Two samples were used to determine the enantiomeric excess: α-pinene, which occurs as a liquid at room temperature and is often used as a chiroptical calibration standard. Furthermore, alanine was used as a prototype chiral molecule investigated in aqueous solution. Alanine is the simplest chiral amino acid, which is also readily available in both enantiomers.
The procedure for processing the enantiomeric excess was first illustrated on spectra in the basic setup (parameters for Savitzky–Golay smoothing, spectral range selection for Raman baseline correction, ROA normalization, artifact correction and EE determination), and the basic observed aspects of the EE determination and possible sources of measurement errors will be explained by these results. Then, the refinement parameters for the spectra processing will be discussed in order to obtain the most accurate and precise results. Finally, we will discuss the question of how the overall accuracy of the EE determination (standard deviation of errors of the EE determination) depends on the required measurement time.
4.1. Basic Processing Routine
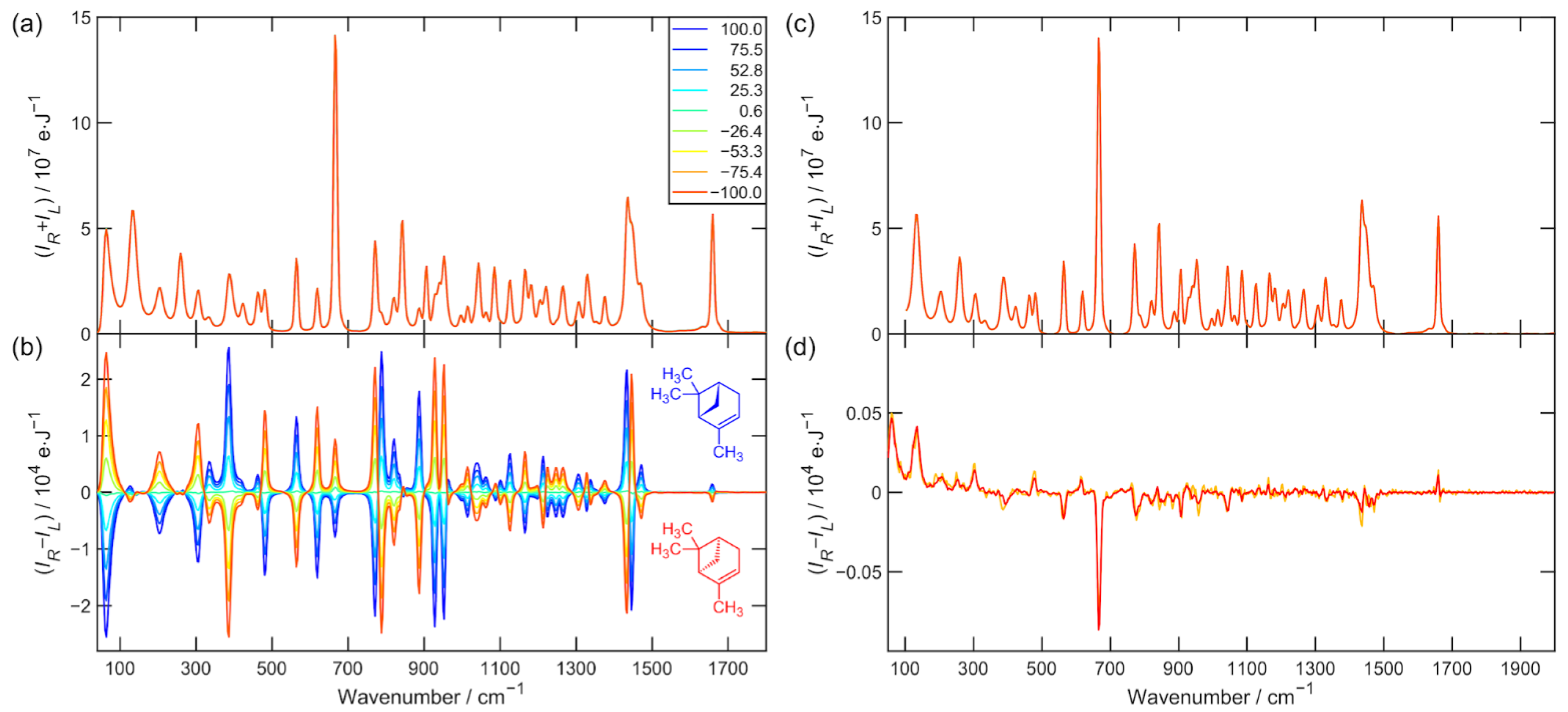
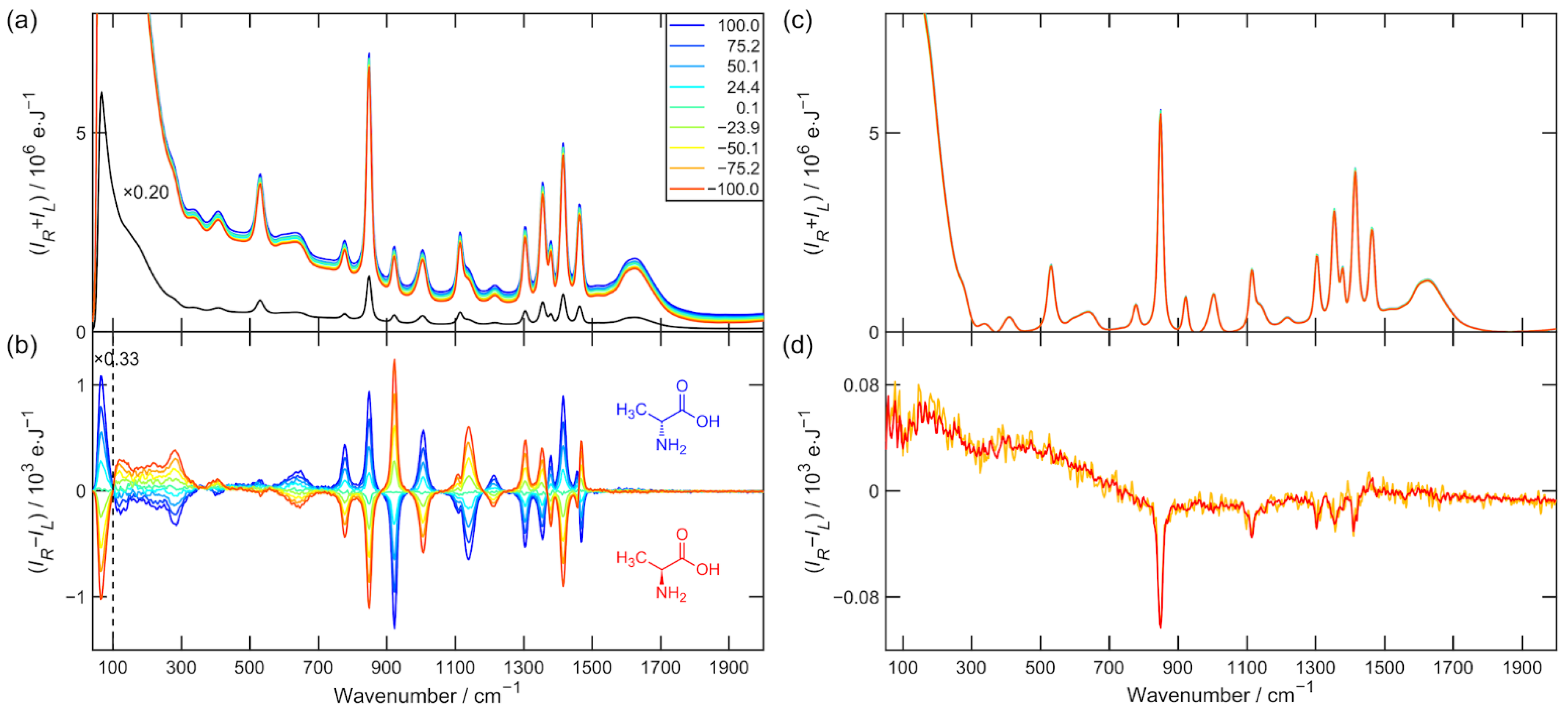
The Raman and ROA raw data used for the further analysis of α-pinene and alanine (nine enantiomeric mixtures) are presented in
Figure 1a,b and
Figure 2a,b. All spectra were then subjected to third-order five-point Savitzky–Golay smoothing. The spectral region 100–2000 cm
−1 was used in all mentioned aspects of data processing.
Figure 1c and
Figure 2c show the Raman spectrum after baseline correction, and
Figure 1d and
Figure 2d show the spectrum of the ROA artifacts determined by Equations (11) and (12). The elevated Raman baseline was effectively corrected by the asymmetric least squares smoothing algorithm taken from [
16]. The aim of this procedure was not to make the baseline as flat as possible but to unify Raman spectra in the data set before the ROA normalization.
