
Phosphorus Compounds of Natural Origin: Prebiotic, Stereochemistry, Application


Abstract
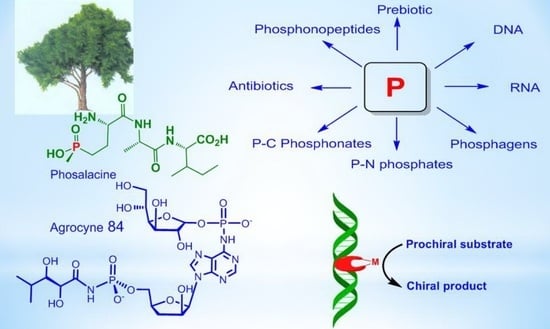
:
1. Introduction
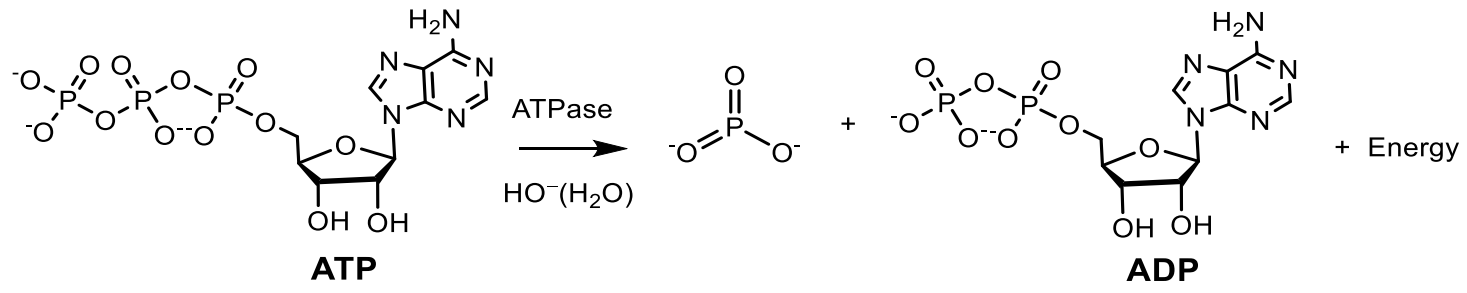
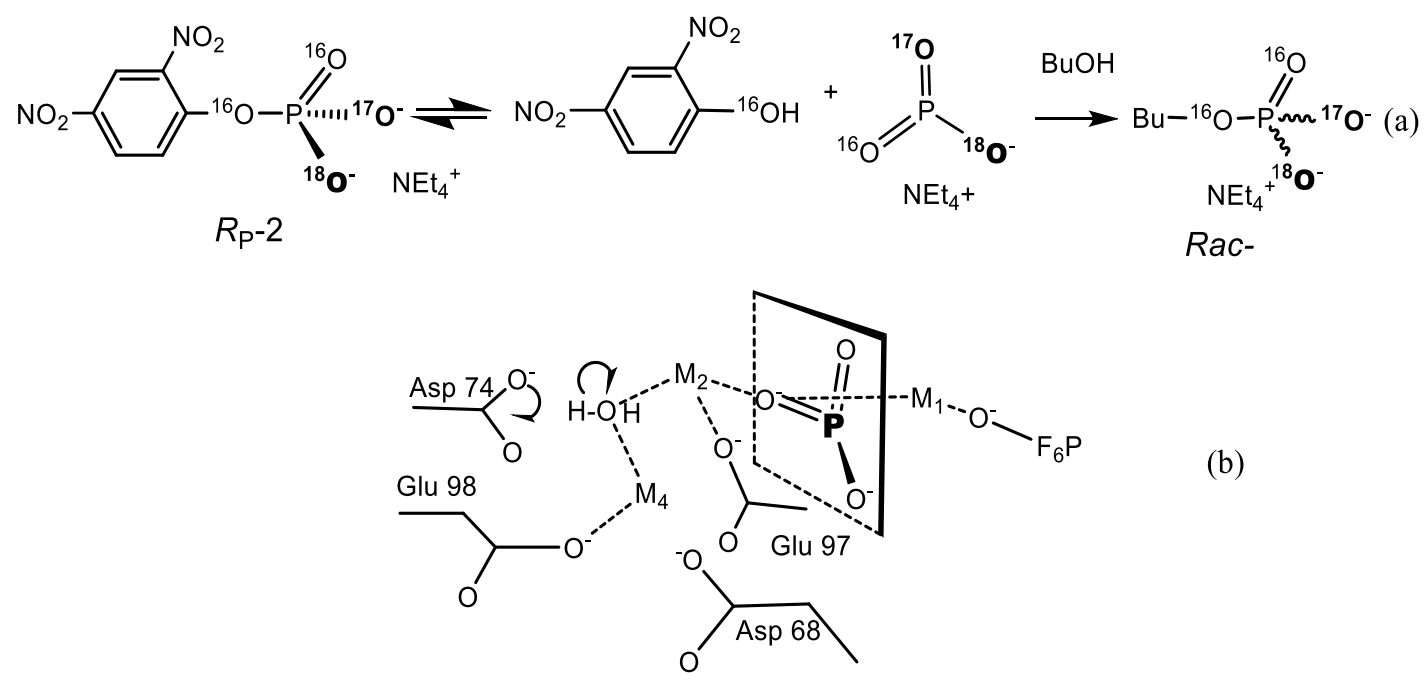
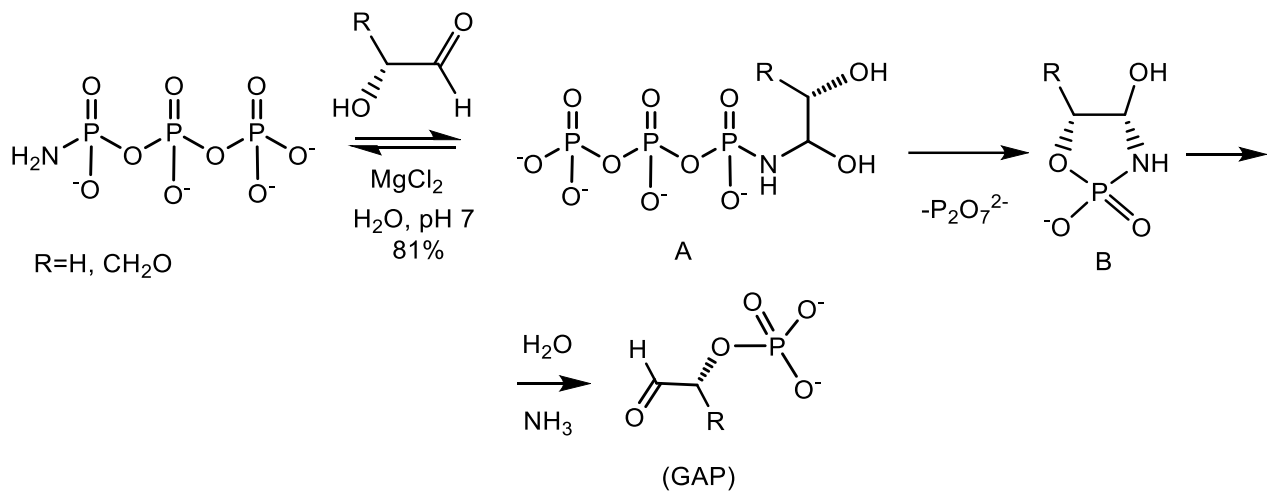
2. The Role of Phosphorus at the Origine and Evolution of Life
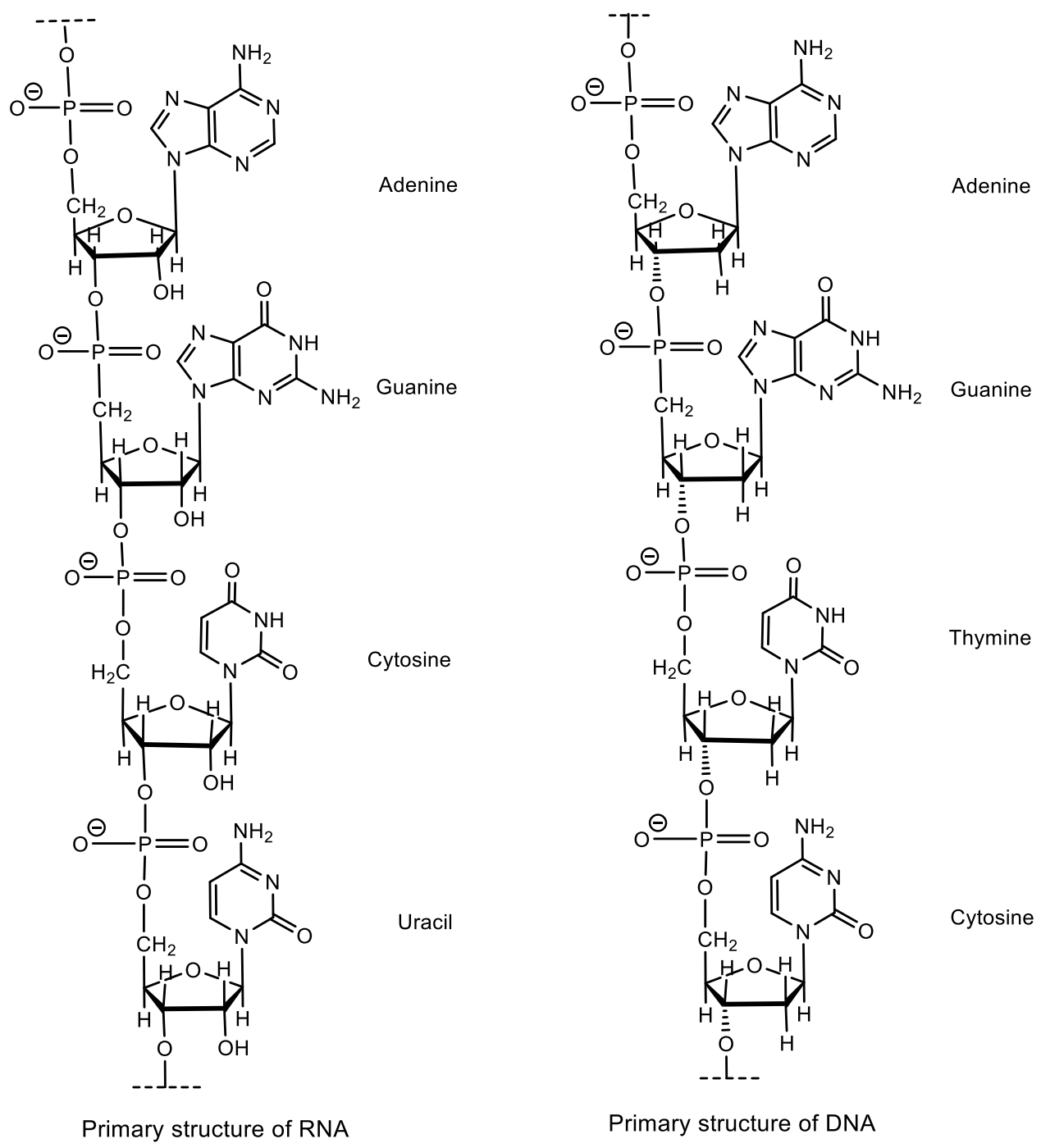
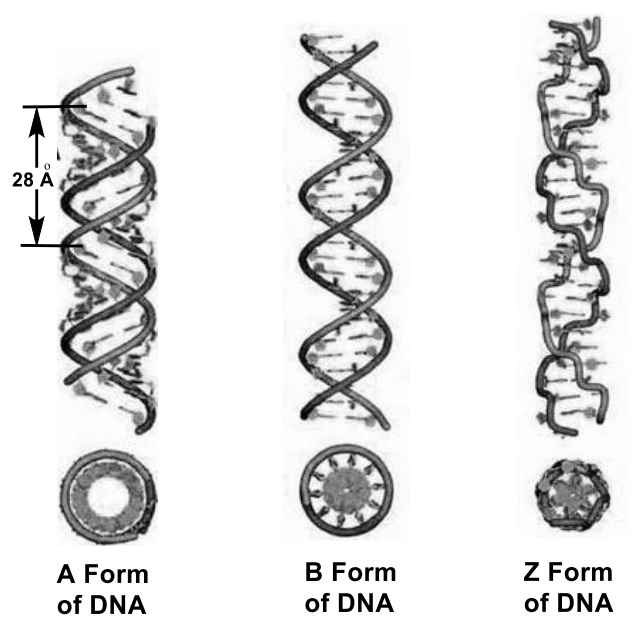
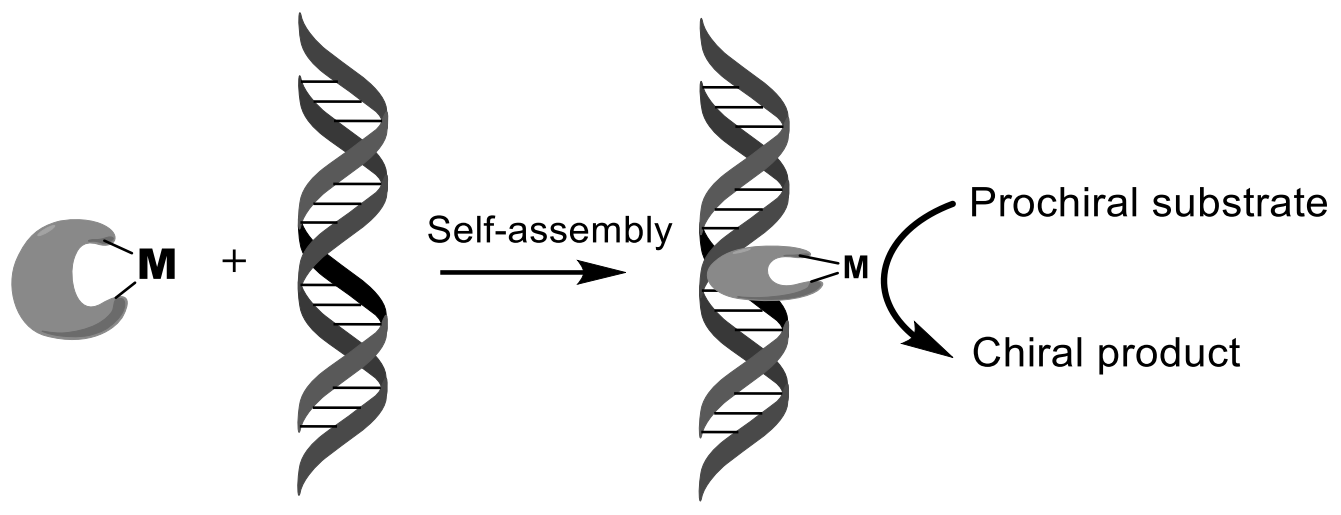
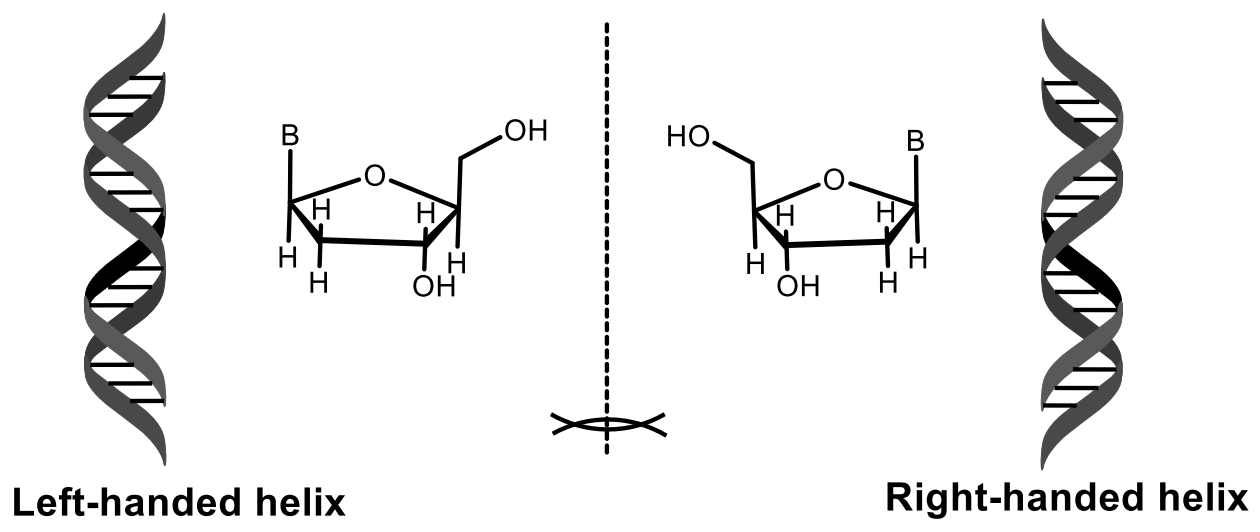
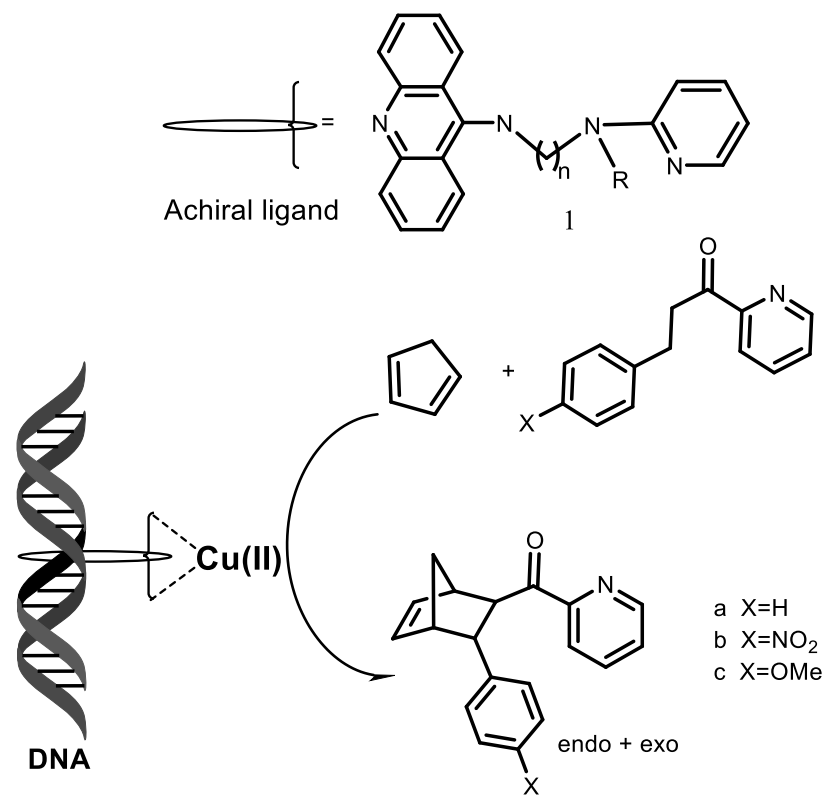
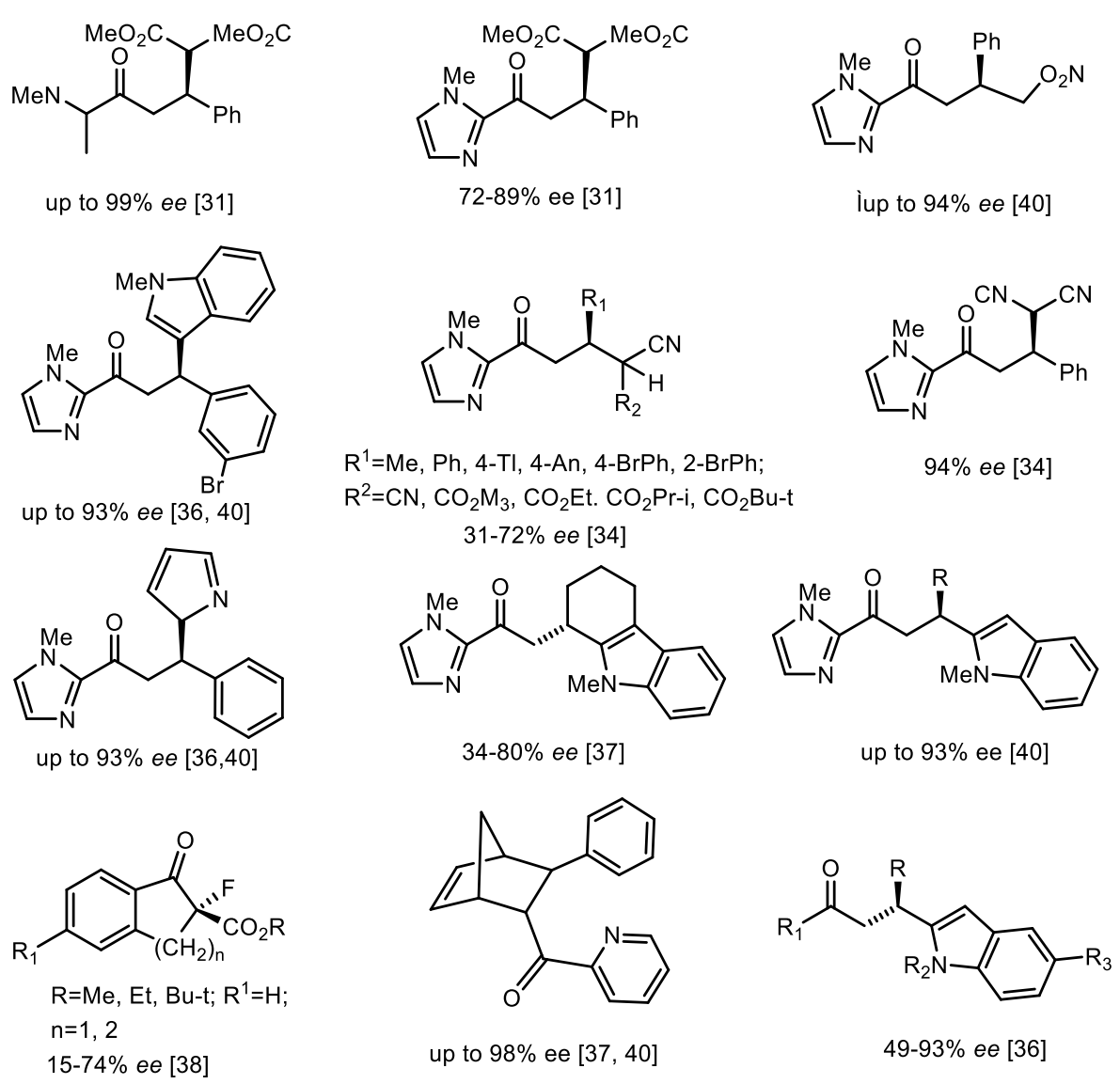
3. Stereochemistry of DNA and RNA
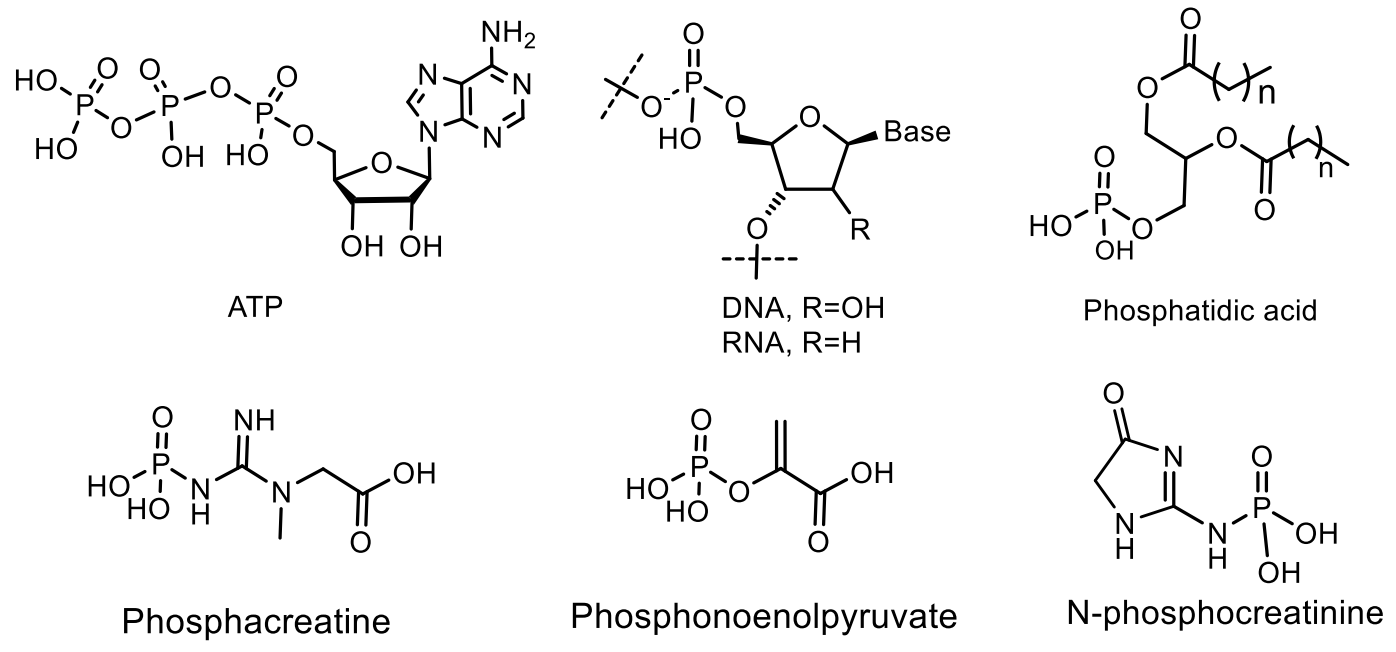
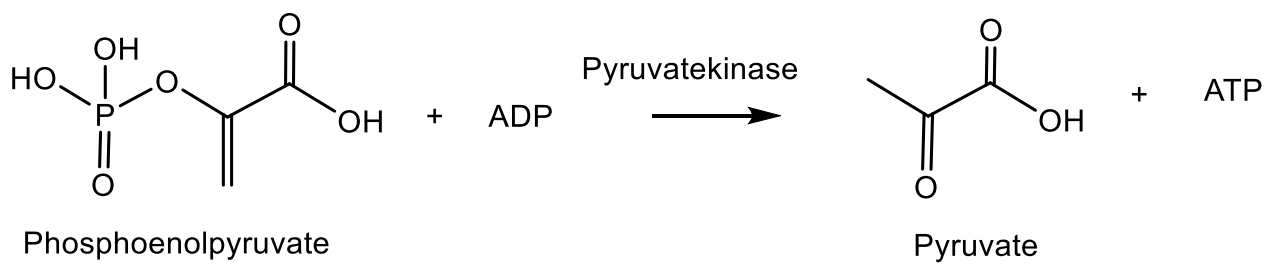
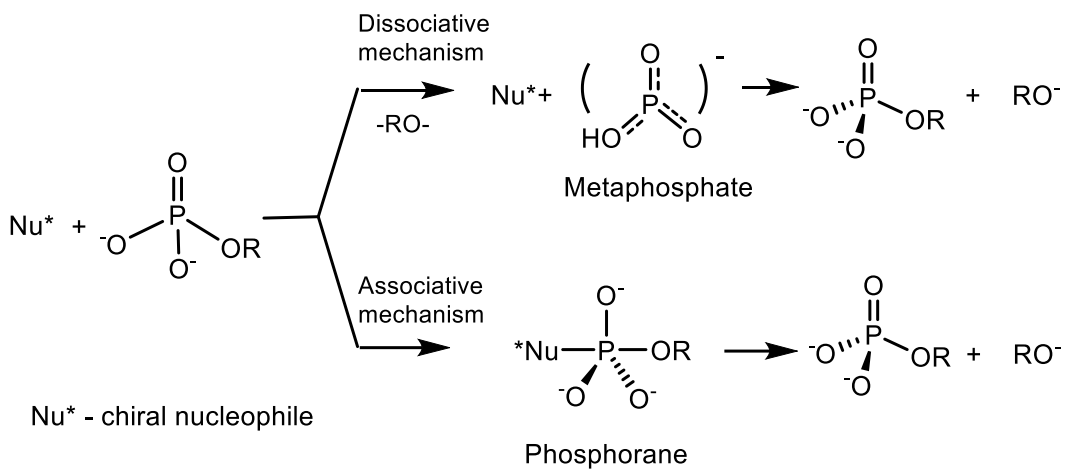
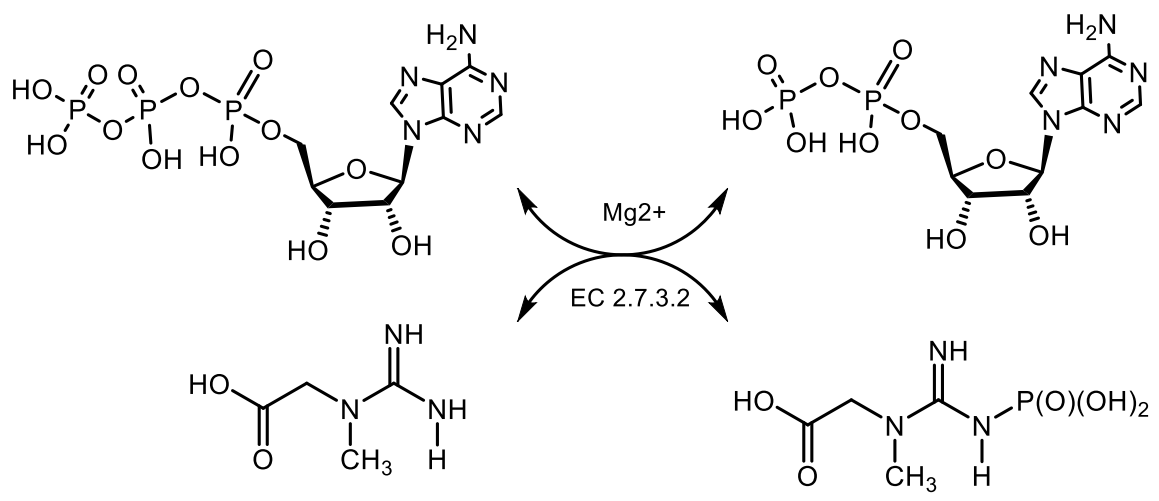
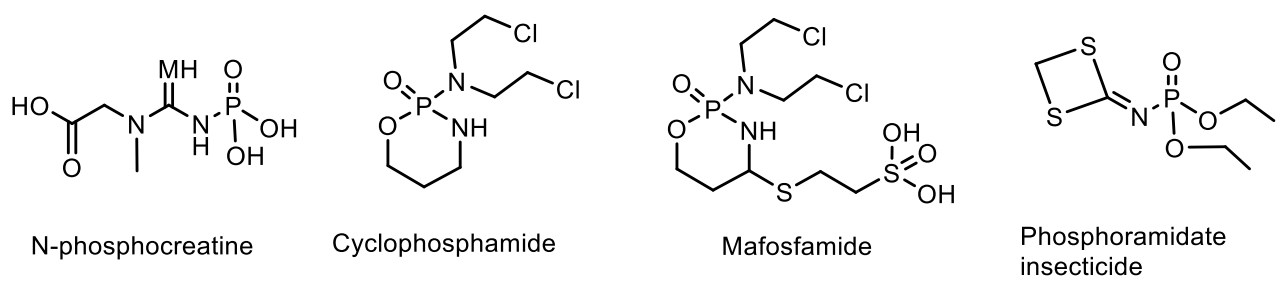
4. Phosphagens
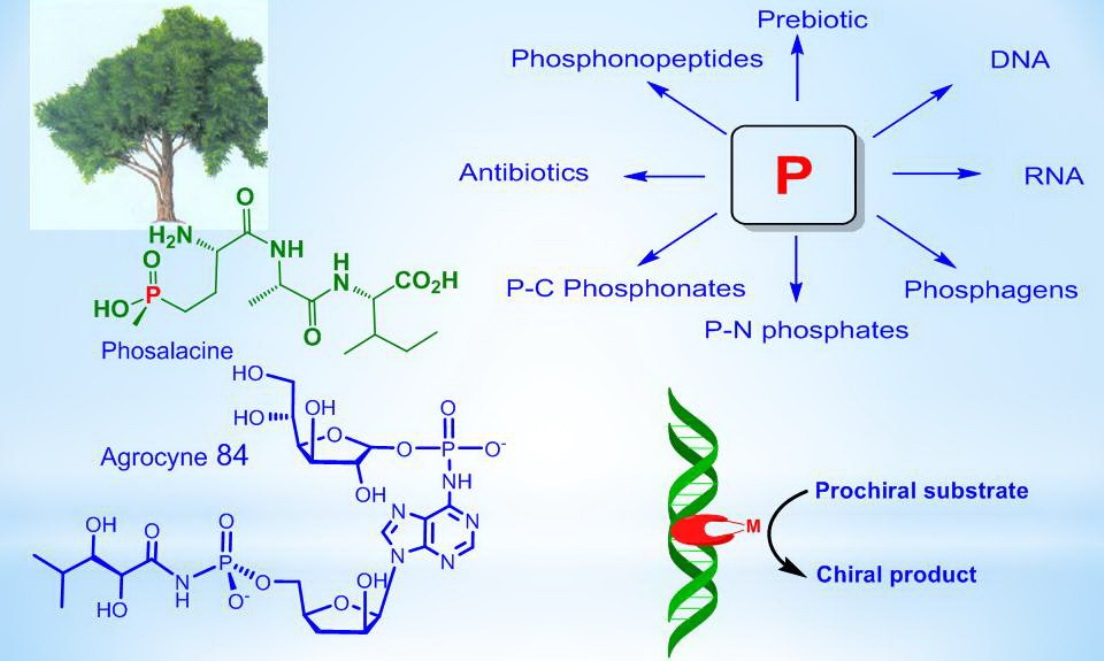
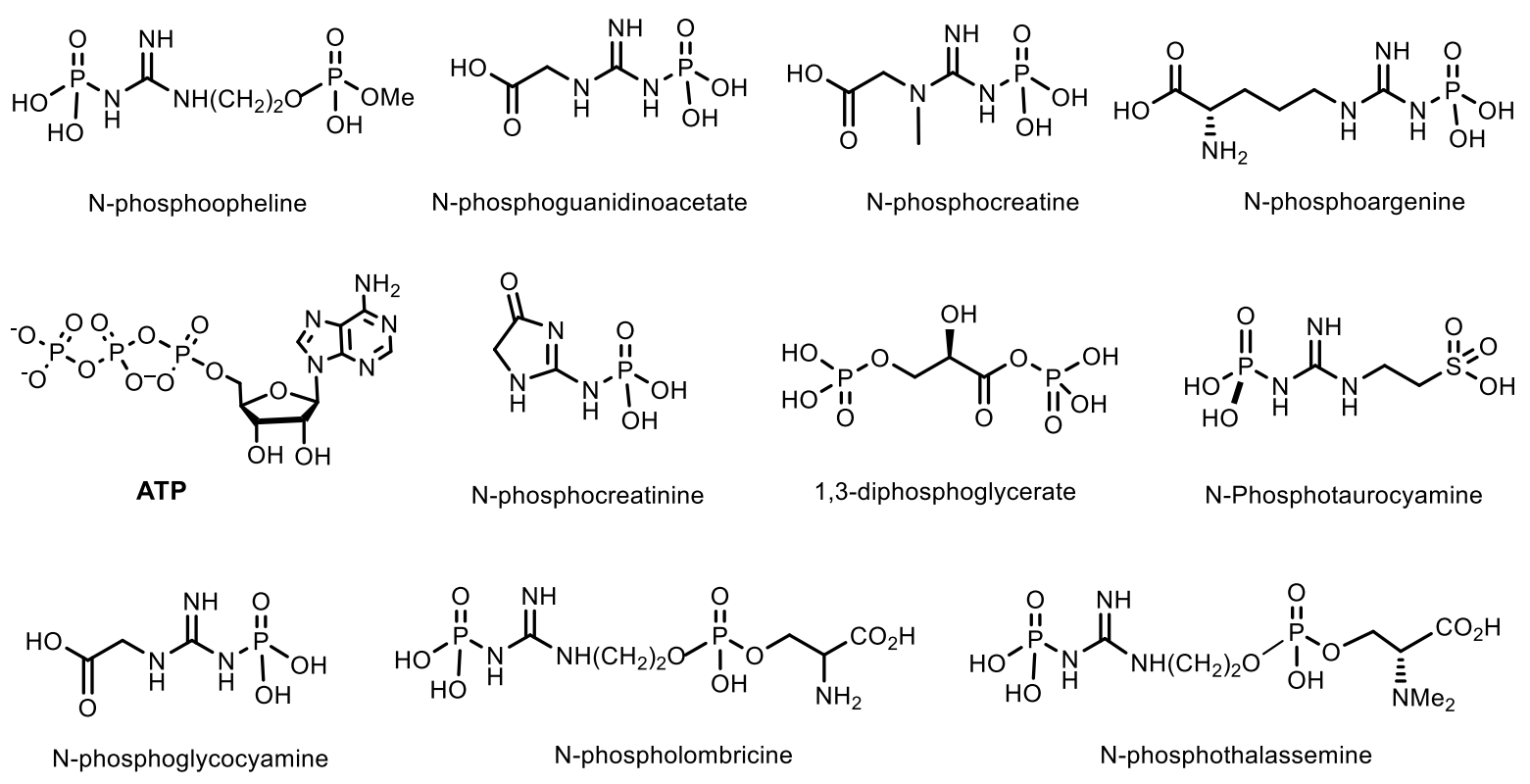
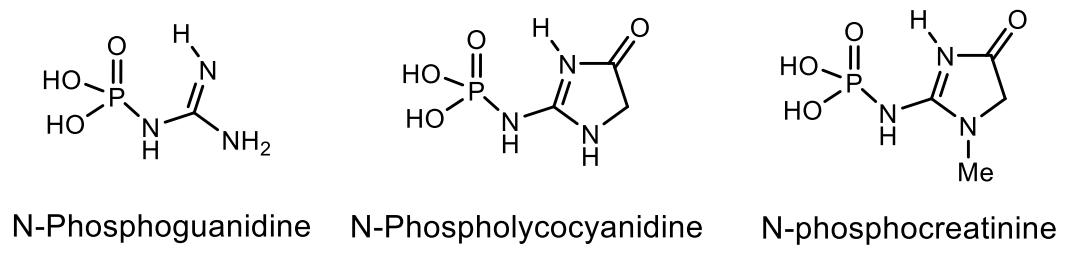
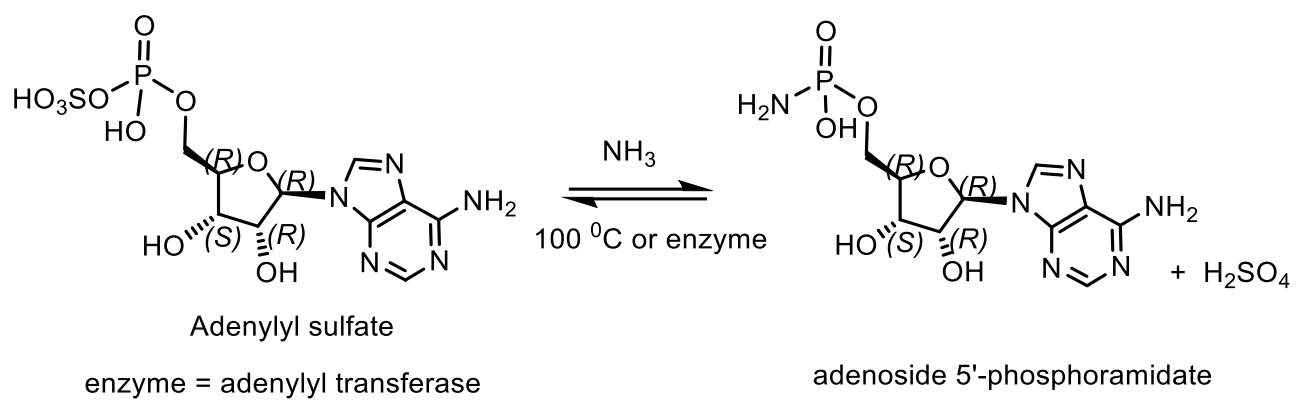
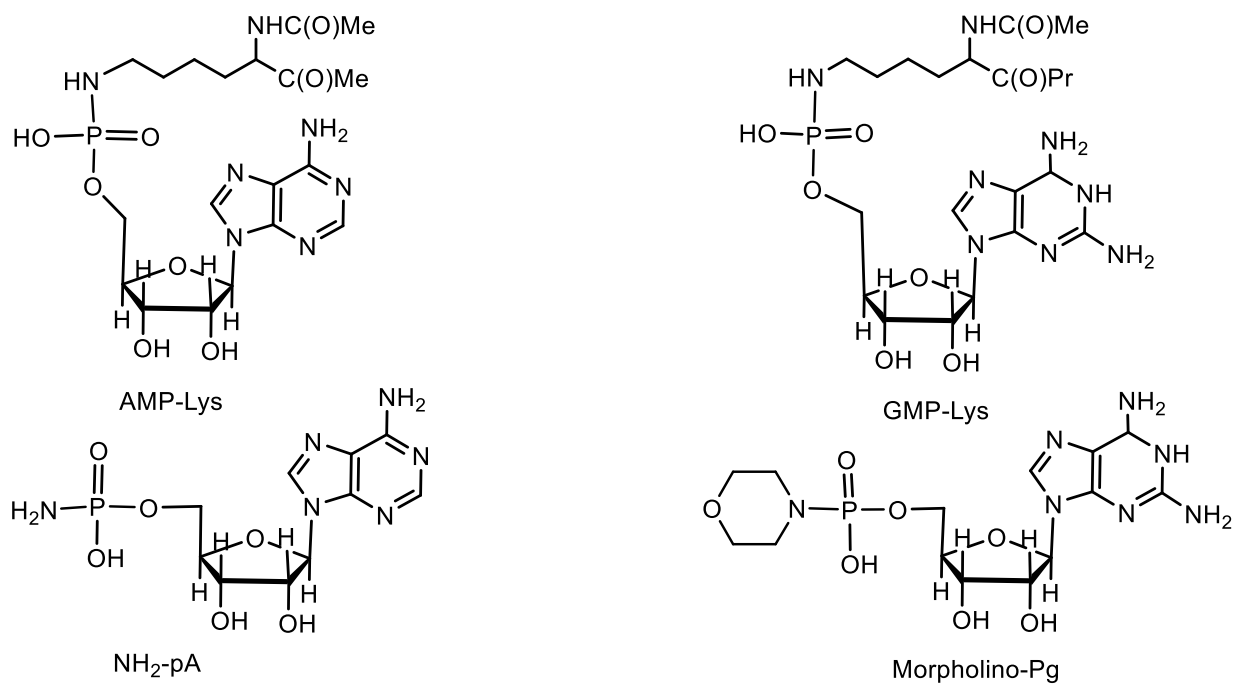
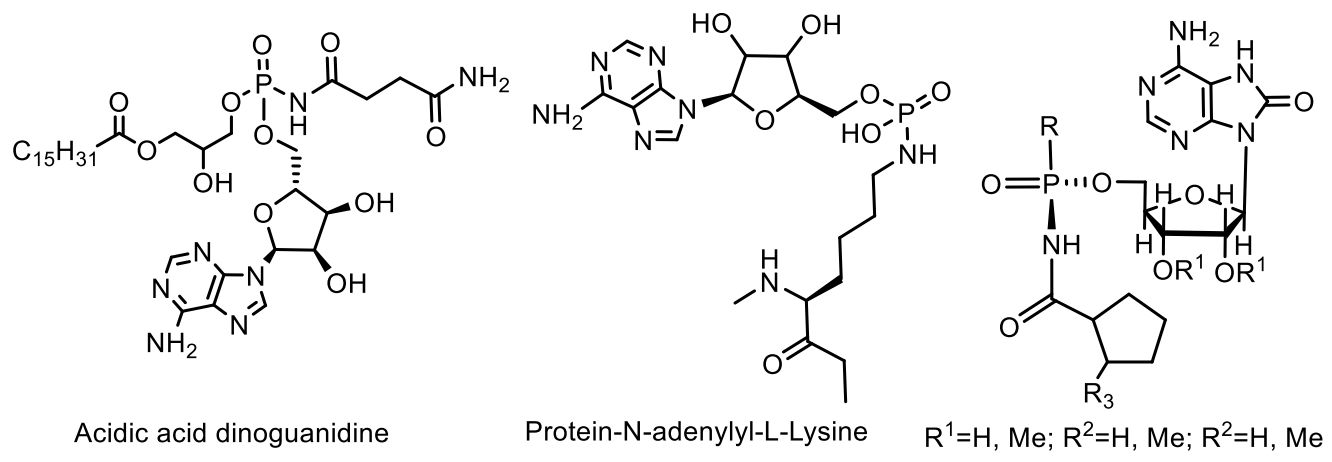
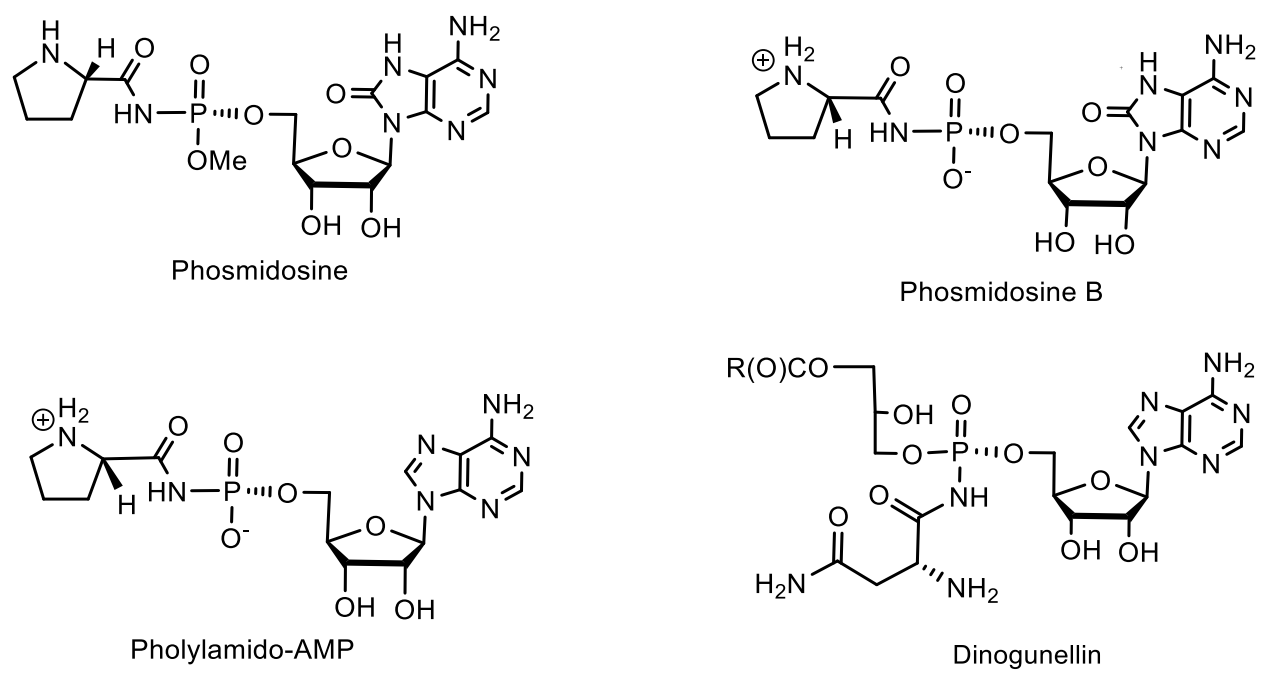
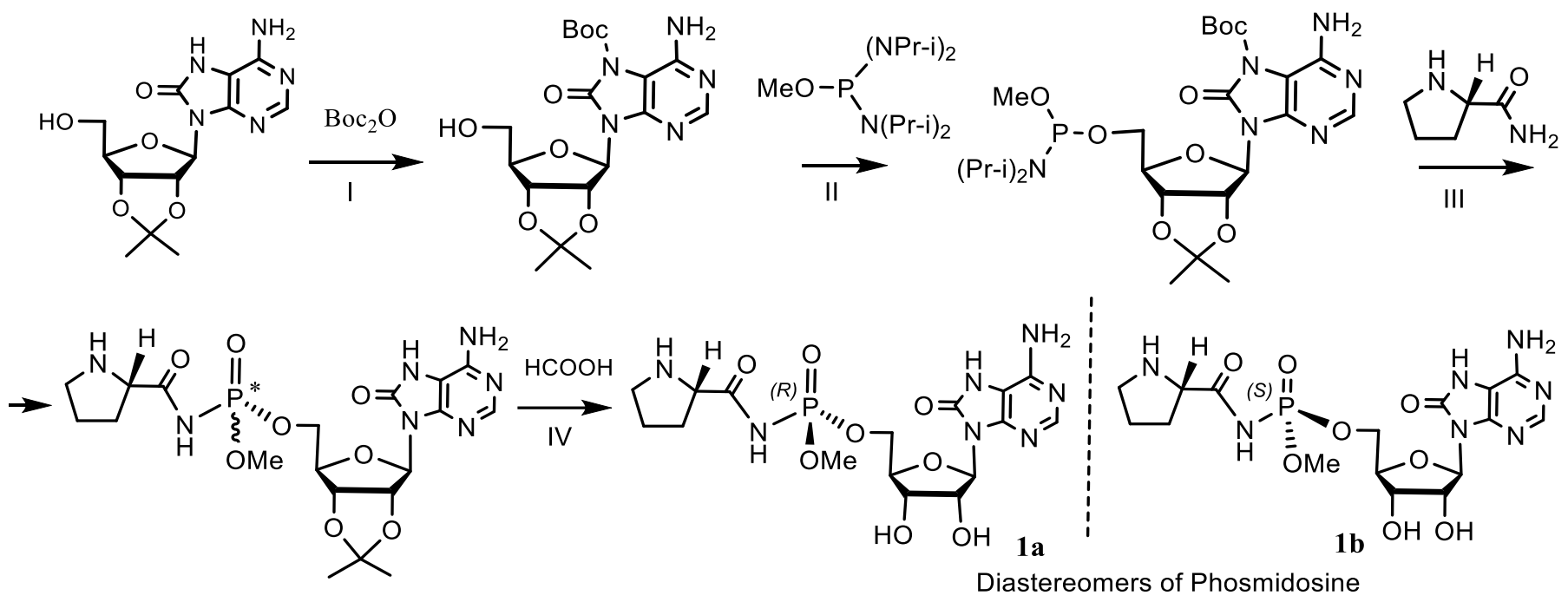
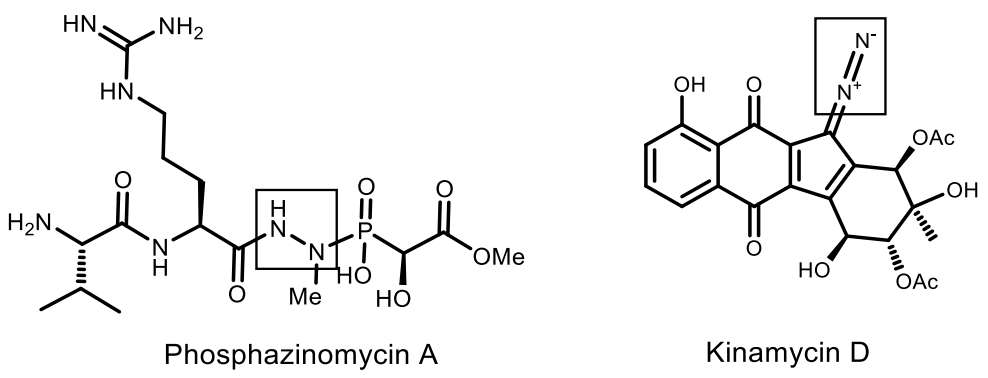
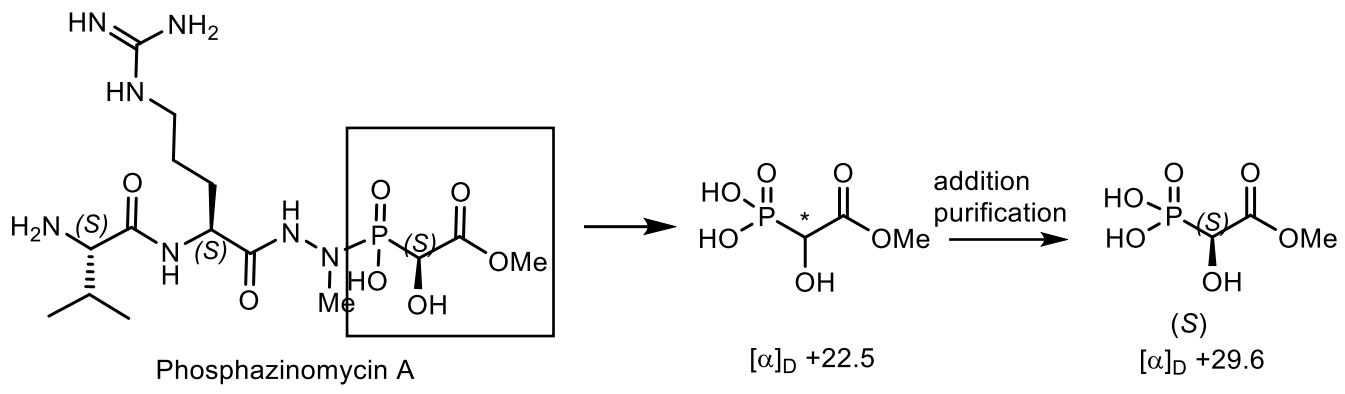
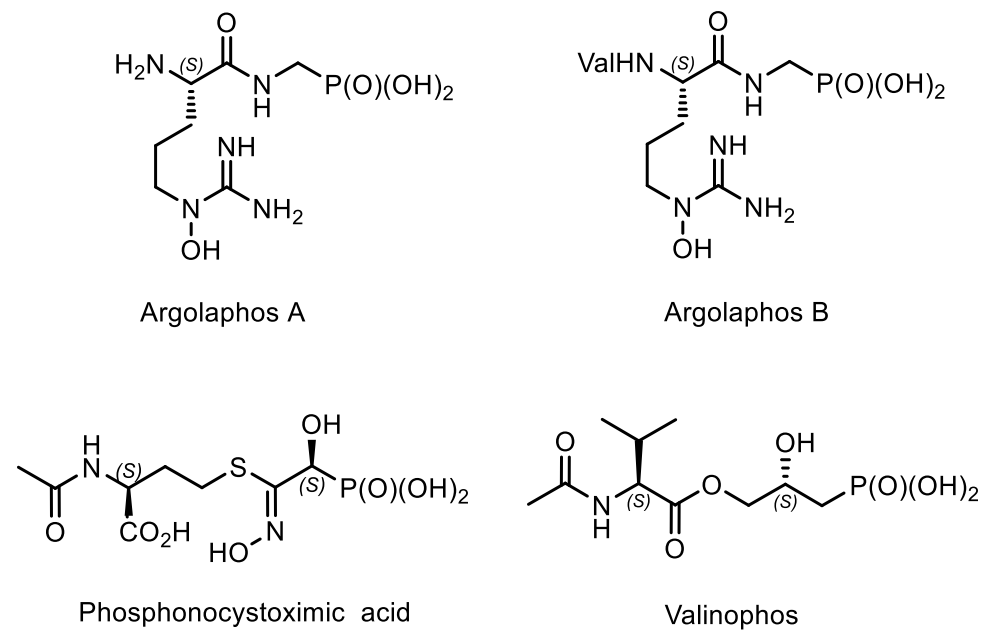
5. Natural Compounds with P-N Bonds
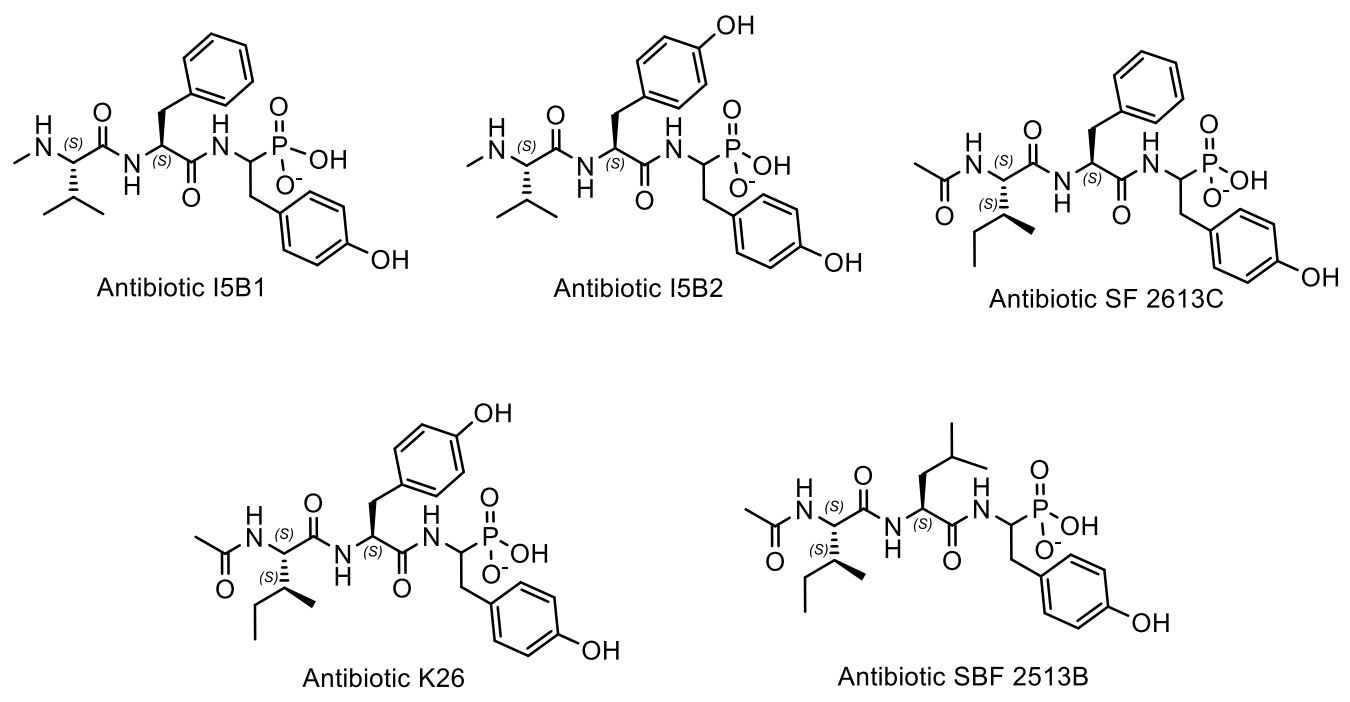
6. Natural Organophosphorus Compounds with P-C Bond
7. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Fernández-García, C.; Coggins, A.J.; Powner, M.W. A Chemist’s Perspective on the Role of Phosphorus at the Origins of Life. Life 2017, 7, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, W.; Abdillah, A.; Palma, J.; Churchill, D.G. Introductory Chapter: Phosphorus—Nature’s Versatile Pentavalent and Tetrahedral Covalent Building Block and Agent for Energy, Disease and Health. In Contemporary Topics about Phosphorus in Biology and Materials; Intech Open: London, UK, 2020; pp. 1–17. [Google Scholar] [CrossRef]
- Lazcano, A.; Miller, S.L. The origin and early evolution of life: Prebiotic chemistry, the pre-RNA world, and time. Cell 1996, 85, 793–798. [Google Scholar] [CrossRef] [Green Version]
- Butusov, M.; Jerneliiv, A. Chapter 1. The Role of Phosphorus in the Origin of Life and in Evolution Phosphorus. In An Element that Could Have Been Called Lucifer; Springer Briefs in Environmental Science: New York, NY, USA, 2013; Volume 9, pp. 1–12. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Rossi, J.-C.; Pascal, R. How Prebiotic Chemistry and Early Life Chose Phosphate. Life 2019, 9, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Westheimer, F. Why nature chose phosphates. Science 1987, 235, 1173–1178. [Google Scholar] [CrossRef] [PubMed]
- Altstein, A.D. Origin of Biological Nucleic Acids and the First Genetic System (The Progene Hypothesis). Transcriptomics 2017, 5, 139–143. [Google Scholar] [CrossRef] [Green Version]
- Goldford, J.E.; Hartman, H.; Smith, T.F.; Segrè, D. Remnants of an ancient metabolism without phosphate. Cell 2017, 168, 1126–1134.e9. [Google Scholar] [CrossRef] [Green Version]
- Mohammed, F.; Chen, K.; Mojica, M.; Conley, M.; Napoline, J.; Butch, C.; Pollet, P.; Krishnamurthy, R.; Liotta, C. A plausible prebiotic origin of glyoxylate: Nonenzymatic transamination reactions of glycine with formaldehyde. Synlett 2016, 28, 93–97. [Google Scholar] [CrossRef] [Green Version]
- Bean, H.D.; Anet, F.A.L.; Gould, I.R.; Hud, N.V. Glyoxylate as a backbone linkage for a prebiotic ancestor of RNA. Orig. Life Evol. Biol. 2006, 36, 39–63. [Google Scholar] [CrossRef]
- Nelson, K.E.; Levy, M.; Miller, S.L. Peptide nucleic acids rather than RNA may have been the first genetic molecule. Proc. Natl. Acad. Sci. USA 2000, 97, 3868–3871. [Google Scholar] [CrossRef] [Green Version]
- Schwartz, A.W. Phosphorus in prebiotic chemistry. Philos. Trans. R. Soc. B. 2006, 361, 1743–1749. [Google Scholar] [CrossRef] [Green Version]
- Dass, A.V.; Jaber, M.; Brack, A.; Foucher, F.; Kee, T.P.; Georgelin, T.; Westall, F. Potential Role of Inorganic Confined Environments in Prebiotic Phosphorylation. Life 2018, 8, 7. [Google Scholar] [CrossRef] [Green Version]
- Pasek, M.A.; Lauretta, D.S. Aqueous corrosion of phosphide minerals from iron meteorites: A highly reactive source of prebiotic phosphorus on the surface of the early earth. Astrobiology 2005, 5, 515–535. [Google Scholar] [CrossRef]
- Bryant, D.E.; Kee, T.P. Direct evidence for the availability of reactive, water soluble phosphorus on the early Earth. H-Phosphinic acid from the Nantan meteorite. Chem. Commun. 2006, 22, 2344–2346. [Google Scholar] [CrossRef]
- Pasek, M.A. Schreibersite on the early earth: Scenarios for prebiotic Phosphorylation. Geosci. Front. 2017, 8, 329–335. [Google Scholar] [CrossRef] [Green Version]
- Kolodiazhnyi, O.I. Stereochemistry of electrophilic and nucleophilic substitutions at phosphorus. Pure Appl. Chem. 2019, 91, 43–57. [Google Scholar] [CrossRef]
- Pasek, M.A.; Kee, T.P.; Bryant, D.E.; Pavlov, A.A.; Lunine, J.I. Production of potentially prebiotic condensed phosphates by phosphorus redox chemistry. Angew. Chem. Int. Ed. 2008, 47, 7918–7920. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Liu, Y.; Gao, X.; Xu, P. The international background of the origin of life. In Phosphorus Chemistry; Walter de Gruyter GmbH: Berlin, Germany, 2019; pp. 1–18. [Google Scholar] [CrossRef]
- Hall, B.G.; Zuzel, T. Evolution of a new enzymatic function by recombination within a gene. Proc. Natl. Acad. Sci. USA 1980, 77, 3529–3533. [Google Scholar] [CrossRef] [Green Version]
- Eldredge, N. Life Pulse Episodes from the Story of the Fossil Record; Pelican Edition: London, UK; New York, NY, USA, 1987. [Google Scholar]
- Skulachev, V.P.; Bogachev, A.V.; Kasparinsky, F.O. Principles of Bioenergetics; Springer Science & Business Media: Berlin/Heidelberg, Germany, 2012; p. 252. ISBN 978-3-642-33430-6. [Google Scholar]
- Bustamante, C.; Bryant, Z.; Smith, S.B. Ten years of tension: Single-molecule DNA mechanics. Nature 2003, 421, 423–427. [Google Scholar] [CrossRef]
- Bruce, A.; Johnson, A.; Lewis, J.; Raff, M.; Roberts, K.; Walters, P. Molecular Biology of the Cell, 4th ed.; Alberts, B., Ed.; Garland Science: New York, NY, USA, 2002; p. 1400, ISBN-13: 978-081533218, ISBN-10: 0815332181. [Google Scholar]
