Y.; Nazarenko, A.Y.; Izatt, R.M. Enantiomeric recognition and separation of chiral organic ammonium salts by chiral pyridino-18-crown-6 ligands. Supramol. Chem. 1993, 1, 267–275. [Google Scholar] [CrossRef]
- Köntös, Z.; Huszthy, P.; Bradshaw, J.S.; Izatt, R.M. Enantioseparation of racemic organic ammonium perchlorates by a silica gel bound optically active di-tert-butylpyridino-18-crown-6 ligand. Tetrahedron Asymmetry 1999, 10, 2087–2099. [Google Scholar] [CrossRef]
- Farkas, V.; Tóth, T.; Orosz, G.; Huszthy, P.; Hollósi, M. Enantioseparation of protonated primary arylalkylamines and amino acids containing an aromatic moiety on a pyridino-crown ether based new chiral stationary phase. Tetrahedron Asymmetry 2006, 17, 1883–1889. [Google Scholar] [CrossRef]
- Kupai, J.; Lévai, S.; Antal, K.; Balogh, G.T.; Tóth, T.; Huszthy, P. Preparation of pyridino-crown ether-based new chiral stationary phases and preliminary studies on their enantiomer separating ability for chiral protonated primary aralkylamines. Tetrahedron Asymmetry 2012, 23, 415–427. [Google Scholar] [CrossRef]
- Németh, T.; Lévai, S.; Kormos, A.; Kupai, J.; Tóth, T.; Balogh, G.T.; Huszthy, P. Preparation and studies of chiral stationary phases containing enantiopure acridino-18-crown-6 ether selectors. Chirality 2014, 26, 651–654. [Google Scholar] [CrossRef]
- Newcomb, M.; Toner, J.L.; Helgeson, R.C.; Cram, D.J. Host–guest complexation. 20. Chiral recognition in transport as a molecular basis for a catalytic resolving machine. J. Am. Chem. Soc. 1979, 101, 4941–4947. [Google Scholar] [CrossRef]
- Szabó, T.; Hirsch, E.; Tóth, T.; Müller, J.; Riethmüller, E.; Balogh, G.T.; Huszthy, P. Synthesis and enantioselective transport studies of optically active lipophilic proton-ionizable crown ethers containing a diarylphosphinic acid unit. Tetrahedron Asymmetry 2015, 15, 650–656. [Google Scholar] [CrossRef]
- Szabó-Szentjóbi, H.; Bagi, P.; Müller, J.; Balogh, G.T.; Tóth, T.; Huszthy, P. Synthesis and enantioselective transport studies of both enantiomers of new chiral proton-ionizable crown ethers containing a diarylphosphinic acid unit. Tetrahedron 2019, 75, 1275–1281. [Google Scholar] [CrossRef]
- Bradshaw, J.S.; Jones, B.A.; Nielsen, R.B.; Spencer, N.O.; Thompson, P.K. The preparation of macrocyclic polyether-diester compounds by transesterification including the preparation of two new nitrogen containing diester-crown compounds. J. Heterocycl. Chem. 1983, 20, 957–962. [Google Scholar] [CrossRef]
- Bradshaw, J.S.; Thompson, P.K.; Izatt, R.M. The preparation of new chiral diphenyl-substituted macrocyclic polyether-diester compounds and their enantiomeric recognition of chiral organic ammonium salts. J. Heterocycl. Chem. 1984, 21, 897–901. [Google Scholar] [CrossRef]