The results of the EE analysis for α-pinene and alanine aqueous solution are listed in
Table 1 and
Table 2, respectively. The columns in these tables have the following meanings:
i is the sample number (not in the order in which the spectra were measured; see Materials and Methods),
nnorm is the ROA normalization factor determined according to Equation (5) after the Raman baseline correction,
is the EE calculated from sample preparation according to Equation (3),
is the EE determined from ROA spectra according to Equation (6),
is the corresponding EE determination error according to Equation (8),
and
are the recalculated EEs and corresponding errors calculated according to Equation (9) based on the assumption that reference
B has a different EE than reference
A and that the sum of errors
is zero. The following four columns
,
,
and
have the same meaning; the artifact spectra according to Equation (13) were subtracted from the experimental ROA spectra to obtain the artifact-free ROA spectra needed for the calculation. The last two columns
and
represent EEs determined by the leave-one-out PLS algorithm applied to baseline corrected and normalized spectra before the systematic artifact correction and their corresponding errors determined by Equation (8), respectively. The bottom line of the tables represents the standard deviation of the errors determined according to Equation (10) for the respective columns.
By comparing the standard deviations of the errors of EE determination and , it can be said that using the assumption that the EE of the “pure” forms A and B need not be the same and that the sum of the errors is zero according to Equation (9) leads to a significant increase in the precision of the EE determination (the standard deviations of the errors of EE determination decreased roughly 1.6× for α-pinene and 9× in the case of alanine in aqueous solution) but not necessarily to improvement of the accuracy, which can be demonstrated by large deviations from −100% of EE of reference B (row i = 9, especially for alanine). Note that absolute values of EE of reference B above 100% are possible, since they are relative to reference A and indicate that reference B has a higher EE than reference A.
The data presented in
Table 1 and
Table 2 for the ROA spectra after subtraction of artifacts and especially the values
and
do not suggest much improvement at first glance. However, the results for the determination of EE (row
i = 9) clearly indicate that there has been a significant improvement in accuracy after artifact subtraction. The artifact spectra of α-pinene and alanine are plotted in
Figure 1d and
Figure 2d. Strongly polarized and also the most intense Raman bands, such as α-pinene breathing vibration 667 cm
−1 or alanine CN and CC stretching vibration 848 cm
−1 are the most artifact prone [
13]. They constitute approximately 10% of the measured ROA signal.
Table 1.
Enantiomeric excess analysis of α-pinene spectra depicted in
Figure 1. See text for detailed descriptions.
Table 1.
Enantiomeric excess analysis of α-pinene spectra depicted in
Figure 1. See text for detailed descriptions.
| i | | | | | | | | | | | | |
|---|
| 1 | 1.000 | 100.00 | 100.00 | 0.00 | 100.00 | 0.00 | 100.00 | 0.00 | 100.00 | 0.00 | 100.34 | −0.34 |
| 2 | 1.005 | 75.49 | 75.32 | 0.17 | 75.52 | 0.21 | 75.28 | 0.21 | 75.49 | 0.21 | 75.43 | 0.06 |
| 3 | 1.004 | 52.79 | 52.69 | 0.11 | 52.86 | 0.17 | 52.62 | 0.18 | 52.79 | 0.18 | 52.72 | 0.07 |
| 4 | 0.997 | 25.28 | 25.21 | 0.07 | 25.38 | 0.17 | 25.11 | 0.17 | 25.28 | 0.17 | 25.15 | 0.13 |
| 5 | 0.994 | 0.59 | 0.64 | −0.06 | 0.72 | 0.08 | 0.51 | 0.08 | 0.59 | 0.08 | 0.50 | 0.08 |
| 6 | 1.004 | −26.39 | −26.22 | −0.17 | −26.22 | 0.00 | −26.38 | −0.01 | −26.39 | −0.01 | −26.45 | 0.05 |
| 7 | 1.000 | −53.28 | −53.00 | −0.27 | −53.07 | −0.07 | −53.21 | −0.07 | −53.28 | −0.07 | −53.33 | 0.06 |
| 8 | 0.996 | −75.41 | −74.96 | −0.45 | −75.17 | −0.21 | −75.19 | −0.21 | −75.40 | −0.21 | −75.34 | −0.07 |
| 9 | 0.995 | −100.00 | −99.39 | −0.61 | −99.73 | −0.34 | −99.65 | −0.35 | −100.00 | −0.34 | −99.80 | −0.20 |
| | | 0.28 | | 0.17 | | 0.18 | | 0.18 | | 0.15 |
Table 2.