- Ussery, D.W. DNA Structure: A-, B- and Z-DNA Helix Families. In Encyclopedia of Life Sciences; Published Online; John Wiley & Sons Ltd.: Hoboken, NJ, USA, 2002; pp. 1–7. [Google Scholar] [CrossRef]
- Tomassi, S.; Montalban, F.F.; Russo, R.; Novellino, E.; Messere, A.; Di Maro, S. Investigation of the Stereochemical-Dependent DNA and RNA Binding of Arginine-Based Nucleopeptides. Symmetry 2019, 11, 567. [Google Scholar] [CrossRef] [Green Version]
- Hickman, A.B.; Dyda, F. DNA Transposition at Work. Chem. Rev. 2016, 116, 12758–12784. [Google Scholar] [CrossRef]
- Liu, J.; Declais, A.-C.; McKinney, S.A.; Ha, T.; Norman, D.G.; Lilley, D.M.J. Stereospecific Effects Determine the Structure of a Four-Way DNA Junction. Chem. Biol. 2005, 12, 217–228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lilley, D.M.J. Structures of helical junctions in nucleic acids. Q. Rev. Biophys. 2000, 33, 109–159. [Google Scholar] [CrossRef] [PubMed]
- Askari, M.S.; Lachance-Brais, C.; Rizzuto, F.J.; Toader, V.; Sleiman, H. Remote control of charge transport and chiral induction along a DNA-metallohelicate. Nanoscale 2019, 11, 11879–11884. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Benedetti, E.; Bethge, L.; Vonhoff, S.; Klussmann, S.; Vasseur, J.-J.; Cossy, J.; Smietana, M.; Arseniyadis, S. DNA vs. Mirror Image DNA: A Universal Approach to Tune the Absolute Configuration in DNA Based Asymmetric Catalysis. Angew. Chem. Int. Ed. 2013, 52, 11546–11549. [Google Scholar] [CrossRef]
- Roelfes, G.; Feringa, B.L. DNA-Based Asymmetric Catalysis. Angew. Chem. Int. Ed. 2005, 44, 3230–3232. [Google Scholar] [CrossRef]
- Coquire, D.; Feringa, B.L.; Roelfes, G. DNA-Based Catalytic Enantioselective Michael Reactions in Water. Angew. Chem. Int. Ed. Engl. 2007, 46, 9308–9311. [Google Scholar] [CrossRef]
- Li, Y.; Wang, C.; Jia, G.; Lu, S.; Li, C. Enantioselective Michael addition reactions in water using a DNA-based catalyst. Tetrahedron 2013, 69, 6585–6590. [Google Scholar] [CrossRef]
- Qiu, H.; Gilroya, J.B.; Manners, I. DNA-induced chirality in water-soluble poly (cobaltoceniumethylene). Chem. Commun. 2013, 49, 42–44. [Google Scholar] [CrossRef]
- Boersma, A.J.; Feringa, B.L.; Roelfes, G. Enantioselective Friedel–Crafts Reactions in Water Using a DNA Based Catalyst. Angew. Chem. Int. Ed. Engl. 2009, 48, 3346–3348. [Google Scholar] [CrossRef]
- Park, S.; Ikehata, K.; Watabe, R.; Hidaka, Y.; Rajendran, A.; Sugiyama, H. Deciphering DNA-based asymmetric catalysis through intramolecular Friedel–Crafts alkylations. Chem. Commun. 2012, 48, 10398–10400. [Google Scholar] [CrossRef] [Green Version]
- Shibata, N.; Yasui, H.; Nakamura, S.; Toru, T. DNA-Mediated Enantioselective Carbon–Fluorine Bond Formation. Synlett 2007, 7, 1153–1157. [Google Scholar] [CrossRef]
- Boersma, A.J.; Coquiere, D.; Geerdink, D.; Rosati, F.; Feringa, B.L.; Roelfes, G. Catalytic enantioselective syn hydration of enones in water using a DNA-based catalyst. Nat. Chem. 2010, 2, 991–995. [Google Scholar] [CrossRef] [Green Version]
- Boersma, A.J.; Megens, R.P.; Feringa, B.L.; Roelfes, G. DNA-based asymmetric catalysis. Chem. Soc. Rev. 2010, 39, 2083–2092. [Google Scholar] [CrossRef] [Green Version]
- Ellington, W.R. Evolution and Physiological Roles of Phosphagen Systems. Annu. Rev. Physiol. 2001, 63, 289–325. [Google Scholar] [CrossRef]
- Webster, C.E. High-Energy Intermediate or Stable Transition State Analogue: Theoretical Perspective of the Active Site and Mechanism of β-Phosphoglucomutase. J. Am. Chem. Soc. 2004, 126, 6840–6841. [Google Scholar] [CrossRef]
- Yamamoto, T. Preferred dissociative mechanism of phosphate monoester hydrolysis in low dielectric environments. Chem. Phys. Lett. 2010, 500, 263–266. [Google Scholar] [CrossRef]
- Lassila, J.K.; Zalatan, J.G.; Herschlag, D. Biological phosphoryl-transfer reactions: Understanding mechanism and catalysis. Annu. Rev. Biochem. 2011, 80, 669–702. [Google Scholar] [CrossRef] [Green Version]
- Westheimer, F.H. Monomeric metaphosphates. Chem. Rev. 1981, 81, 313–326. [Google Scholar] [CrossRef]
- Keglevich, G.; Kovács, T.; Csatlós, F. The Deoxygenation of Phosphine Oxides under Green Chemical Conditions. Heteroat. Chem. 2015, 26, 199–205. [Google Scholar] [CrossRef]
- Kolodiazhnyi, O.I.; Kolodiazhna, A.O. Nucleophilic substitution at phosphorus: Stereochemistry and Mechanisms. Tetrahedron Asymmetry 2017, 28, 1651–1674. [Google Scholar] [CrossRef]
- Kolodiazhna, A.O.; Kolodiazhnyi, O.I. Asymmetric Electrophilic Reactions in Phosphorus Chemistry. Symmetry 2020, 12, 108. [Google Scholar] [CrossRef] [Green Version]
- Duarte, F.; Barrozo, A.; Åqvist, J.; Williams, N.H.; Kamerlin, S.C.L. The Competing Mechanisms of Phosphate Monoester Dianion Hydrolysis. J. Am. Chem. Soc. 2016, 138, 10664–10673. [Google Scholar] [CrossRef] [Green Version]
- Duarte, F.; Åqvist, J.; Williams, N.H.; Kamerlin, S.C.L. Resolving Apparent Conflicts between Theoretical and Experimental Models of Phosphate Monoester Hydrolysis. J. Am. Chem. Soc. 2015, 137, 1081–1093. [Google Scholar] [CrossRef]
- Bhengu, T.T.; Wood, T.P.; Shumane, M.; Tafesse, F. Detection of metaphosphate intermediates in reaction solutions of phosphate esters: Chromatographic and spectroscopic studies. Phosph. Sulf. Silicon Relat. Elem. 2017, 192, 1144–1152. [Google Scholar] [CrossRef]
- Henchman, M.; Viggiano, A.A.; Paulson, J.F.; Freedman, A.; Wormhoudt, J. Thermodynamic and kinetic properties of the metaphosphate anion, PO3-, in the gas phase. J. Am. Chem. Soc. 1985, 107, 1453–1455. [Google Scholar] [CrossRef]
- Range, K.; McGrath, M.J.; Lopez, X.; York, D.M. The Structure and Stability of Biological Metaphosphate, Phosphate, and Phosphorane Compounds in the Gas Phase and in Solution. J. Am. Chem. Soc. 2004, 126, 1654–1665. [Google Scholar] [CrossRef]
- Lahiri, S.D.; Zhang, G.; Dunaway-Mariano, D.; Allen, K.N. The Pentacovalent Phosphorus Intermediate of a Phosphory Transfer Reaction. Science 2003, 299, 2067–2071. [Google Scholar] [CrossRef] [Green Version]
- Buchwald, S.L.; Friedman, J.M.; Knowles, J.R. Stereochemistry of Nucleophilic Displacement on Two Phosphoric Monoesters and a Phosphoguanidine: The Role of Metaphosphate. J. Am. Chem. Soc. 1984, 106, 4911–4916. [Google Scholar] [CrossRef]