- Davidson, R.B.; Bradshaw, J.S.; Jones, B.A.; Dalley, N.K.; Christensen, J.J.; Izatt, R.M. Enantiomeric recognition of organic ammonium salts by chiral crown ethers based on the pyridino-18-crown-6 structure. J. Org. Chem. 1984, 49, 353–357. [Google Scholar] [CrossRef]
- Huszthy, P.; Oue, M.; Bradshaw, J.S.; Zhu, C.Y.; Wang, T.; Dalley, N.K.; Curtis, J.C.; Izatt, R.M. New Symmetrical Chiral Dibenzyl- and Diphenyl-Substituted Diamido-, Dithionoamido-, Diaza-, and Azapyridino-18-crown-6 Ligands. J. Org. Chem. 1992, 57, 5383–5394. [Google Scholar] [CrossRef]
- Izatt, R.M.; Wang, T.; Hathaway, J.K.; Zhang, X.X.; Curtis, J.C.; Bradshaw, J.S.; Zhu, C.Y.; Huszthy, P. Factors Influencing Enantiomeric Recognition of Primary Alkylammonium Salts by Pyridino-18-crown-6 Type Ligands. J. Incl. Phenom. Mol. Recognit. Chem. 1994, 17, 157–175. [Google Scholar] [CrossRef]
- Izatt, R.M.; Zhang, X.X.; Huszthy, P.; Zhu, C.Y.; Hathaway, J.K.; Wang, T.; Bradshaw, J.S. A Thermodynamic Study of Enantiomeric Recognition of Organic Ammonium Cations by Pyridino-18-Crown-6 Type Ligands in Methanol and a 1: 1 Methanol-1,2-Dichloroethane Mixture at 25.0 °C. J. Incl. Phenom. Mol. Recognit. Chem. 1994, 18, 353–367. [Google Scholar] [CrossRef]
- Bradshaw, J.S.; Huszthy, P.; Redd, J.T.; Zhang, X.X.; Wang, T.; Hathaway, J.K.; Young, J.; Izatt, R.M. Enantiomeric recognition of chiral ammonium salts by chiral pyridino- and pyrimidino-18-crown-6 ligands: Effect of structure and solvents. Pure Appl. Chem. 1995, 67, 691–695. [Google Scholar] [CrossRef]
- Wang, T.; Bradshaw, J.S.; Huszthy, P.; Izatt, R.M. Various aspects of enantiomeric recognition of (S,S)-dimethylpyridino-18-crown-6 by several organic ammonium salts. Supramol. Chem. 1996, 6, 251–255. [Google Scholar] [CrossRef]
- Jones, B.A.; Bradshaw, J.S.; Brown, P.R.; Christensen, J.J.; Izatt, R.M. Preparation of macrocyclic polyether-thiono diester and -thiono tetraester ligands containing either the pyridine subcyclic unit or the oxalyl moiety, their complexes, and their reductive desulfurization to crown ethers. J. Org. Chem. 1983, 48, 2635–2639. [Google Scholar] [CrossRef]
- Kupai, J.; Huszthy, P.; Székely, K.; Tóth, T.; Párkányi, L. Synthesis of new enantiopure dimethyl- and diisobutyl-substituted pyridino-18-crown-6 ethers containing a halogen atom or a methoxy group at position 4 of the pyridine ring for enantiomeric recognition studies. Arkivoc 2011, ix, 77–93. [Google Scholar] [CrossRef] [Green Version]
- Tóth, T.; Huszthy, P.; Kupai, J.; Nyitrai, J. Synthesis of new enantiopure dimethyl-substituted pyridino-18- crown-6 ether type macrocycles containing different substituents at position 4 of the pyridine ring for enantiomeric recognition studies. Arkivoc 2008, 3, 66–79. [Google Scholar] [CrossRef] [Green Version]
- Hashizume, M.; Tobey, S.; Lynch, V.M.; Anslyn, E.V. Synthesis and Evaluation of a Cyclophane Receptor for Acetic Acid. Supramol. Chem. 2002, 14, 511–517. [Google Scholar] [CrossRef]