Enantiomeric excess analysis results of the alanine spectra depicted in
Figure 2. See text for detailed descriptions.
Table 2.
Enantiomeric excess analysis results of the alanine spectra depicted in
Figure 2. See text for detailed descriptions.
| i | | | | | | | | | | | | |
|---|
| 1 | 1.000 | 100.00 | 100.00 | 0.00 | 100.00 | 0.00 | 100.00 | 0.00 | 100.00 | 0.00 | 100.13 | −0.13 |
| 2 | 1.021 | 75.23 | 74.24 | 0.99 | 74.74 | 0.51 | 74.73 | 0.50 | 75.23 | 0.50 | 74.78 | 0.45 |
| 3 | 1.009 | 50.08 | 48.84 | 1.23 | 49.09 | 0.25 | 49.78 | 0.30 | 50.08 | 0.30 | 49.89 | 0.19 |
| 4 | 1.028 | 24.37 | 23.13 | 1.24 | 22.87 | −0.25 | 24.62 | −0.25 | 24.37 | −0.25 | 24.74 | −0.38 |
| 5 | 1.003 | 0.08 | −1.88 | 1.96 | −1.89 | −0.01 | 0.20 | −0.12 | 0.08 | −0.12 | 0.23 | −0.15 |
| 6 | 1.035 | −23.93 | −26.33 | 2.41 | −26.37 | −0.04 | −23.88 | −0.05 | −23.93 | −0.05 | −23.91 | −0.02 |
| 7 | 1.020 | −50.12 | −53.24 | 3.12 | −53.08 | 0.16 | −50.24 | 0.12 | −50.12 | 0.12 | −50.40 | 0.28 |
| 8 | 1.027 | −75.21 | −78.26 | 3.04 | −78.67 | −0.41 | −74.85 | −0.37 | −75.21 | −0.37 | −74.93 | −0.29 |
| 9 | 1.022 | −100.00 | −103.74 | 3.74 | −103.95 | −0.20 | −99.87 | −0.13 | −100.00 | −0.13 | −100.09 | 0.09 |
| | | 2.28 | | 0.26 | | 0.26 | | 0.26 | | 0.26 |
The determination of EE by PLS is quite robust to artifacts in ROA spectra. ROA artifact correction had minimal influence on the results obtained from the PLS method, and therefore these results are not shown in the tables. The PLS method gives only slightly better results than the simple artifact reduction method that we described. A major advantage of our method may be that the stated accuracies are achieved for independent pairs of spectra (only two spectra are needed), one of which was chosen as reference A.
Coincidentally, the determined EE of reference B for both α-pinene and alanine is close to 100% within the error margin. Therefore, correction of the enantiomeric purity of reference B according to Equation (9) does not yield a significant improvement. However, if the EEs of the two “pure” forms were different, we believe that this correction may represent a substantial improvement in the results.
The importance of Raman baseline correction and ROA normalization is described by comparing
Table 1 and
Table 2 with
Table 3 and
Table 4, where the Raman baseline correction and ROA normalization were not applied (
nnorm factor was set to unity). The results show that, unless the baseline correction and the resulting normalization of the ROA spectra is performed, it is not possible to reduce the standard deviation of the errors of EE below 0.26% for α-pinene and approximately 1% for alanine.
4.2. Optimization of Parameters for EE Determination
The first of the parameters whose optimal values that we attempted to find were the Savitzky–Golay smoothing parameters. The results of the determination of the standard deviation of the EE determination errors after artifact correction
, together with the coefficient
cB1 indicating the determined EE of reference
B, are shown in
Table 5. Other parameters were set as in the previous section.
The results show that a slight reduction in the standard deviation was achieved for mild smoothing; however, the overall accuracy of the EE determination was not very dependent on the smoothing parameters as long as the smoothing and subsequent resolution degradation was not overly significant.