- Catrina, I.E.; Czyryca, P.G.; Hengge, A.C. Isotope effects on enzymatic and nonenzymatic reactions of phosphorothioates. Nukleonika 2002, 47, S17–S23. [Google Scholar]
- Cullis, P.M.; Nicholls, D. The existence of monomeric metaphosphate in hydroxylic solvent: A positional isotope exchange study. J. Chem. Soc. Chem. Commun. 1987, 10, 783–785. [Google Scholar] [CrossRef]
- Kiani, F.A.; Fischer, S. Catalytic strategy used by the myosin motor to hydrolyze ATP. Proc. Natl. Acad. Sci. USA 2014, 111, E2947–E2956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedman, J.M.; Freeman, S.; Knowles, J.R. The quest for free metaphosphate in solution. Racemization at phosphorus in the transfer of the phospho group from aryl phosphate monoesters to tert-butyl alcohol in acetonitrile or in tert-butyl alcohol. J. Am. Chem. Soc. 1988, 110, 1268–1275. [Google Scholar] [CrossRef]
- Choe, J.Y.; Iancu, C.V.; Fromm, H.J.; Honzatko, R. Metaphosphate in the active site of fructose-1,6-bisphosphatase B. J. Biol. Chem. 2003, 278, 16015–16020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molecular System Bioenergetics: Energy for Life; Saks, V. (Ed.) Wiley-VCH: Weinheim, Germany, 2007; p. 633. ISBN 978-3-527-31787-5. [Google Scholar]
- Wyss, M.; Kaddurah-Daouk, R. Creatine and Creatinine Metabolism. Physiol. Rev. 2000, 80, 1107–1213. [Google Scholar] [CrossRef]
- Iyengar, M.R.; Coleman, D.W.; Butler, T.M. Phosphocreatinine, a high-energy phosphate in muscle, spontaneously forms phosphocreatine and creatinine under physiological conditions. J. Biol. Chem. 1985, 260, 7562–7567. [Google Scholar] [CrossRef]
- VanWagenen, B.C.; Larsen, R.; Cardellina, J.H.; Randazzo, D.; Lidert, Z.C.; Swithenbank, C. Ulosantoin, a potent insecticide from the sponge Ulosa ruetzleri. J. Org. Chem. 1993, 58, 335–337. [Google Scholar] [CrossRef]
- Karki, M.; Gibard, C.; Bhowmik, S.; Krishnamurthy, R. Nitrogenous Derivatives of Phosphorus and the Origins of Life: Plausible Prebiotic Phosphorylating Agents in Water. Life 2017, 7, 32. [Google Scholar] [CrossRef] [Green Version]
- Krishnamurthy, R.; Guntha, S.; Eschenmoser, A. Regioselective ex-phosphorylation of aldoses in aqueous solution. Angew. Chem. Int. Ed. Engl. 2000, 39, 2281–2285. [Google Scholar] [CrossRef]
- Anastasi, C.; Buchet, F.F.; Crowe, M.A.; Helliwell, M.; Raftery, J.; Sutherland, J.D. The Search for a Potentially Prebiotic Synthesis of Nucleotides via Arabinose-3-phosphate and Its Cyanamide Derivative. Chem. Eur. J. 2008, 14, 2375–2388. [Google Scholar] [CrossRef]
- Fankhauser, H.; Schiff, J.A.; Garber, L.J. Purification and properties of adenylyl sulphate:ammonia adenylyltransferase from Chlorella catalysing the formation of adenosine 50-phosphoramidate from adenosine 50-phosphosulphate and ammonia. Biochem. J. 1981, 195, 545–560. [Google Scholar] [CrossRef] [Green Version]
- Fankhauser, H.; Berkowitz, G.A.; Schiff, J.A. A nucleotide with the properties of adenosine 50 phosphoramidate from Chlorella cells. Biochem. Biophys. Res. Commun. 1981, 101, 524–532. [Google Scholar] [CrossRef]
- Wojdyla-Mamon, A.M.; Guranowski, A. Adenylylsulfate-ammonia adenylyltransferase activity is another inherent property of Fhit proteins. Biosci. Rep. 2015, 35, e00235–e00243. [Google Scholar] [CrossRef]
- Guranowski, A.; Wojdyla, A.M.; Rydzik, A.M.; Stepinski, J.; Jemielity, J. Plant nucleoside 5’-phosphoramidate hydrolase; simple purification from yellow lupin (Lupinus luteus) seeds and properties of homogeneous enzyme. Acta Biochim. Pol. 2011, 58, 131–136. [Google Scholar] [CrossRef] [Green Version]
- Bretes, E.; Wojdyla-Mamon, A.M.; Kowalska, J.; Jemielity, J.; Kaczmarek, R.; Baraniak, J.; Guranowski, A. Hint2, the mitochondrial nucleoside 50-phosphoramidate hydrolase; properties of the homogeneous protein from sheep (Ovis aries) liver. Acta Biochim. Pol. 2013, 60, 249–254. [Google Scholar] [CrossRef] [Green Version]
- Rossomando, E.F.; Hadjimichael, J. Characterization and cAMP inhibition of a lysyl-(N-ε-5′-phospho) adenosyl phosphoamidase in Dictyostelium discoideum. Int. J. Biochem. 1986, 18, 481–484. [Google Scholar] [CrossRef]
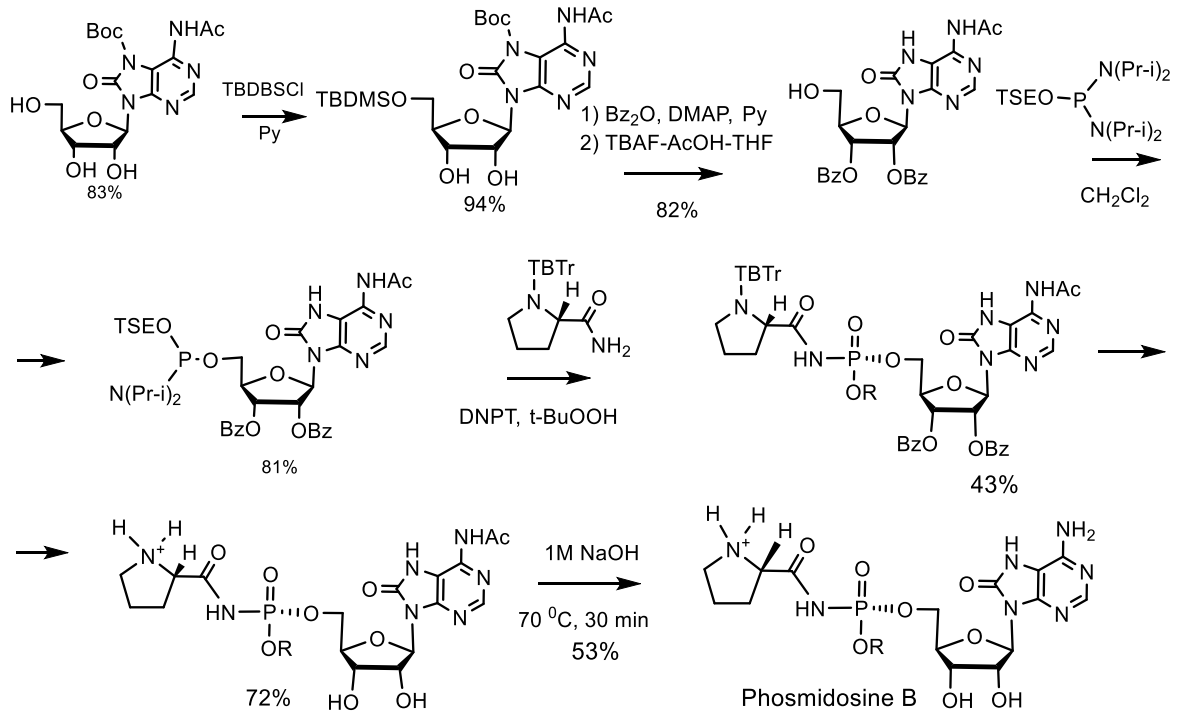
- Moriguchi, T.; Asai, N.; Okada, K.; Seio, K.; Sasaki, T.; Sekine, M. First synthesis and anticancer activity of phosmidosine and its related compounds. J. Org. Chem. 2002, 67, 3290–3300. [Google Scholar] [CrossRef]
- Ozga, M.; Dolot, R.; Janicka, M.; Kaczmarek, R.; Krakowiak, A. Histidine Triad Nucleotide-binding Protein 1 (HINT-1) Phosphoramidase Transforms Nucleoside 5′-O-Phosphorothioates to Nucleoside 5′-O-Phosphates. J. Biol. Chem. 2010, 285, 40809–40818. [Google Scholar] [CrossRef] [Green Version]
- Brenner, C. Hint, Fhit, GalT: Function, structure, evolution, and mechanism of three branches of the histidine triad superfamily of nucleotide hydrolases and transferases. Biochemistry 2002, 41, 9003–9014. [Google Scholar] [CrossRef] [Green Version]
- Bieganowski, P.; Garrison, P.N.; Hodawadekar, S.C.; Faye, G.; Barnes, L.D.; Brenner, C. Adenosine monophosphoramidase activity of Hint and Hnt1 supports function of Kin28, Ccl1, and Tfb3. J. Biol. Chem. 2002, 277, 10852–10860. [Google Scholar] [CrossRef] [Green Version]
- Hadjimichael, J.; Rossomando, E.F. Isolation and characterization of the protein phosphoamidates formed by a membrane bound adenylyl transferase reaction in Dictyostelium discoideum. Int. J. Biochem. 1991, 23, 535–539. [Google Scholar] [CrossRef]
- Hatano, M.; Hashimoto, Y. Properties of a toxic phospholipid in the northern blenny roe. Toxicon 1974, 12, 231–236. [Google Scholar] [CrossRef]
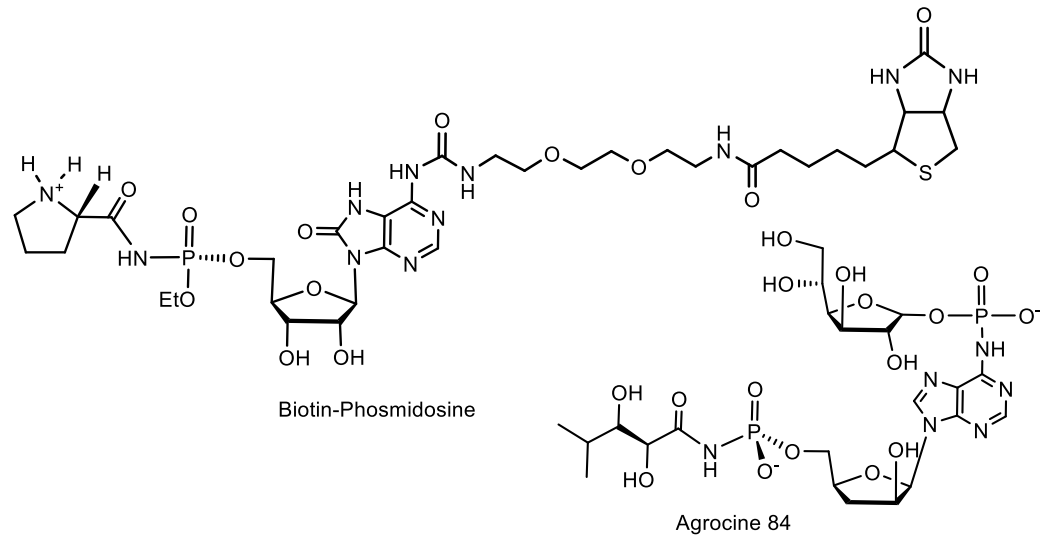
- Sekine, M.; Moriguchi, T.; Wada, T.; Seio, K. Total Synthesis of Agrocin 84 and Phosmidosine as Naturally Occurring Nucleotidic Antibiotics Having P-N Bond Linkages. J. Synth. Org. Chem. Jpn. 2001, 59, 1109–1120. [Google Scholar] [CrossRef] [Green Version]
- Phillips, D.R.; Uramoto, M.; Isono, K.; McCloskey, J.A. Structure of the antifungal nucleotide antibiotic phosmidosine. J. Org. Chem. 1993, 58, 854–859. [Google Scholar] [CrossRef]