- Hommes, P.; Fischer, C.; Lindern, C.; Zipse, H.; Reissig, H.-U. Unprecedented Strong Lewis Bases—Synthesis and Methyl Cation Affinities of Dimethylamino-Substituted Terpyridines. Angew. Chem. Int. Ed. 2014, 53, 7647–7651. [Google Scholar] [CrossRef]
- Bhattacharya, S.; Snehalatha, K.; George, S.K. Synthesis of Some Copper(II)-Chelating (Dialkylamino)pyridine Amphiphiles and Evaluation of Their Esterolytic Capacities in Cationic Micellar Media. J. Org. Chem. 1998, 63, 27–35. [Google Scholar] [CrossRef]
- Horváth, G.; Rusa, C.; Köntös, Z.; Gerencsér, J.; Huszthy, P. A new Efficient Method for the Preparation of 2,6-Pyridinedimethyl Ditosylates from Dimethyl 2,6-Pyridinedicarboxylates. Synth. Commun. 1999, 29, 3719–3731. [Google Scholar] [CrossRef]
- Horváth, G.; Huszthy, P. Chromatographic enantioseparation of racemic α-(1-naphthyl)ethylammonium perchlorate by a Merrifield resin-bound enantiomerically pure chiral dimethylpyridino-18-crown-6 ligand. Tetrahedron Asymmetry 1999, 10, 4573–4583. [Google Scholar] [CrossRef]
- Nakajima, N.; Ubukata, M. Preparation of nitriles from primary amides under Swern oxidation conditions. Tetrahedron Lett. 1997, 38, 2099–2102. [Google Scholar] [CrossRef]
- Maloney, K.M.; Kuethe, J.T.; Linn, K. A Practical, One-Pot Synthesis of Sulfonylated Pyridines. Org. Lett. 2011, 13, 102–105. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Wang, X.; Pan, B.-B.; Su, X.-C. Single-armed phenylsulfonated pyridine derivative of DOTA is rigid and stable paramagnetic tag in protein analysis. Chem. Commun. 2016, 52, 11535–11538. [Google Scholar] [CrossRef]
- Nguyen, V.D.; Nguyen, V.T.; Haug, G.C.; Dang, H.T.; Arman, H.D.; Ermler, W.C.; Larionov, O.V. Rapid and Chemodivergent Synthesis of N-Heterocyclic Sulfones and Sulfides: Mechanistic and Computational Details of the Persulfate-Initiated Catalysis. ACS Catal. 2019, 9, 4015–4024. [Google Scholar] [CrossRef]
- Christensen, J.J.; Ruckman, J.; Eatough, D.J.; Izatt, R.M. Determination of equilibrium constants by titration calorimetry: Part I. Introduction to titration calorimetry. Thermochim. Acta 1972, 3, 203–218. [Google Scholar] [CrossRef]
- Kaljurand, I.; Kütt, A.; Sooväli, L.; Rodima, T.; Mäemets, V.; Leito, I.; Koppel, I.A. Extension of the Self-Consistent Spectrophotometric Basicity Scale in Acetonitrile to a Full Span of 28 pKa Units: Unification of Different Basicity Scales. J. Org. Chem. 2005, 70, 1019–1028. [Google Scholar] [CrossRef]
- Hansch, C.; Leo, A.; Taft, R.W. A survey of Hammett substituent constants and resonance and field parameters. Chem. Rev. 1991, 91, 165–195. [Google Scholar] [CrossRef]
- Riddick, J.A.; Bunger, W.B.; Sakano, T.K. Organic Solvents. In Techniques of Chemistry, 4th ed.; Weissberger, A., Ed.; Wiley-Interscience: New York, NY, USA, 1986; Volume 2. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ligand | Substituent (Y) | σpa | Guest | log K b | Δ log K | ΔH c (kJ mol−1) | ΔS d (J mol−1 K−1) |