Another important set of parameters was the spectral range selection for Raman baseline correction, ROA normalization, artifact correction and EE determination. For the evaluations, we used the standard deviation of the EE determination errors after artifact correction
again with the coefficient
cB1, and the results are shown in
Table 6 for α-pinene and in
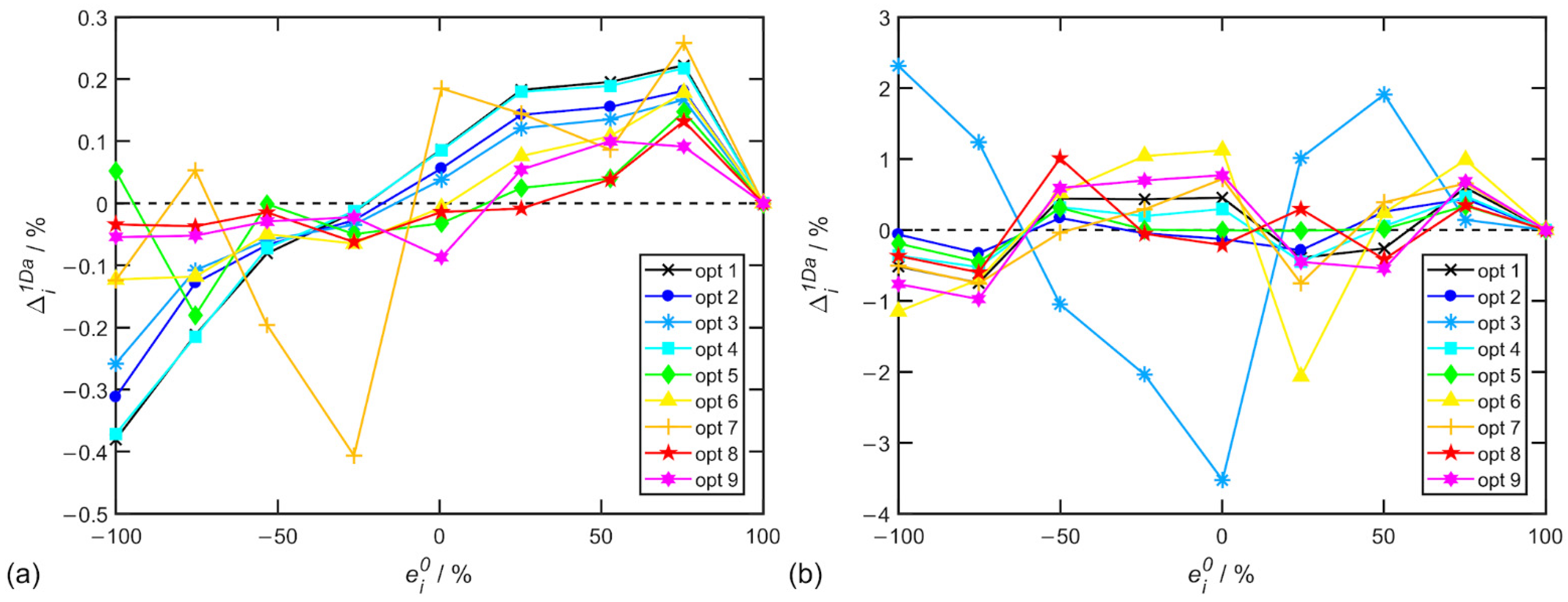
Table 7 for alanine. Individual EE determination errors for options in
Table 6 and
Table 7 are depicted in
Figure 3.
The spectral ranges were chosen considering the presence of dominant artifacts at 667 cm−1 for α-pinene and 848 cm−1 for alanine, which divide the spectrum into approximately two halves. For α-pinene, it was also important to include a relatively isolated polarized band around 1660 cm−1 and a region towards 2000 cm−1 where bands corresponding to fundamental vibrations do not occur; however, this region can be important for the correct determination of the background in Raman spectra.
These results confirm that the appropriate choice of spectral range for baseline correction and ROA normalization can lead to a substantial reduction in EE determination errors.
Table 6 shows that, if the spectral range used for normalization of α-pinene’s spectra is limited to 1500–1800 cm
−1, i.e., around the relatively isolated spectral band of 1660 cm
−1, the most accurate results can be obtained.