- Roberts, W.P.; Tate, M.E.; Kerr, A. Agrocin 84 is a 6-N-phosphoramidate of an adenine nucleotide analogue. Nature 1977, 265, 379–381. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-G.; Park, B.K.; Kim, S.-U.; Choi, D.; Nahm, B.H.; Moon, J.S. Bases of biocontrol: Sequence predicts synthesis and mode of action of agrocin 84, the Trojan Horse antibiotic that controls crown gall. Proc. Natl. Acad. Sci. USA 2006, 103, 8846–8851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pertusati, F.; McGuigana, C. Diastereoselective synthesis of P-chirogenic phosphoramidate prodrugs of nucleoside analogues (ProTides) via copper catalysed reaction. Chem. Commun. 2015, 51, 8070–8073. [Google Scholar] [CrossRef] [Green Version]
- Tate, M.E.; Murphy, P.J.; Roberts, W.P.; Kerr, A. Adenine N6-substituent of agrocin 84 determines its bacteriocin-like specificity. Nature 1979, 280, 697–699. [Google Scholar] [CrossRef] [PubMed]
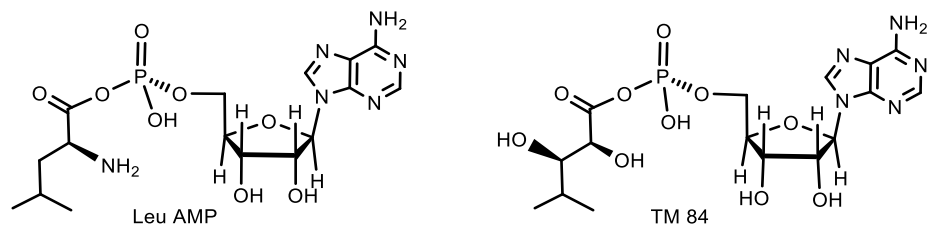
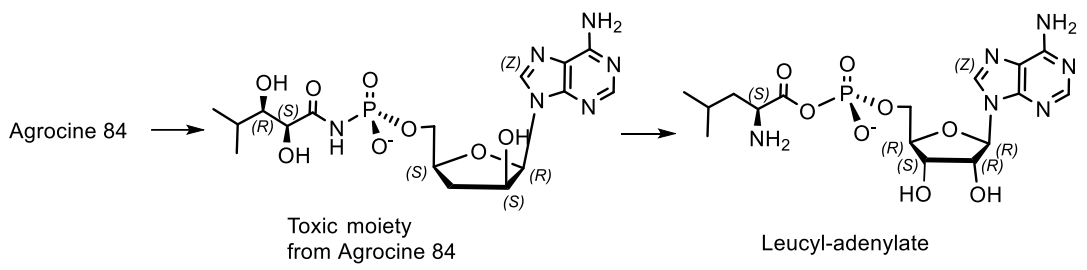
- Reader, J.S.; Ordoukhanian, P.T.; Kim, J.-G.; de Crecy-Lagard, V.; Hwang, I.; Farrand, S.; Schimmel, P. Major Biocontrol of Plant Tumors Targets tRNA Synthetase. Science 2005, 309, 1533 LP–1553 LP. [Google Scholar] [CrossRef] [Green Version]
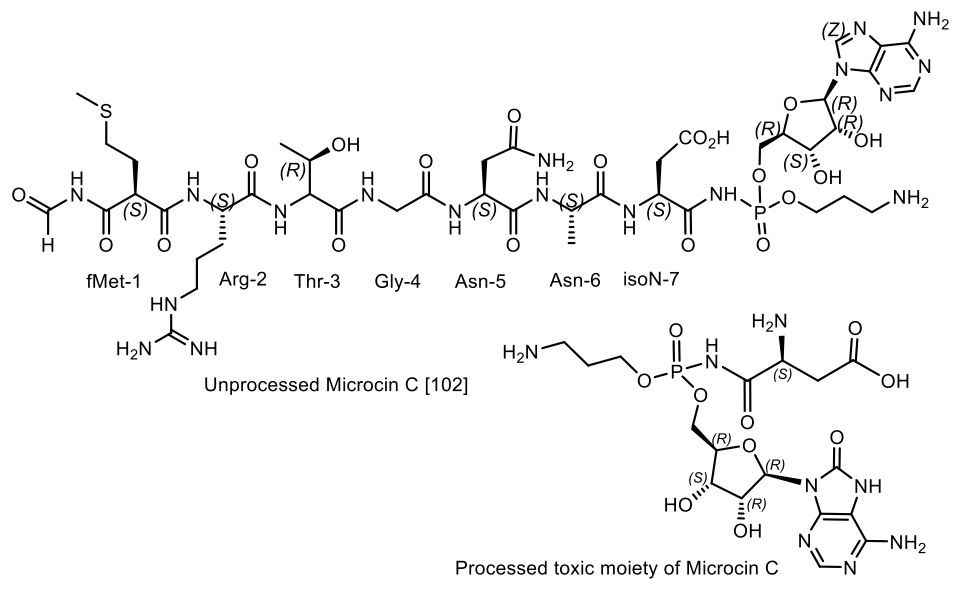
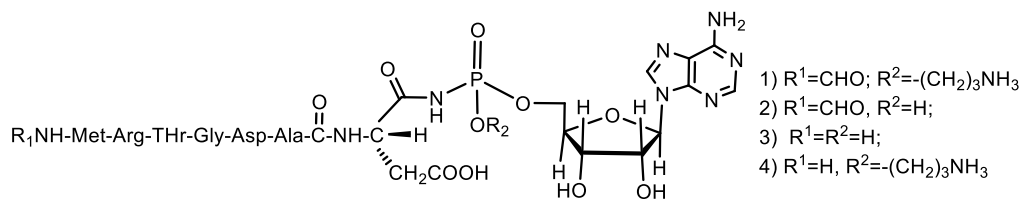
- Chopra, S.; Palencia, A.; Virus, C.; Tripathy, A.; Temple, B.R.; Velazquez-Campoy, A.; Cusack, S.; Reader, J.S. Plant tumour biocontrol agent employs a tRNA-dependent mechanism to inhibit leucyl-tRNA synthetase. Nat. Commun. 2013, 4, 1417–1430. [Google Scholar] [CrossRef] [Green Version]
- Chopra, S.; Palencia, A.; Virus, C.; Schulwitz, S.; Temple, B.R.; Cusack, S.; Reader, J. Structural characterization of antibiotic self-immunity tRNA synthetase in plant tumour biocontrol agent. Nat. Commun. 2016, 7, 12928–12941. [Google Scholar] [CrossRef] [Green Version]
- Taguchi, H.; Ohkubo, A.; Sekine, M.; Seio, K.; Kakeya, H.; Osada, H. Synthesis and Biological Properties of New Phosmidosine Analogs Having an N-Acylsulfamate. Nucleosides Nucleotides Nucleic Acids 2006, 25, 647–654. [Google Scholar] [CrossRef]
- Sekine, M.; Okada, K.; Seio, K.; Kakeya, H.; Osada, H.; Obata, T.; Sasaki, T. Synthesis of Chemically Stabilized Phosmidosine Analogues and the Structure-Activity Relationship of Phosmidosine. J. Org. Chem. 2004, 69, 314–326. [Google Scholar] [CrossRef]
- Sekine, M.; Okada, K.; Seio, K.; Obata, T.; Sasaki, T.; Kakeya, H.; Osada, H. Synthesis of a biotin-conjugate of phosmidosine O-ethyl ester as a G1 arrest antitumor drug. Bioorganic. Med. Chem. 2004, 12, 6343–6349. [Google Scholar] [CrossRef]
- Paz-Yepes, J.; Brahamsha, B.; Palenik, B. Role of a microcin-C-like biosynthetic gene cluster in allelopathic interactions in marine Synechococcus. Proc. Natl. Acad. Sci. USA 2013, 110, 12030–12035. [Google Scholar] [CrossRef] [Green Version]
- Hengralph, N.C.K.; Jack, W. Chapter 13—Microcins. In Handbook of Biologically Active Peptides; Kastin, A.J., Ed.; Academic Press: Cambridge, MA, USA, 2006; pp. 75–81. [Google Scholar] [CrossRef]
- Rebuffat, S. Chapter 20—Microcins. In Handbook of Biologically Active Peptides: Bacterial/Antibiotic Peptides, 2nd ed.; Kastin, A., Ed.; Academic Press: Boston, MA, USA, 2013; pp. 129–137. ISBN 978-0-12-385095-9. [Google Scholar]
- Kazakov, T.; Vondenhoff, G.H.; Datsenko, K.A.; Novikova, M.; Metlitskaya, A.; Wanner, B.L.; Severinov, K. Escherichia coli Peptidase A, B, or N Can Process Translation Inhibitor Microcin, C. J. Bacteriol. 2008, 190, 2607–2610. [Google Scholar] [CrossRef] [Green Version]
- Baquero, F.; Lanza, V.F.; Baquero, M.-R.; del Campo1, R.; Bravo-Vázquez, D.A. Microcins in Enterobacteriaceae: Peptide Antimicrobials in the Eco-Active Intestinal Chemosphere. Front. Microbiol. 2019, 10, 2261. [Google Scholar] [CrossRef]
- Vondenhoff, G.H.M.; Van Aerschot, A. Microcin C: Biosynthesis, Mode of Action, and Potential as a Lead in cAntibiotics Development. Nucleosides Nucleotides Nucleic Acids 2011, 30, 465–474. [Google Scholar] [CrossRef]
- Kulikovsky, A.; Tsibulskaya, D.; Piskunova, J.; Severinov, K.; Serebryakva, M.; Dubiley, S. Microcin C-like peptide-adenylate antibiotic from Rubidus massiliensis. In FEBS Open Bio; WILEY: Hoboken, NJ, USA, 2018; Volume 8, p. 280. [Google Scholar]
- Tsibulskaya, D.; Mokina, O.; Kulikovsky, A.; Piskunova, J.; Severinov, K.; Serebryakova, M.; Dubiley, S. The Product of Yersinia pseudotuberculosis mcc Operon Is a Peptide-Cytidine Antibiotic Activated Inside Producing Cells by the TldD/E Protease. J. Am. Chem. Soc. 2017, 139, 16178–16187. [Google Scholar] [CrossRef]
- Duquesne, S.; Destoumieux-Garzón, D.; Peduzzi, J.; Rebuffat, S. Microcins, gene-encoded antibacterial peptides from enterobacteria. Nat. Prod. Rep. 2007, 24, 708–734. [Google Scholar] [CrossRef]
- Serebryakova, M.; Tsibulskaya, D.; Mokina, O.; Kulikovsky, A.; Nautiyal, M.; Van Aerschot, A.; Severinov, K.; Dubiley, S. A Trojan-Horse Peptide-Carboxymethyl-Cytidine Antibiotic from Bacillus amyloliquefaciens. J. Am. Chem. Soc. 2016, 138, 15690–15698. [Google Scholar] [CrossRef]
- Severinov, K.; Nair, S.K. Microcin C: Biosynthesis and mechanisms of bacterial resistance. Future Microbiol. 2012, 7, 281–289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
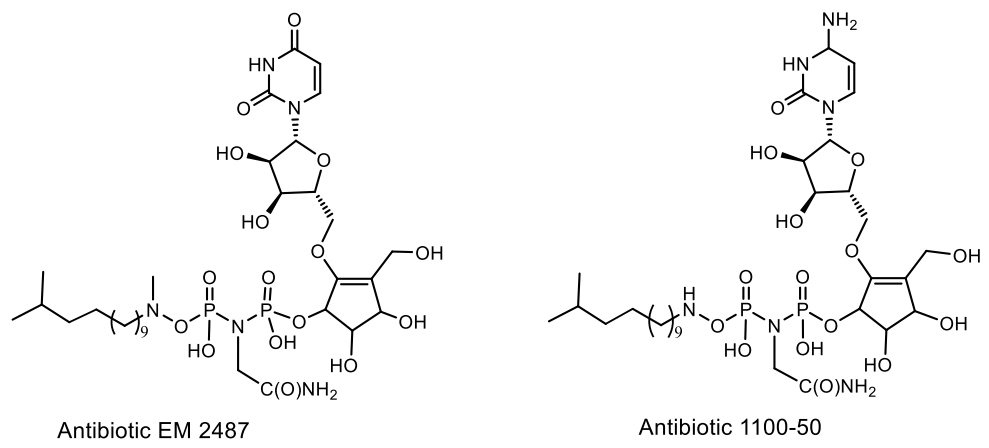
- Takatsu, T.; Horiuchi, N.; Ishikawa, M.; Wanibuchi, K.; Moriguchi, T.; Takahashi, S. 1100-50, a novel nematocide from Streptomyces lavendulae SANK 64297. J. Antibiot. 2003, 56, 306–309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baba, M.; Okamoto, M.; Takeuchi, H. Inhibition of human immunodeficiency virus type 1 replication in acutely and chronically infected cells by EM2487, a novel substance produced by a Streptomyces species. Antimicrob. Agents Chemother. 1999, 43, 2350–2355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takeuchi, H.; Asai, N.; Tanabe, K.; Kozaki, T.; Fujita, M.; Sakai, T.; Okuda, A.; Naruse, N.; Yamamoto, S.; Sameshima, T. EM2487, a Novel Anti-HIV-1 Antibiotic, Produced by Streptomyces sp. Mer-2487. J. Antibiot. 1999, 52, 971–982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Liu, Y.; Gao, X.; Xu, P. Chemistry, Biochemistry, Organic Chemistry, Industrial Chemistry, Biochemical Engineering, Life Sciences, Evolutionary Biology; Walter de Gruyter GmbH: Berlin, Germany; Munich, Germany; Boston, MA, USA, 2018; ISBN 9783110562552. [Google Scholar]
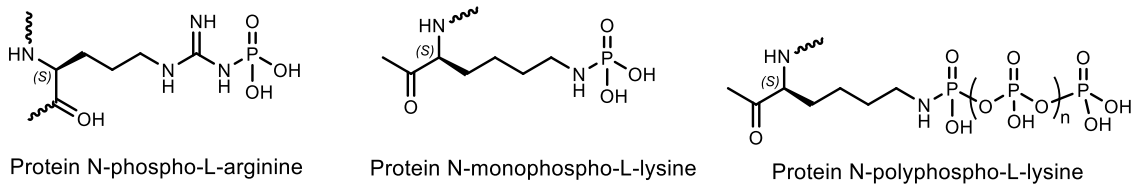
- Cohen, P. The origins of protein phosphorylation. Nat. Cell Biol. 2002, 4, E127–E130. [Google Scholar] [CrossRef]