|---|---|---|---|---|---|---|---|
| (K in M−1) | (K in M−1) | ||||||
| (S,S)-2 | MeO | −0.27 | (R)-PEA | 5.84 | 0.23 | −46.0 | −42.2 |
| (S)-PEA | 5.61 | −40.8 | −29.0 | ||||
| (R)-NEA | 5.95 | 0.25 | −48.9 | −50.0 | |||
| (S)-NEA | 5.70 | −39.5 | −23.5 | ||||
| (S,S)-3 | Ph | −0.01 | (R)-PEA | 5.59 | 0.23 | −43.0 | −37.3 |
| (S)-PEA | 5.36 | −41.8 | −37.6 | ||||
| (R)-NEA | 5.72 | 0.32 | −48.1 | −51.8 | |||
| (S)-NEA | 5.40 | −41.9 | −37.3 | ||||
| (S,S)-4 | H | 0.00 | (R)-PEA | 5.56 | 0.24 | −44.6 | −43.3 |
| (S)-PEA | 5.32 | −39.8 | −31.7 | ||||
| (R)-NEA | 5.62 | 0.30 | −45.8 | −46.0 | |||
| (S)-NEA | 5.32 | −39.8 | −31.7 | ||||
| (S,S)-5 | Cl | 0.23 | (R)-PEA | 4.83 | 0.19 | −43.7 | −54.3 |
| (S)-PEA | 4.64 | −40.7 | −47.7 | ||||
| (R)-NEA | 5.00 | 0.25 | −44.5 | −53.7 | |||
| (S)-NEA | 4.75 | −40.2 | −44.0 | ||||
| (S,S)-6 | CN | 0.66 | (R)-PEA | 4.49 | 0.17 | −40.2 | −48.9 |
| (S)-PEA | 4.32 | −37.1 | −41.7 | ||||
| (R)-NEA | 4.73 | 0.32 | −44.1 | −57.4 | |||
| (S)-NEA | 4.41 | −38.4 | −44.3 | ||||
| (S,S)-7 | SO2Me | 0.72 | (R)-PEA | 4.43 | 0.15 | −39.2 | −46.7 |
| (S)-PEA | 4.28 | −33.9 | −31.8 | ||||
| (R)-NEA | 4.78 | 0.37 | −42.0 | −49.5 | |||
| (S)-NEA | 4.41 | −36.1 | −36.6 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Szemenyei, B.; Novotny, B.; Zsolnai, S.; Miskolczy, Z.; Biczók, L.; Takács, A.; Kollár, L.; Drahos, L.; Móczár, I.; Huszthy, P. Push or Pull for a Better Selectivity? A Study on the Electronic Effects of Substituents of the Pyridine Ring on the Enantiomeric Recognition of Chiral Pyridino-18-Crown-6 Ethers. Symmetry 2020, 12, 1795. https://doi.org/10.3390/sym12111795
Szemenyei B, Novotny B, Zsolnai S, Miskolczy Z, Biczók L, Takács A, Kollár L, Drahos L, Móczár I, Huszthy P. Push or Pull for a Better Selectivity? A Study on the Electronic Effects of Substituents of the Pyridine Ring on the Enantiomeric Recognition of Chiral Pyridino-18-Crown-6 Ethers. Symmetry. 2020; 12(11):1795. https://doi.org/10.3390/sym12111795
Chicago/Turabian StyleSzemenyei, Balázs, Balázs Novotny, Sarolta Zsolnai, Zsombor Miskolczy, László Biczók, Attila Takács, László Kollár, László Drahos, Ildikó Móczár, and Péter Huszthy. 2020. "Push or Pull for a Better Selectivity? A Study on the Electronic Effects of Substituents of the Pyridine Ring on the Enantiomeric Recognition of Chiral Pyridino-18-Crown-6 Ethers" Symmetry 12, no. 11: 1795. https://doi.org/10.3390/sym12111795
APA StyleSzemenyei, B., Novotny, B., Zsolnai, S., Miskolczy, Z., Biczók, L., Takács, A., Kollár, L., Drahos, L., Móczár, I., & Huszthy, P. (2020). Push or Pull for a Better Selectivity? A Study on the Electronic Effects of Substituents of the Pyridine Ring on the Enantiomeric Recognition of Chiral Pyridino-18-Crown-6 Ethers. Symmetry, 12(11), 1795. https://doi.org/10.3390/sym12111795