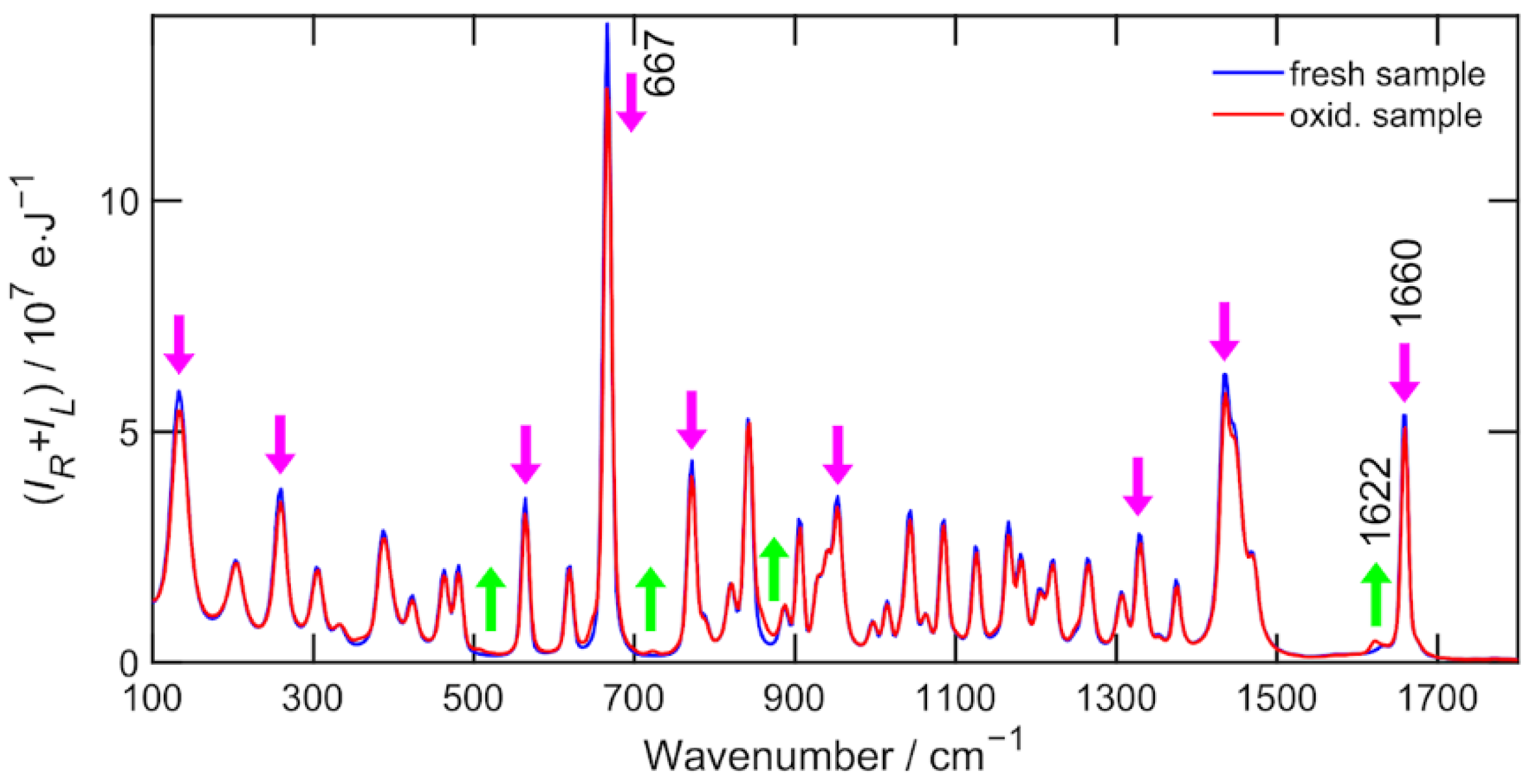
However, two caveats must be added. First, α-pinene is a substance that undergoes air oxidation over longer time scales of weeks and one of the most pronounced changes is in the vicinity of the 1660 cm
−1 band (see
Figure A1). Second, the region 1700 cm
−1 and above, while free of fundamental vibrations, contains a number of low-intensity but significant bands from combinatorial vibrations (ref. [
15]) that make proper baseline correction not an easy task.
Not overly surprisingly, the exclusion of the bands carrying the largest artifacts leads to an increase in the accuracy of the EE determination, for example, limiting the EE determination to the 700–1500 cm−1 range for α-pinene. However, it was surprising that even higher accuracy of EE determination for α-pinene was achieved for another drastic reduction of the spectral range for EE determination to only a few bands in the 700–940 cm−1 region. Admittedly, these bands achieve a large ROA to Raman ratio and are little burdened by artifacts.