- Besant, P.G.; Attwood, P.V.; Piggott, M.J. Focus on phosphoarginine and phospholysine. Curr. Protein Pept. Sci. 2009, 10, 536–550. [Google Scholar] [CrossRef]
- Vlastaridis, P.; Kyriakidou, P.; Chaliotis, A.; de Peer, Y.V.; Oliver, S.G.; Amoutzias, G.D. Estimating the total number of phosphoproteins and phosphorylation sites in eukaryotic proteomes. Gigascience 2017, 6, 1–11. [Google Scholar] [CrossRef]
- Guerra-Castellano, A.; Baños-Jaime, B.; Díaz-Quintana, A.; González-Arzola, K.; Ángel De la Rosa, M.; Díaz-Moreno, I. Exploring protein phosphorylation by combining computational approaches and biochemical methods. Comput. Struct. Biotechnol. J. 2020, 18, 1852–1863. [Google Scholar] [CrossRef]
- Piggott, M.J.; Attwood, P.V. Focus on O-phosphohydroxylysine, O-phosphohydroxyproline, N 1-phosphotryptophan and S-phosphocysteine. Amino Acids 2017, 49, 1309–1323. [Google Scholar] [CrossRef]
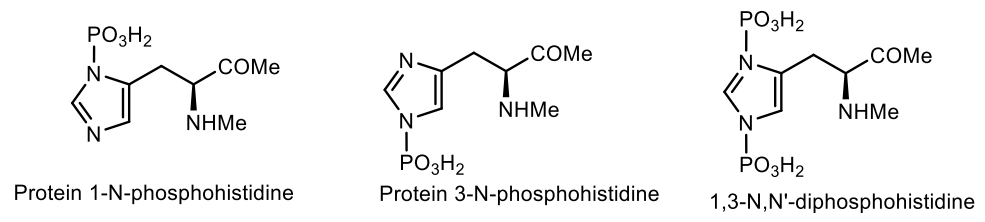
- Gonzalez-Sanchez, M.B.; Lanucara, F.; Helm, M.; Eyers, C.E. Attempting to rewrite History: Challenges with the analysis of histidine-phosphorylated peptides. Biochem. Soc. Trans. 2013, 41, 1089–1095. [Google Scholar] [CrossRef] [Green Version]
- Attwood, P.V.; Piggott, M.J.; Zu, X.L.; Besant, P.G. Focus on phosphohistidine. Amino Acids 2007, 32, 145–156. [Google Scholar] [CrossRef]
- Cheng, C.M.; Liu, X.H.; Li, Y.M.; Ma, Y.; Tan, B.; Wan, R.; Zhao, Y.F. N-Phosphoryl Amino Acids and Biomolecular Origins. Review Paper in Honor of the 50th Anniversary of the Publication of “A Production of Amino Acids under Possible Primitive Earth Conditions” (Miller, 1953). Orig. Life Evol. Biosph. 2004, 34, 455–464. [Google Scholar] [CrossRef]
- Guillaume, H.A.; Perich, J.W.; Johns, R.B.; Tregear, G.W. Synthesis of N(1)-phosphorylated tryptophan derivatives. J. Org. Chem. 1989, 54, 1664–1668. [Google Scholar] [CrossRef]
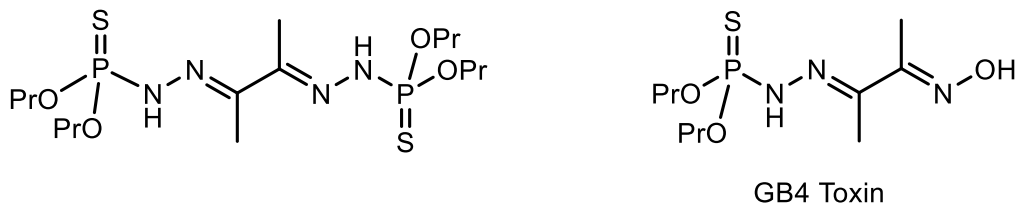
- Alam, M.; Sanduja, R.; Hossain, M.B.; Van der Helm, D. Gymnodinium breve toxins. 1. Isolation and X-ray structure of O,O-dipropyl (E)-2-(1-methyl-2-oxopropylidene) phosphorohydrazidothioate (E)-oxime from the red tide dinoflagellate Gymnodinium breve. J. Am. Chem. Soc. 1982, 104, 5232–5234. [Google Scholar] [CrossRef]
- Abraham, S.P.; Hoang, T.D.; Alam, M.; Jones, E.B.G. Chemistry of the cytotoxic principles of the ma rine fungus Lignincola laevis. Pure Appl. Chem. 1994, 66, 2391–2394. [Google Scholar] [CrossRef]
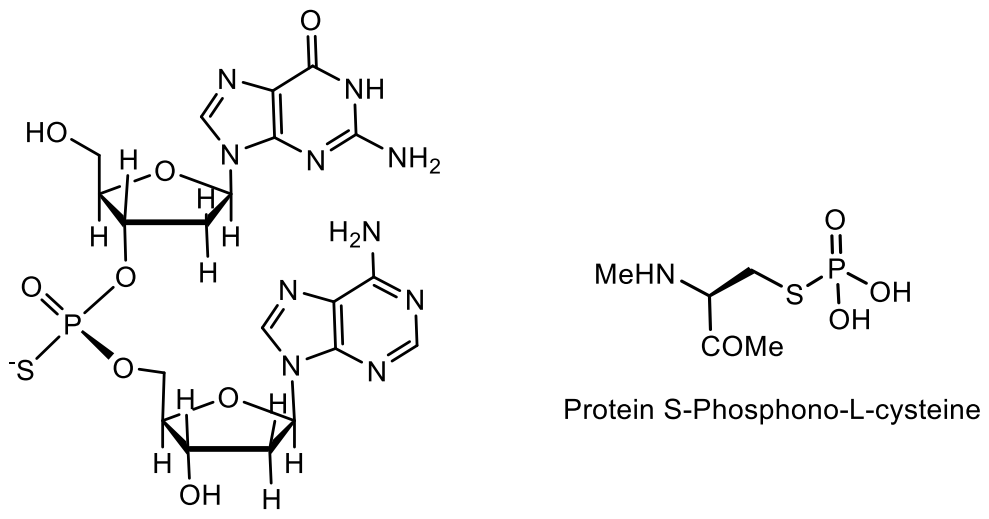
- Tong, T.; Chen, S.; Wang, L.; Tang, Y.; Ryu, J.Y.; Jiang, S.; Wu, X. Occurrence, evolution, and functions of DNA phosphorothioate epigenetics in bacteria. Proc. Natl. Acad. Sci. USA 2018, 115, E2988–E2996. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Chen, S.; Vergin, K.L.; Giovannoni, S.J.; Chan, S.W.; DeMott, M.S.; Taghizadeh, K.; Cordero, O.X.; Cutler, M.; Timberlake, S. DNA phosphorothioation is widespread and quantized in bacterial genomes. Proc. Natl. Acad. Sci. USA 2011, 108, 2963–2968. [Google Scholar] [CrossRef] [Green Version]
- Crooke, S.T.; Seth, P.P.; Vickers, T.A.; Liang, X.-H. The Interaction of Phosphorothioate-Containing RNA Targeted Drugs with Proteins Is a Critical Determinant of the Therapeutic Effects of These Agents. J. Am. Chem. Soc. 2020, 142, 14754–14771. [Google Scholar] [CrossRef]
- Wang, L.; Chen, S.; Xu, T.; Taghizadeh, K.; Wishnok, J.S. Phosphorothioation of DNA in bacteria by dnd genes. Nat. Chem. Biol. 2007, 3, 709–710. [Google Scholar] [CrossRef]
- Wang, L.; Jiang, S.; Deng, Z.; Dedon, P.C.; Chen, S. DNA phosphorothioate modification—A new multi-functional epigenetic system in bacteria. FEMS Microbiol. Rev. 2019, 43, 109–122. [Google Scholar] [CrossRef] [Green Version]
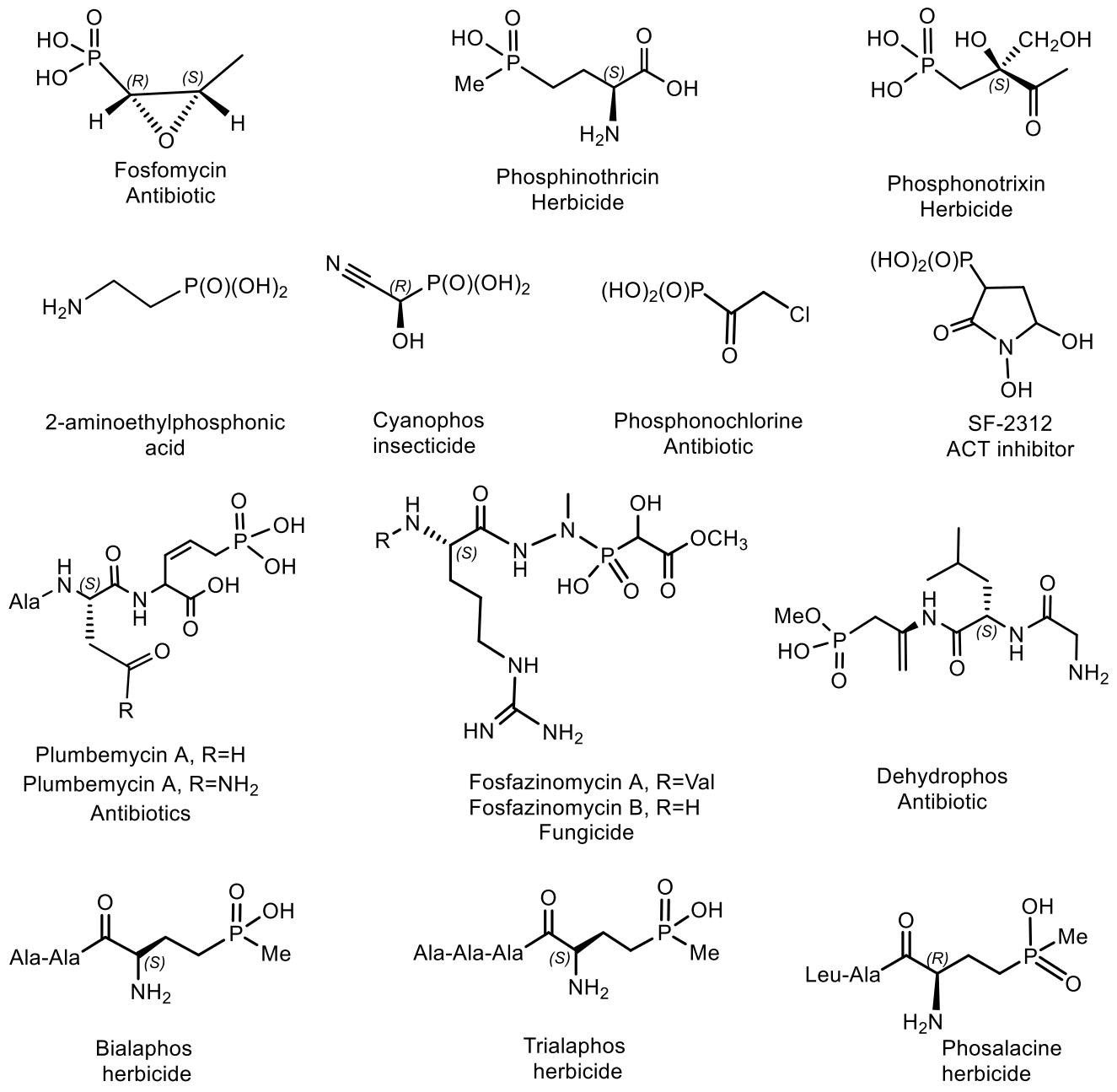
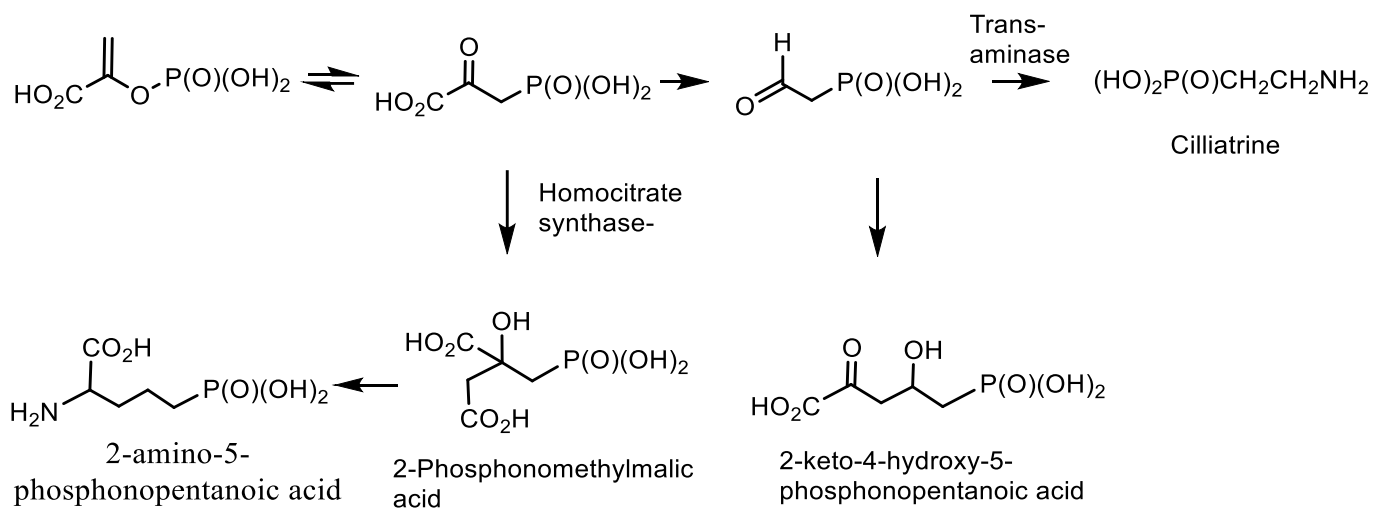
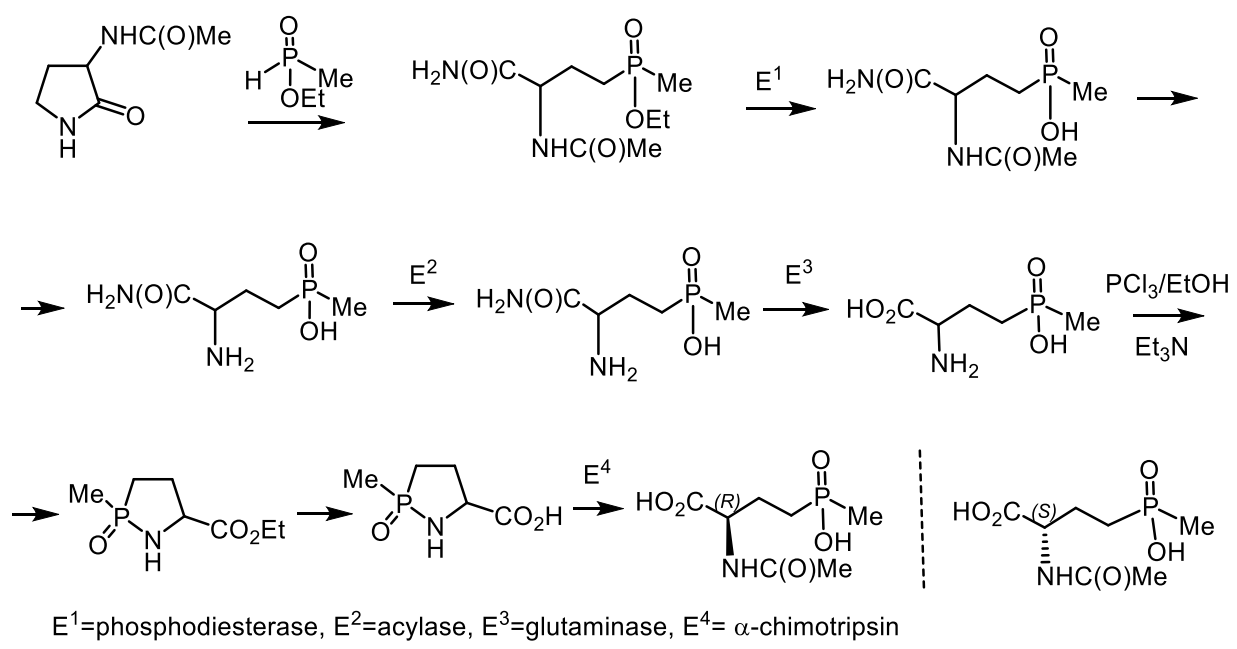
- Horsman, G.P.; Zechel, D.L. Phosphonate Biochemistry. Chem. Rev. 2017, 117, 5704–5783. [Google Scholar] [CrossRef] [PubMed]
- Metcalf, W.W.; van der Donk, W.A. Biosynthesis of phosphonic and phosphinic acid natural products. Annu. Rev. Biochem. 2009, 78, 65–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villarreal-Chiu, J.F.; Quinn, J.P.; McGrath, J.W. The genes and enzymes of phosphonate metabolism by bacteria, and their distribution in the marine environment. Front. Microbiol. 2012, 3, 19–32. [Google Scholar] [CrossRef] [Green Version]
- Hudson, H.; Kukhar, V.P. Aminophosphonic and Aminophosphinic Acids: Chemistry and Biological Activity; John Wiley: New York, NY, USA, 2000; p. 660. ISBN 978-0-471-89149-9. [Google Scholar]
- Mikołajczyk, M. Phosphonate reagents and building blocks in the synthesis of bioactive compounds, natural products and medicines. Pure Appl. Chem. 2019, 91, 811–838. [Google Scholar] [CrossRef]
- Ordonez, M.; Viveros-Ceballos, J.L.; Cativiela, C.; Sayago, F.J. An update on the stereoselective synthesis of α-aminophosphonic acids and derivatives. Tetrahedron 2015, 71, 1745–1784. [Google Scholar] [CrossRef]
- Mikołajczyk, M.; Łyżwa, P.; Drabowicz, J. Asymmetric addition of dialkyl phosphite and diamido phosphite anions to chiral, enantiopure sulfinimines: A new, convenient route to enantiomeric α-aminophosphonic acids. Tetrahedron Asym. 1997, 8, 3991–3994. [Google Scholar] [CrossRef]
- Kafarski, P. Phosphonates: Their Natural Occurrence and Physiological Role. In Contemporary Topics about Phosphorus in Biology and Materials; Churchill, D.G., Čolović, B., Milhofer, H.F., Eds.; Intechopen Publ.: London, UK, 2020; pp. 1–19. [Google Scholar] [CrossRef]
- Kolodiazhnyi, O.I. Asymmetric synthesis of hydroxyphosphonates. Tetrahedron Asymm. 2005, 16, 3295–3341. [Google Scholar] [CrossRef]
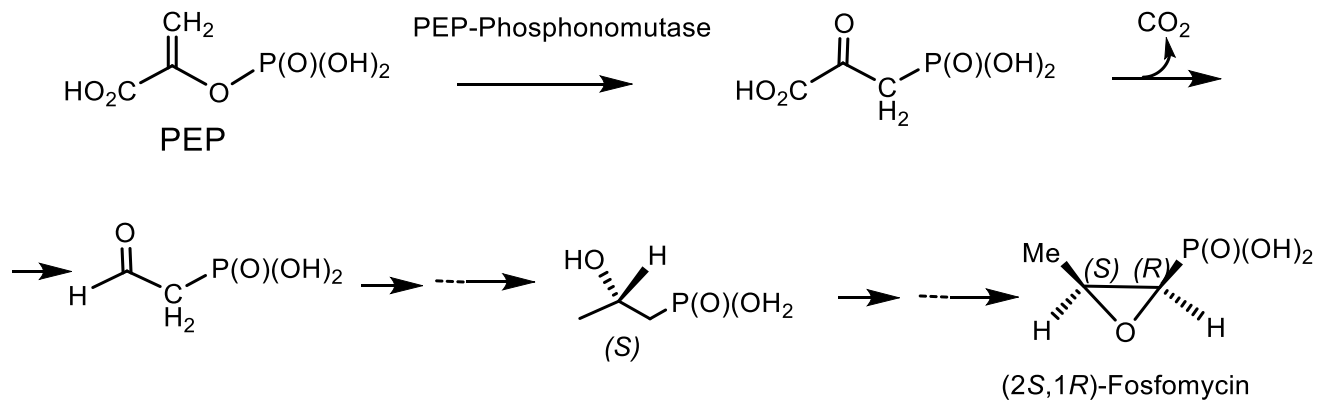
- Falagas, M.E.; Vouloumanou, K.; Samonis, G.; Vardakas, K.Z. Fosfomycin. Clin. Microbiol. Rev. 2016, 29, 321–347. [Google Scholar] [CrossRef] [Green Version]
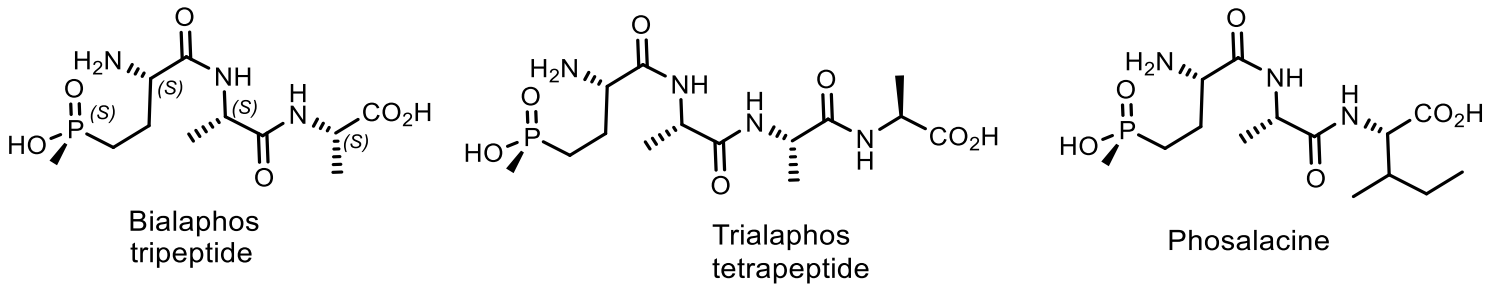
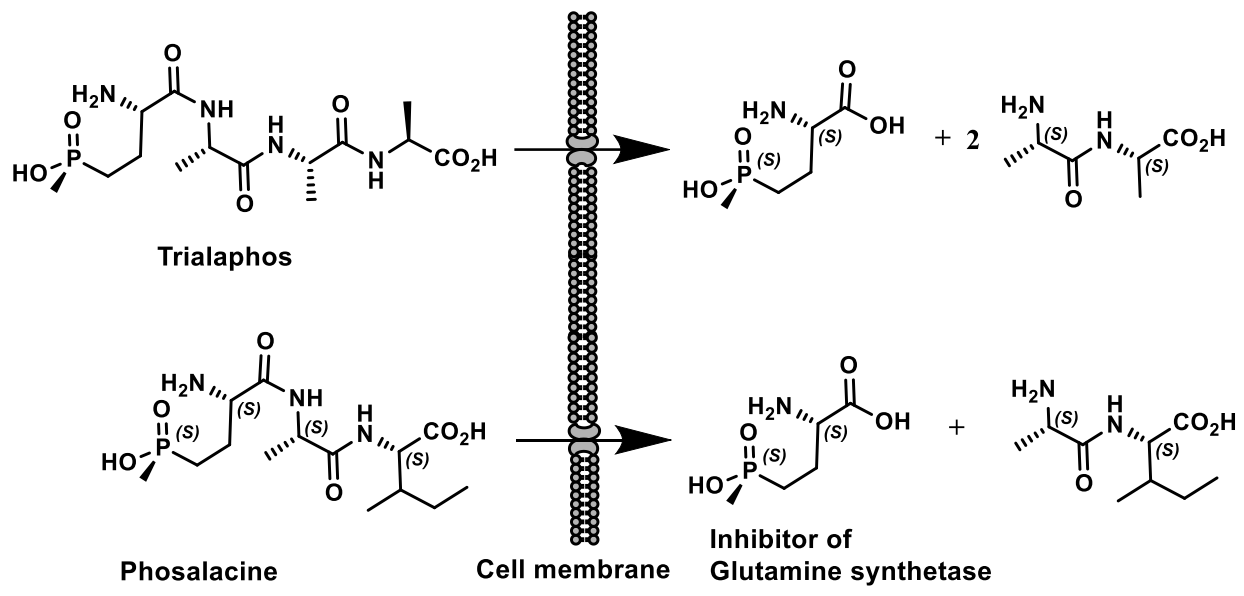
- Murakami, T.; Anzai, H.; Imai, S.; Satoh, A.; Nagaoka, K.; Thompson, C.J. The bialaphos biosynthetic genes of Streptomyces hygroscopicus: Molecular cloning and characterization of the gene cluster. MGG Mol. Gen. Genet. 1986, 205, 42–50. [Google Scholar] [CrossRef]
- Wu, G.; Yuan, M.; Wei, L.; Zhang, Y.; Lin, Y.; Zhang, L.; Liu, Z. Characterization of a Novel Cold-Adapted Phosphinothricin N-Acetyltransferase From the Marine Bacterium Rhodococcus Sp. Strain YM12. J. Mol. Catal. B Enzym. 2014, 104, 23–28. [Google Scholar] [CrossRef]
- Omura, S.; Murata, M.; Hanaki, H.; Hinotozawa, K.; Oiwa, R.; Tanaka, H. Phosalacine, a new herbicidal antibiotic containing phosphinothricin. Fermentation, isolation, biological activity and mechanism of action. J. Antibiot. 1984, 37, 829–835. [Google Scholar] [CrossRef]
- Kato, H.; Nagayama, K.; Abe, H.; Kobayashi, R.; Ishihara, E. Isolation, Structure and Biological Activity of Trialaphos. Agr. Biol. Chem. 1991, 55, 1133–1134. [Google Scholar] [CrossRef]
- Bougioukou, D.J.; Mukherjee, S.; van der Donk, W.A. Revisiting the biosynthesis of dehydrophos reveals a tRNA-dependent pathway. Proc. Natl. Acad. Sci. USA 2013, 110, 10952–10957. [Google Scholar] [CrossRef] [Green Version]
- Lell, B.; Ruangweerayut, R.; Wiesner, J.; Missinou, M.A.; Schindler, A.; Baranek, T.; Hintz, M.; Hutchinson, D.; Jomaa, H.; Kremsner, P.G. Fosmidomycin, a novel chemotherapeutic agent for malaria. Antimicrob. Agents Chemother 2003, 47, 735–738. [Google Scholar] [CrossRef] [Green Version]
- Gahungu, M.; Arguelles-Arias, A.; Fickers, P.; Zervosen, A.; Joris, B.; Damblon, C. Synthesis and biological evaluation of potential threonine synthase inhibitors: Rhizocticin A and Plumbemycin, A. Bioorg. Med. Chem. 2013, 21, 4958–4967. [Google Scholar] [CrossRef]
- Leonard, P.G.; Satani, N.; Maxwell, D.; Lin, Y.-H.; Hammoudi, N.; Peng, Z.F. SF2312 is a natural phosphonate inhibitor of enolase. Nat. Chem. Biol. 2016, 12, 1053–1058. [Google Scholar] [CrossRef]
- Takeuchi, M.; Nakajima, M.; Ogita, T.; Inukai, M.; Kodama, K.; Furuya, K.; Nagaki, H.; Haneishi, T. Fosfonochlorin, a new antibiotic with spheroplast forming activity. J Antibiot 1989, 42, 198–205. [Google Scholar] [CrossRef] [Green Version]
- Cioni, J.P.; Doroghazi, J.R.; Kou-San, J.; Yu, X.; Evans, B.S.; Lee, J.; Metcalf, W.W. Cyanohydrin Phosphonate Natural Product from Streptomyces regensis. J. Nat. Prod. 2014, 77, 243–249. [Google Scholar] [CrossRef]
- Takahashi, E.; Kimura, T.; Nakamura, K.; Arahira, M.; Iida, M. Phosphonothrixin, a novel herbicidal antibiotic produced by Saccharothrix sp. ST-888. I. Taxonomy, fermentation, Hitakaisolation and biological properties. J. Antibiot. 1995, 48, 1124–1129. [Google Scholar] [CrossRef]
- Hidaka, T.; Seto, H.; Imai, S. Biosynthesis mechanism of carbon-phosphorus bond formation. Isolation of carboxyphosphonoenolpyruvate and its conversion to phosphinopyruvate. J. Am. Chem. Soc. 1989, 111, 8012–8013. [Google Scholar] [CrossRef]
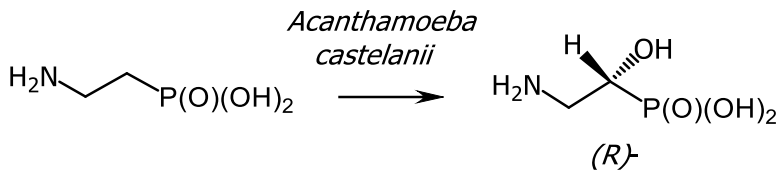
- Hammerschmidt, F. Biosynthese von Naturstoffen mit einer P—C-Bindung, IV. Synthese der (R)- und (S)-(2-Amino[2-D1]ethyl)phosphonsäure und Hydroxylierung zu (2-Amino-1-hydroxyethyl)phosphonsäure in Acanthamoeba castellanii (Neff). Liebigs Ann. Chem. 1988, 961–964. [Google Scholar] [CrossRef]
- Harnrnerschrnidt, F. Synthese der (R)- und (S)-(2-Amino[2-D1]ethy1)phosphonsaure und Hydroxylierung zu (2-Amino-1-hydroxyethy1)phosphonsaure in Acanthamoeba castellanii (Neff). Lieb Ann. Chem. 1988, 1988, 961–964. [Google Scholar] [CrossRef]
- Peck, S.C.; van der Donk, W. Phosphonate biosynthesis and catabolism: A treasure trove for unusual enzymology. Curr. Opin. Chem. Biol. 2013, 17, 580–588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walsh, C.T. The Chemical Biology of Phosphorus; RSC: Cambridge, UK, 2020; p. 546. ISBN 139781839162022. [Google Scholar]
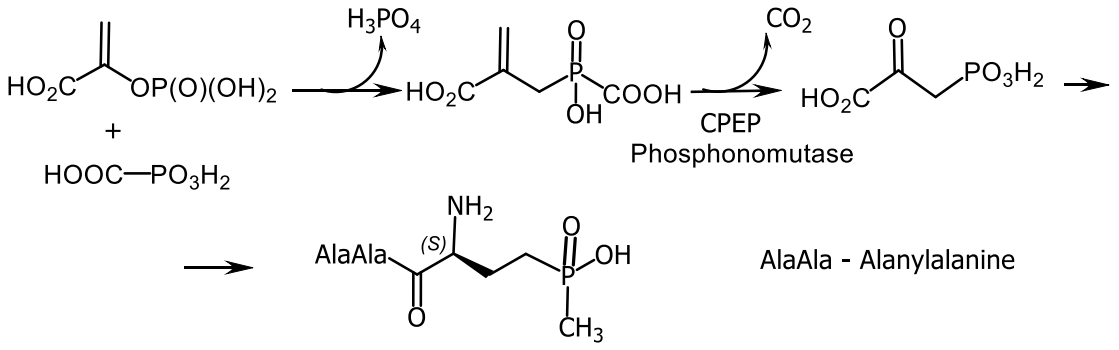
- Blodgett, J.A.V.; Thomas, P.M.; Li, G.; Velasquez, J.E.; van der Donk, W.A. Unusual transformations in the biosynthesis of the antibiotic phosphinothricin tripeptide. Nat. Chem. Biol. 2007, 3, 480–485. [Google Scholar] [CrossRef] [Green Version]
- McQueney, M.S.; Lee, S.L.; Swartz, W.H.; Ammon, H.L.; Mariano, P.S.; Dunaway-Mariano, D. Evidence for an intramolecular, stepwise reaction pathway for PEP phosphomutase catalyzed phosphorus-carbon bond formation. J. Org. Chem. 1991, 56, 7121–7130. [Google Scholar] [CrossRef]
- McQueney, M.S.; Lee, S.L.; Bowman, E.; Mariano, P.S.; Dunaway-Mariano, D. A remarkable pericyclic mechanism for enzyme-catalyzed phosphorus-carbon bond formation. J. Am. Chem. Soc. 1989, 111, 6885–6887. [Google Scholar] [CrossRef]
- Freeman, S.; Seidel, H.M.; Schwalbe, C.H.; Knowles, J.R. Phosphonate biosynthesis: The stereochemical course of phosphoenolpyruvate phosphomutase. J. Am. Chem. Soc. 1989, 111, 9233–9234. [Google Scholar] [CrossRef]
- Hammerschmidt, F. Incorporation of ~-Methyl-*H,. methionine and 2-Hydroxy-’80. hydroxyethylphosphonic Acid into Fosfomycin in Streptomyces frudiue-An Unusual Methyl Transfer. Angew. Chem. Intern. Ed. Engl. 1994, 106, 334–342. [Google Scholar] [CrossRef]
- Behrman, E.J.; Gopalan, V. Phosphoenolpyruvate. Biochem. Mol. Biol. 2008, 36, 323–324. [Google Scholar] [CrossRef]
- Petkowski, J.J.; Bains, W.; Seager, S. Natural Products Containing ‘Rare’ Organophosphorus Functional Groups. Molecules 2019, 24, 866. [Google Scholar] [CrossRef] [Green Version]
- Kolodiazhnyi, O.I. Enzymatic synthesis of organophosphorus compounds. Russ. Chem. Rev. 2011, 80, 883–910. [Google Scholar] [CrossRef]
- Simov., B.P.; Wuggenig., F.; Lammerhofer, M.; Lindner, W.; Zarbl, E.; Hammerschmidt, F. Indirect Evidence for the Biosynthesis of (1S,2S)-1,2-Epoxypropylphosphonic Acid as a Co-Metabolite of Fosfomycin (1R,2S)-1,2-Epoxypropylphosphonic Acid. by Streptomyces fradiae. Eur. J. Org. Chem. 2002, 1139–1142. [Google Scholar] [CrossRef]
- Munos, J.W.; Moon, S.-J.; Mansoorabadi, S.O.; Chang, W.; Hong, L.; Yan, F.; Liu, A.; Liu, H.-W. Purification and Characterization of the Epoxidase Catalyzing the Formation of Fosfomycin from Pseudomonas syringae. Biochemistry 2008, 47, 8726–8735. [Google Scholar] [CrossRef] [Green Version]
- Yan, F.; Moon, S.-J.; Liu, P.; Zhao, Z.; Lipscomb, J.D.; Liu, A.; Liu, H.-W. Determination of the Substrate Binding Mode to the Active Site Iron of (S)-2-Hydroxypropylphosphonic Acid Epoxidase Using 17O-Enriched Substrates and Substrate Analogues. Biochemistry 2007, 46, 12628–23638. [Google Scholar] [CrossRef] [Green Version]