Therefore, the whole reliable spectral region 100–2000 cm−1 can be used as the first choice; however, an appropriate choice of spectral ranges can lead to a substantial increase in the accuracy of the EE determination.
4.3. Dependence of EE Determination Accuracy on Measurement Time
In the last section, we attempted to use the measured data to investigate how the accuracy of the EE determination depends on the measurement time of the ROA spectra. In other words, we try to answer the question of how long ROA measurements are needed to be able to achieve a certain accuracy of EE determination.
The ROA spectra are generally shot-noise limited [
13]. As the signal-to-noise ratio increases with a square root of the exposition time, it is reasonable to describe the standard deviation of EE determination errors as:
where
is the standard deviation expected for the unit exposition time
t and
is the limit of the standard deviation for infinite time measurement, which can be interpreted as a residual systematic error.
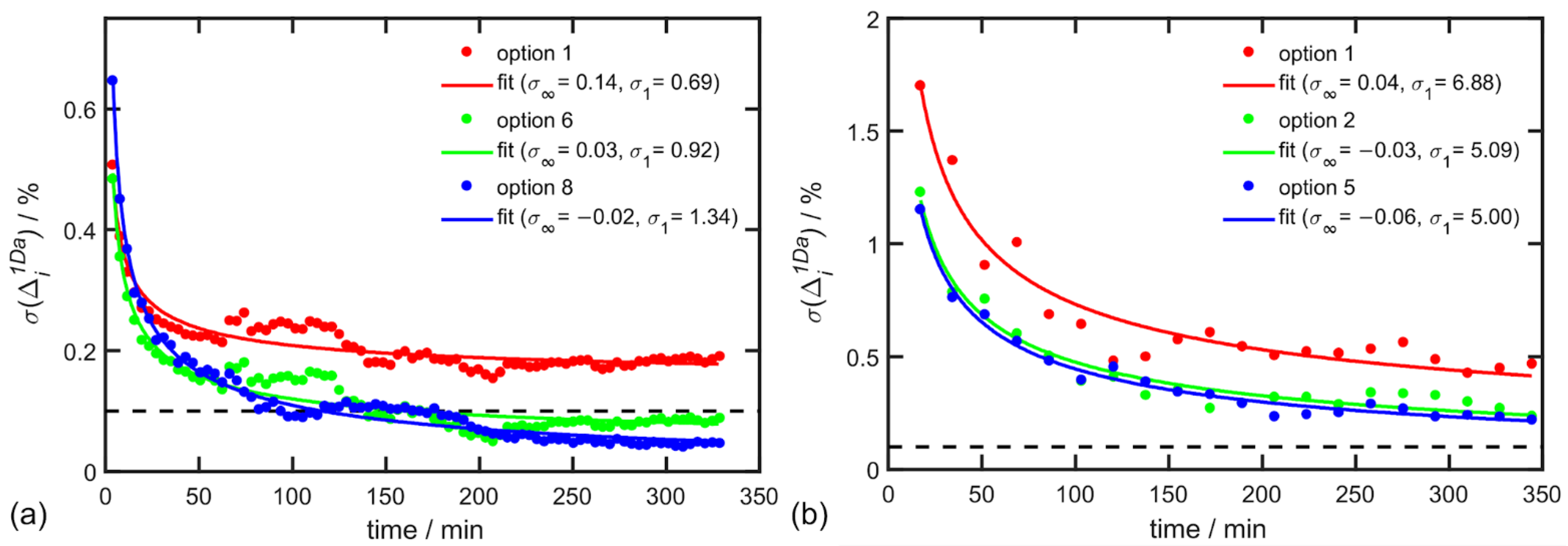
Raman and ROA spectra were exported periodically during the experiment, for α-pinene every 3.91 min and for alanine every 17.2 min. The time dependence of
was then calculated for every cumulative step for three different combinations of parameters listed in
Table 6 and
Table 7 and results are depicted in
Figure 4.
In the case of α-pinene, the EE accuracy of 0.1% was already achieved after 100 min of exposition time and accuracy of 0.05% after 330 min of exposition time for the best choice of parameters (option 8 in
Table 6). A similar convergence was confirmed by alanine in aqueous solution but with a lower accuracy ~0.22% after 345 min. The worse accuracy for alanine is caused by the ten-times weaker ROA signal in comparison to α-pinene in the analyzed spectral region.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}