- Kramer, G.J.; Mohd, A.; Schwager, S.L.U.; Masuyer, G.; Ravi Acharya, K.; Sturrock, E.D.; Bachmann, B.O. Interkingdom Pharmacology of Angiotensin-I Converting Enzyme Inhibitor Phosphonates Produced by Actinomycetes. ACS Med. Chem. Lett 2014, 5, 346–351. [Google Scholar] [CrossRef]
- Ohba, K.; Sato, Y.; Sasaki, T.; Sezaki, M. Studies on a new phosphonic acid antibiotic SF2312.II isolation, physicochemical properties and structure. Sci. Rep. 1986, 25, 18–22. [Google Scholar]
- Okuhara, M.; Kuroda, Y.; Goto, T.; Okamoto, M.; Terano, H.; Kohsaka, M. Studies on a new phosphonic acid antibiotic III. Isolation and characterisation of FRFR-31564, FR-32863 and FR-33289. J. Antibiot. 1980, 33, 24–28. [Google Scholar] [CrossRef] [Green Version]
- Kaya, K.; Morrison, L.F.; Codd, G.A.; Metcalf, J.S.; Sano, T.; Takagi, H. A novel biosurfactant,2-Acyloxyethylphosphonate, isolated from Waterblooms of Aphanizomenon flos-aquae. Molecules 2006, 11, 539–548. [Google Scholar] [CrossRef] [Green Version]
- Bayer, E.; Gugel, K.H.; Hägele, K.; Hagenmaier, H.; Jessipow, S.; König, W.A.; Zähner, H. Metabolic Products of Microorganisms.98. Phosphinothricin and Phosphinothricyl-Alanyl-Alanine. Helv. Chim. Acta 1972, 55, 224–239. [Google Scholar] [CrossRef]
- Donn, G.; Köcher, H. Inhibitors of Glutamine Synthetase. In Herbicide Classes in Development; Springer: Berlin/Heidelberg, Germany, 2002; pp. 87–101. [Google Scholar]
- Mowbray, S.L.; Kathiravan, M.K.; Pandey, A.A.; Odell, L.R. Inhibition of Glutamine Synthetase: A Potential Drug Target in Mycobacterium Tuberculosis. Molecules 2014, 19, 13161–13176. [Google Scholar] [CrossRef] [Green Version]
- Natchev, I.A. Enzymatic synthesis of D,D,L, and Lphosphinothricin and their cyclic analogs. Bull. Chem. Soc. Jpn. 1988, 61, 3699–3704. [Google Scholar] [CrossRef] [Green Version]
- Kolodiazhnyi, O.I. Asymmetric Synthesis in Organophosphorus Chemistry; John Wiley: Weinheim, Germany, 2016; p. 400. [Google Scholar]
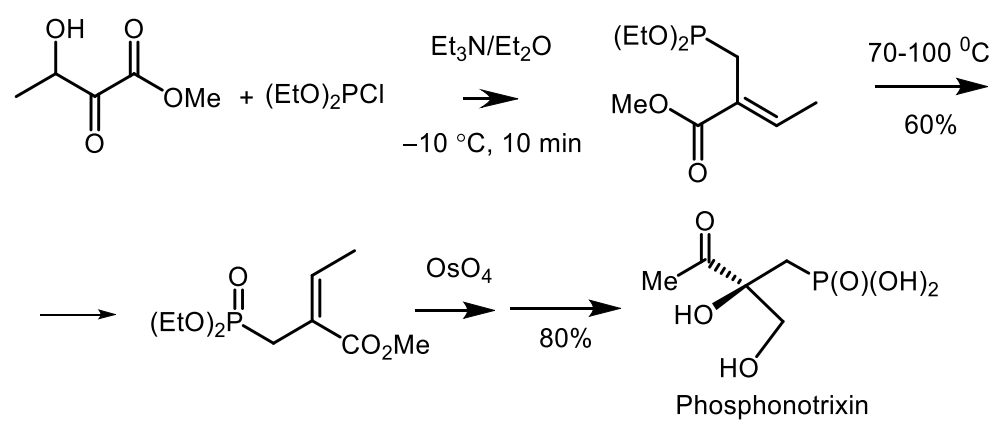
- Nakamura, K.; Yamamura, S. Enantioseleetive Synthesis of Phosphonothrixin and Its Absolute Stereochemistry. Tetrahedron Lett. 1997, 38, 437–438. [Google Scholar] [CrossRef]
- Chênevert, R.; Simard, M.; Bergeron, J.; Dasser, M. Chemoenzymatic formal synthesis of (S)-(−)-phosphonotrixin. Tetrahedron Asymmetry 2004, 15, 1889–1892. [Google Scholar] [CrossRef]
- Field, S.C. Total synthesis of (±)-Phosphonothrixin. Tetrahedron Lett. 1998, 39, 6621–6624. [Google Scholar] [CrossRef]
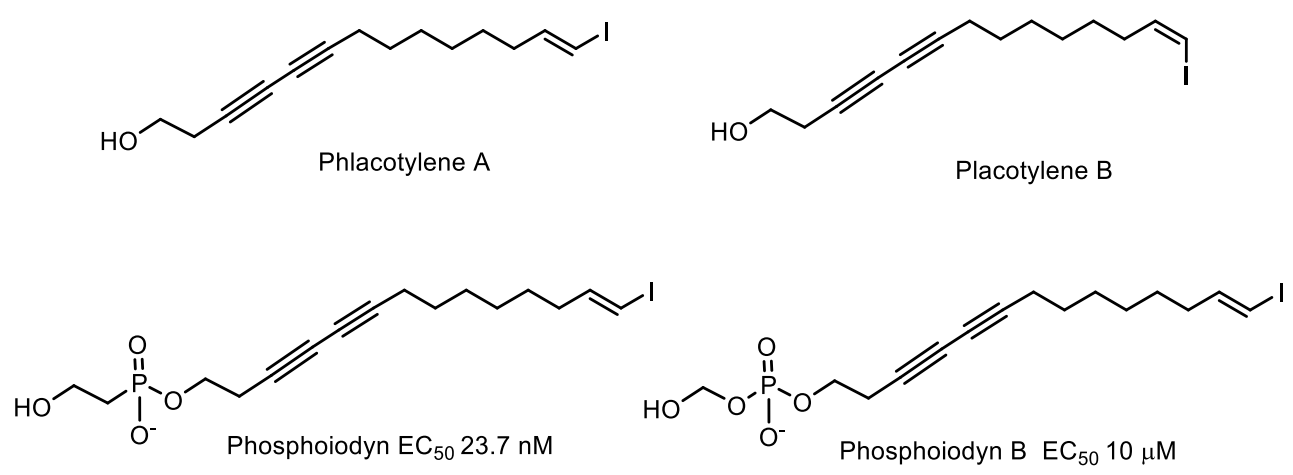
- Kim, H.; Chin, J.; Choi, H.; Baek, K.; Lee, T.-G. Phosphoiodyns A and B, Unique Phosphorus-Containing Iodinated Polyacetylenes from a Korean Sponge Placospongia sp. Org. Lett. 2013, 15, 100–103. [Google Scholar] [CrossRef]
- Kinarivala, N.; Suh, J.H.; Botros, M.; Webb, P.; Trippier, P.C. Pharmacophore elucidation of phosphoiodyn A—Potent and selective peroxisome proliferator-activated receptor β/δ agonists with neuroprotective activity. Bioorg. Med. Chem. Lett. 2016, 26, 1889–1893. [Google Scholar] [CrossRef]
- Tamari, M.; Ogawa, M.; Kametaka, M. A New Bile Acid Conjugate, Ciliatocholic Acid, from Bovine Gall Bladder Bile. J. Biochem. 1976, 80, 371–377. [Google Scholar] [CrossRef]
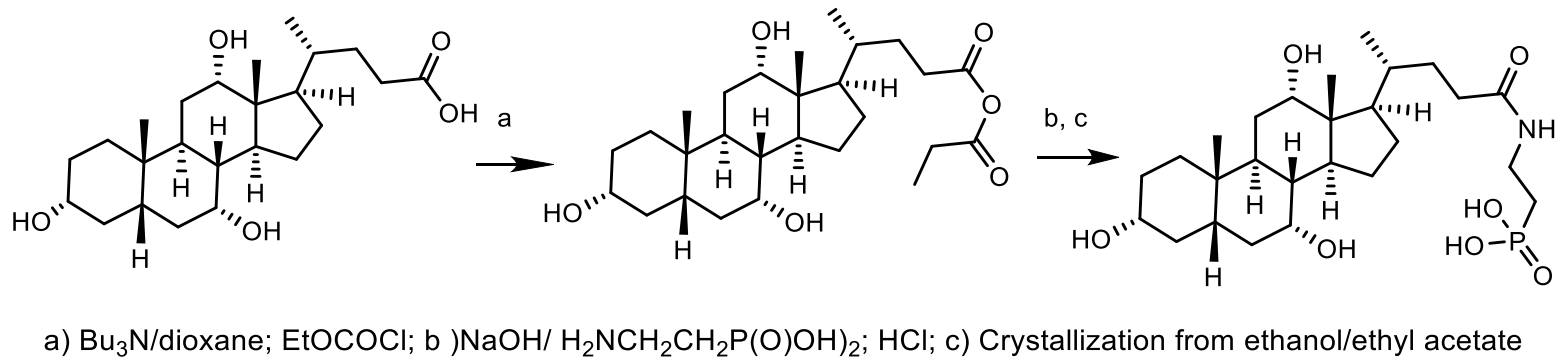
- Katayama, N.; Tsubotani, S.; Nozaki, Y.; Harada, S.; Ono, H. Fosfadecin and fosfocytocin, new nucleotide antibiotics produced by bacteria Affiliations expand PMID: 2182591. J. Antibiot. 1990, 43, 238–246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
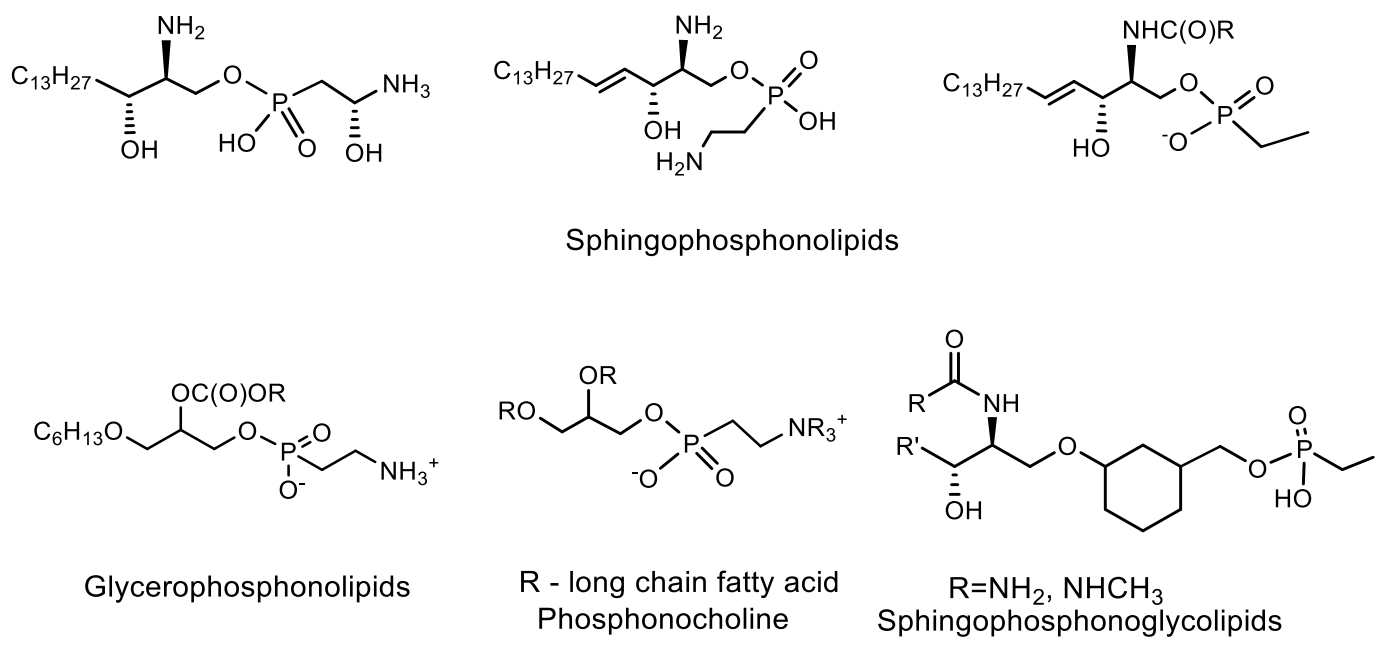
- Moschidis, M.C. Phosphonolipids. Prog. Lipid Res. 1984, 23, 223–246. [Google Scholar] [CrossRef]
- Rouser, G.; Kritchevsky, G.; Heeler, D.; Lieaer, E. Lipid composition of beef brain, beef liver, and the sea anemone: Two approaches to quantitative fractionation of complex lipid mixture. J. Am. Oil Chem. Soc. 1963, 40, 425–454. [Google Scholar] [CrossRef]
- Mukhamedova, K.S.; Glushenkova, A.I. Natural phosphonolipids. Chem. Nat. Compd. 2000, 36, 329–341. [Google Scholar] [CrossRef]
- Marrs, E.C.L.; Varadi, L.; Bedernjak, A.F.; Day, K.M.; Gray, M.; Jones, A.L.; Cummings; Anderson, S.P.; Anderson, R.J.; Perry, J.D. Phosphonopeptides Revisited, in an Era of Increasing Antimicrobial Resistance. Molecules 2020, 25, 1445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Z.; Kwo-Kwang; Wang, A.; van der Donk, W.A. New insights into the biosynthesis of fosfazinomycin. Chem. Sci. 2016, 7, 5219–5223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goettge, M.N.; Cioni, J.P.; Ju, K.-S.; Pallitsch, K.; Metcalf, W.W. PcxL and HpxL are flavin-dependent, oxime-forming N-oxidases in phosphonocystoximic acid biosynthesis in Streptomyces. J. Biol. Chem. 2018, 293, 6859–6868; [Google Scholar] [CrossRef] [Green Version]
- Rapp, C.; Jung, G.; Kugler, M.; Loeffler, W. Rhizocticins—New phosphono- oligopeptides with antifungal activity. Eur. J. Org. Chem. 1988, 7, 655–661. [Google Scholar] [CrossRef]
- Pallitsch, K.; Happl, B.; Stieger, C. Determination of the Absolute Configuration of (−)-Hydroxynitrilaphos and Related Biosynthetic Questions. Chem. Eur. J. 2017, 23, 15655–15665. [Google Scholar] [CrossRef]
- Borisova, S.A.; Circello, B.T.; Zhang, J.K.; van der Donk, W.W.; Metcalf, W.W. Biosynthesis of rhizocticins, antifungal phosphonate oligopeptides produced by Bacillus subtilis ATCC6633. Chem. Biol. 2010, 17, 28–37. [Google Scholar] [CrossRef] [Green Version]
- Evans, B.S.; Zhao, C.; Gao, J.; Evans, C.M.; Ju, K.-S.; Doroghazi, J.R.; Metcalf, W.W. Discovery of the antibiotic phosacetamycin via a new mass spectrometry-based method for phosphonic acid detection. ACS Contemporary Topics about Phosphorus in Biology and Materials. Chem. Biol. 2013, 8, 908–913. [Google Scholar] [CrossRef] [Green Version]
- Kugler, M.; Loeffler, W.; Rapp, C.; Kern, A.; Jung, G. Rhizocticin A, an antifungal phosphono-oligopeptide of Bacillus subtilis ATCC 6633: Biological properties. Archiv. Microbiol. 1990, 153, 276–281. [Google Scholar] [CrossRef]
- Johnson, R.D.; Kastner, R.M.; Larsen, S.H.; Ose, E.E. Antibiotic A53868 and Process for Production Thereof. U.S. Patent 4,482,488, 13 November 1988. [Google Scholar]
- Gunji, S.; Arima, K.; Beppu, T. Screening of antifungal antibiotics according to activities inducing morphological abnormalities. Agric. Biol. Chem. 1983, 41, 2016–2069. [Google Scholar] [CrossRef]
- Gao, J.; Ju, K.-S.; Yu, X.; Velásques, J.E.; Mukherjee, S.; Lee, J.; Van Der Donk, W.A. Use of phosphonate methyltransferase in the identification of the fosfazinomycin biosynthetic gene cluster. Angew. Chem. 2014, 53, 1334–1337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schneemann, I.; Nagel, K.; Labes, A.; Wiese, J.; Imhoff, J.F. Comprehensivei of marine Actinobacteria associated with the sponge Halichondria panacea. Appl. Environ. Microbiol. 2010, 76, 3702–3714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, K.-K.A.; Ng, T.L.; Wang, P.; Huang, Z.; Balskus, E.P.; van der Donk, W.A. Glutamic acid is a carrier for hydrazine during the biosyntheses of fosfazinomycin and kanamycin. Nat. Commun. 2018, 9, 3687–3698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
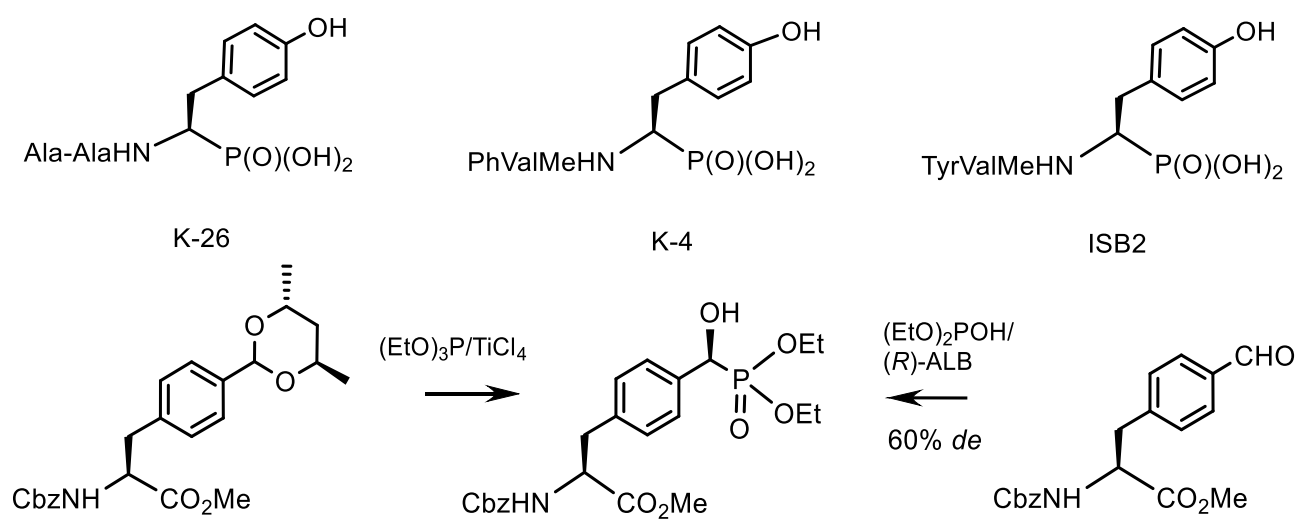
- Yamato, M.; Koguchi, T.; Okachi, R.; Yamada, K.; Nakayama, K.; Kase, H.; Karasawa, A.; Shuto, K. K-26, a novel inhibitor of angiotensin I converting enzyme produced by an actinomycete K-26. J. Antibiot. 1986, 39, 44–52. [Google Scholar] [CrossRef] [Green Version]
- Ju, K.-S.; Gao, J.; Doroghazi, J.R.; Wang, K.-K.A.; Thibodeaux, C.J. Discovery of phosphonic acid natural products by mining the genomes of 10,000 actinomycetes. Proc. Natl. Acad. Sci. USA 2015, 112, 12175–12180. [Google Scholar] [CrossRef] [Green Version]
- Huang, Z.A.; Wang, K.-K.; Lee, J.; van der Donk, W. A Biosynthesis of fosfazinomycin is a convergent process. Chem. Sci. 2015, 6, 1282–1287. [Google Scholar] [CrossRef] [Green Version]
- Yokomatsu, T.; Yamagishi, T.; Matsumoto, K.; Shibuya, S. Stereocontrolled synthesis of hydroxymethylene phosphonate analogues of phosphorylated tyrosine and their conversion to monofluoromethylene phosphonate analogues. Tetrahedron 1996, 52, 11725–11738. [Google Scholar] [CrossRef]
- Masuyer, G.; Cozier, G.E.; Kramer, G.J.; Bachmann, B.O.; Acharya, K.R. Crystal structure of a peptidyl-dipeptidase K-26-DCP from Actinomycete in complex with its natural inhibitor. FEBS J. 2016, 283, 4357–4369. [Google Scholar] [CrossRef] [Green Version]
- Kido, Y.; Hamakado, T.; Anno, M.; Miyagawa, E.; Motoki, Y.; Wakamiya, T. Isolation and characterization of I5B2, a new phosphorus containing inhibitor of angiotensin i converting enzyme produced by actinomadura sp. J. Antibiot. 1984, 37, 965–969. [Google Scholar] [CrossRef] [Green Version]
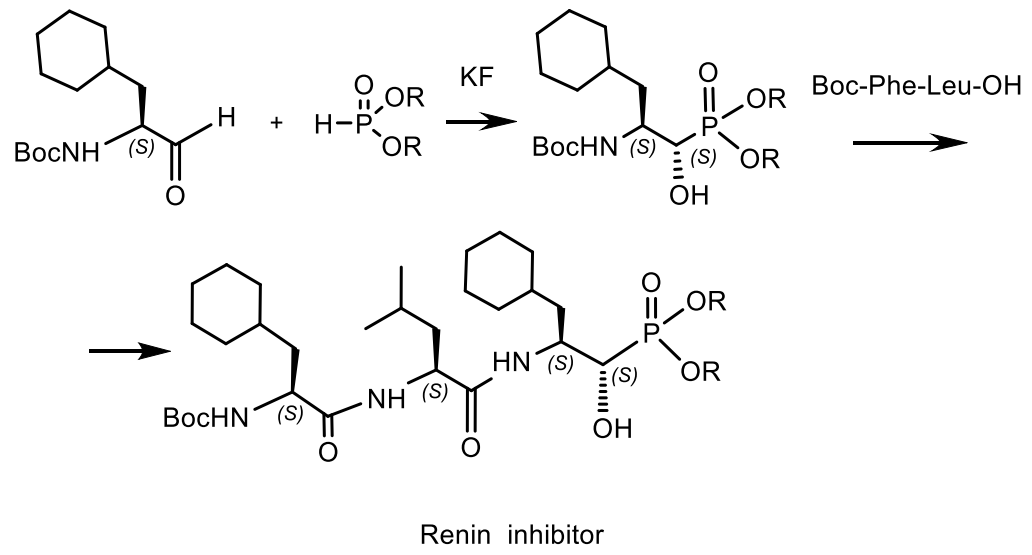
- Patel, D.V.; Rielly-Gauvin, K.; Ryono, D.E.; Free, C.A.; Lynn Rogers, W. Hydroxy Phosphinyl-Based Inhibitors of Human Renin. J. Med. Chem. 1995, 38, 4557–4569. [Google Scholar] [CrossRef]
- Allen, J.G.; Atherton, F.R.; Hall, M.J.; Hassall, C.H.; Holmes, S.W.; Lambert, R.W.; Nisbet, L.J.; Ringrose, P.S. Phosphonopeptides, a new class of synthetic antibacterial agents. Nature 1978, 272, 56–58. [Google Scholar] [CrossRef]
- Kafarski, P. Phosphonopeptides containing free phosphonic groups: Recent advances. RSC Adv. 2020, 10, 25898–25910. [Google Scholar] [CrossRef]
- Lamberth, C. Naturally occurring amino acid derivatives with herbicidal, fungicidal or insecticidal activity. Amino Acids 2016, 48, 929–940. [Google Scholar] [CrossRef]
- Hoagland, R.E. Naturally Occurring Carbon—Phosphorus Compounds as Herbicides. Biol. Act. Nat. Prod. 1988, 13, 182–210. [Google Scholar] [CrossRef]
- Blodgett, J.A.V.; Zhang, J.K.; Yu, X.; Metcalf, W.W. Conserved biosynthetic pathways for phosalacine, bialaphos and newly discovered phosphonic acid natural products. J. Antibiot. 2016, 69, 15–25. [Google Scholar] [CrossRef] [Green Version]
- Hunt, A.; Elzey, T. Revised structure of A53868A. J. Antibiot. 1988, 41, 802. [Google Scholar] [CrossRef] [Green Version]
- Dragicevic, M.; Platiša, J.; Nikolic, R.; Todorovic, S.; Bogdanovic, M.; Mitic, N.; Simonovic, A. Herbicide phosphinothricin causes direct stimulation hormesis Formerly Nonlinearity in Biology, Toxicology, and Medicine. Dose-Response 2013, 11, 344–360. [Google Scholar] [CrossRef]
- Schwartz, D.; Berger, S.; Heinzelmann, E.; Muschko, K.; Welzel, K.; Wohlleben, W. Biosynthetic Gene Cluster of the Herbicide Phosphinothricin Tripeptide from Streptomyces viridochromogenes Tu494. Appl. Environ. Microbiol. 2004, 70, 7093–7102. [Google Scholar] [CrossRef] [Green Version]
- Timsit, Y. DNA Self-Assembly: From Chirality to Evolution. Int. J. Mol. Sci. 2013, 14, 8252–8270. [Google Scholar] [CrossRef] [Green Version]
- Tajti, Á.; Keglevich, G. 3. The Importance of Organophosphorus Compounds as Biologically Active Agents. In Organophosphorus Chemistry; Walter de Gruyter GmbH: Boston, Germany; Boston, MA, USA, 2018; ISBN 978-3-11-053453-5. [Google Scholar] [CrossRef]
- Mikulík, K.; Melčová, M.; Zídková, J. Antibacterial peptides from thermophilic bacteria. Int. J. Eng. Res. Sci. 2017, 3, 46–57. [Google Scholar] [CrossRef]





































































{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
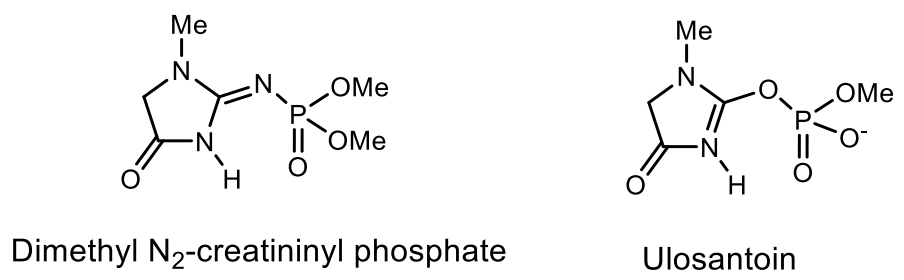{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Acid | pKa(1) | pKa(2) | pKa(3) |
|---|---|---|---|
| Phosphoric | 2.12 | 7.2 | 12.3 |
| Citric | 3.22 | 4.8 | 6.4 |
 | |||||
| Entry | R1 | R2 | R3 | ee
[%] with D-DNA | ee
[%] with L-DNA |
| 1 | MeO | H | Me | 89 | 90 |
| 2 | H | H | Me | 76 | 76 |
| 3 | H | Me | o-BrC6H4 | 76 | 80 |
| 4 | MeO | H | 4-ClC6H4 | 72 | 74 |
 | R1 | R2 | Name |
| Me |  | Phosmidosine | |
| H |  | Phosmidosine B | |
| Me | H | Phosmidosine C |
| Type of tumor cells | IC50 (μM) | |
|---|---|---|
| Phosmidosine 1a | Phosmidosine 1b | |
| KB | 3.6 | 3.2 |
| KATO-III | 2.6 | 1.9 |
| MKN-28 | 9.6 | 8.5 |
| MKN-45 | 2.9 | 2.5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kolodiazhnyi, O.I. Phosphorus Compounds of Natural Origin: Prebiotic, Stereochemistry, Application. Symmetry 2021, 13, 889. https://doi.org/10.3390/sym13050889
Kolodiazhnyi OI. Phosphorus Compounds of Natural Origin: Prebiotic, Stereochemistry, Application. Symmetry. 2021; 13(5):889. https://doi.org/10.3390/sym13050889
Chicago/Turabian StyleKolodiazhnyi, Oleg I. 2021. "Phosphorus Compounds of Natural Origin: Prebiotic, Stereochemistry, Application" Symmetry 13, no. 5: 889. https://doi.org/10.3390/sym13050889
APA StyleKolodiazhnyi, O. I. (2021). Phosphorus Compounds of Natural Origin: Prebiotic, Stereochemistry, Application. Symmetry, 13(5), 889. https://doi.org/10.3390/sym13050889