
Impact of Molecular Symmetry/Asymmetry on Insulin-Sensitizing Treatments for Type 2 Diabetes

 , , and
, , and 
Abstract
:1. Introduction
2. Use of Symmetrical Compounds
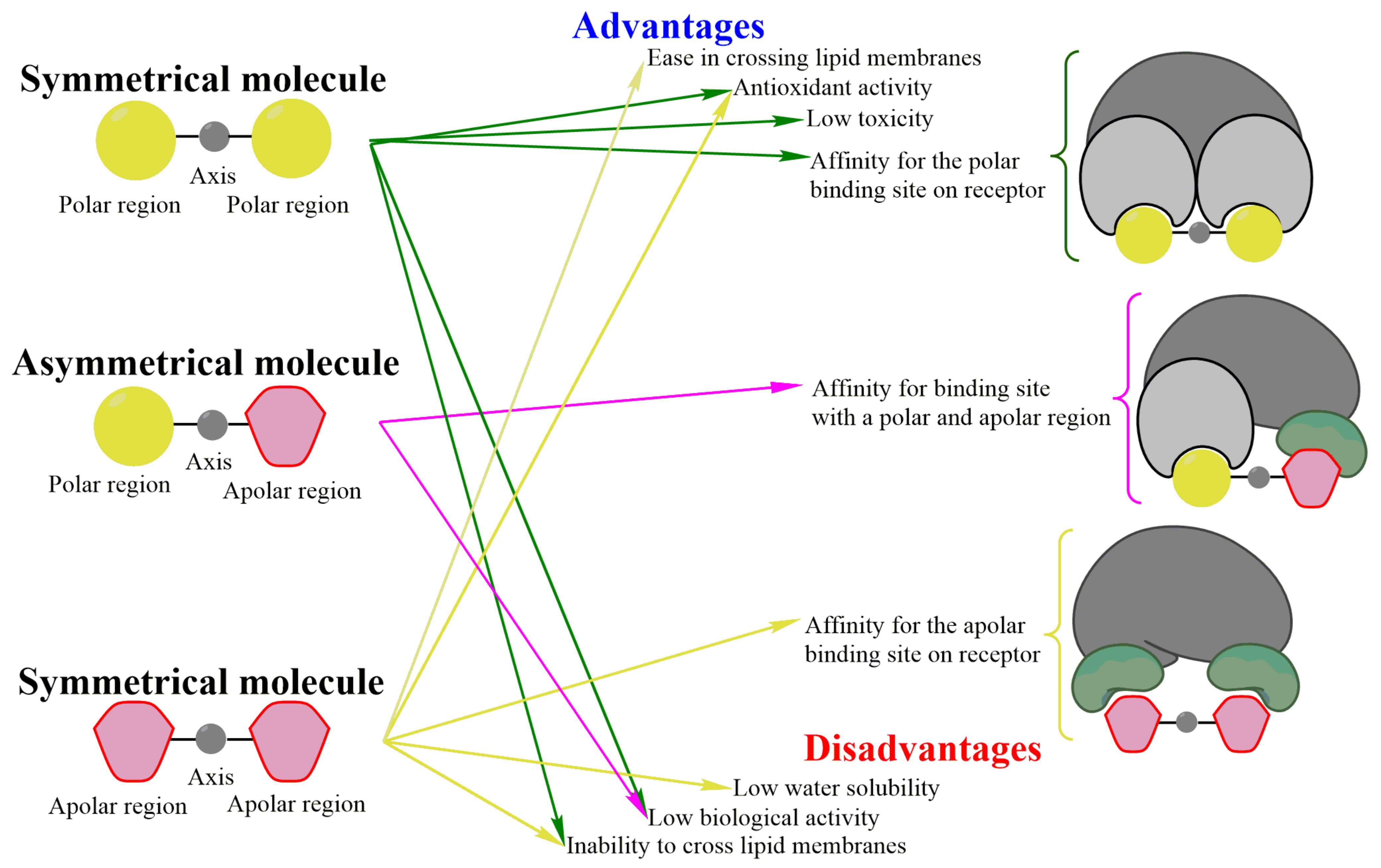
2.1. Advantages of Molecular Symmetry
2.1.1. Low Toxicity and Good Antioxidant Activity
2.1.2. Affinity of Ligands for Their Receptors
2.2. Disadvantages of Molecular Symmetry
2.3. Symmetry: An Advantage or Disadvantage for Ligand-Receptor Interactions?
3. Use of Asymmetrical Molecules: Analogs and Derivatives
3.1. Advantages of Molecular Asymmetry
3.2. Disadvantages of Molecular Asymmetry
4. Theoretical Studies (In Silico) of Symmetrical and Asymmetrical Insulin Sensitizers
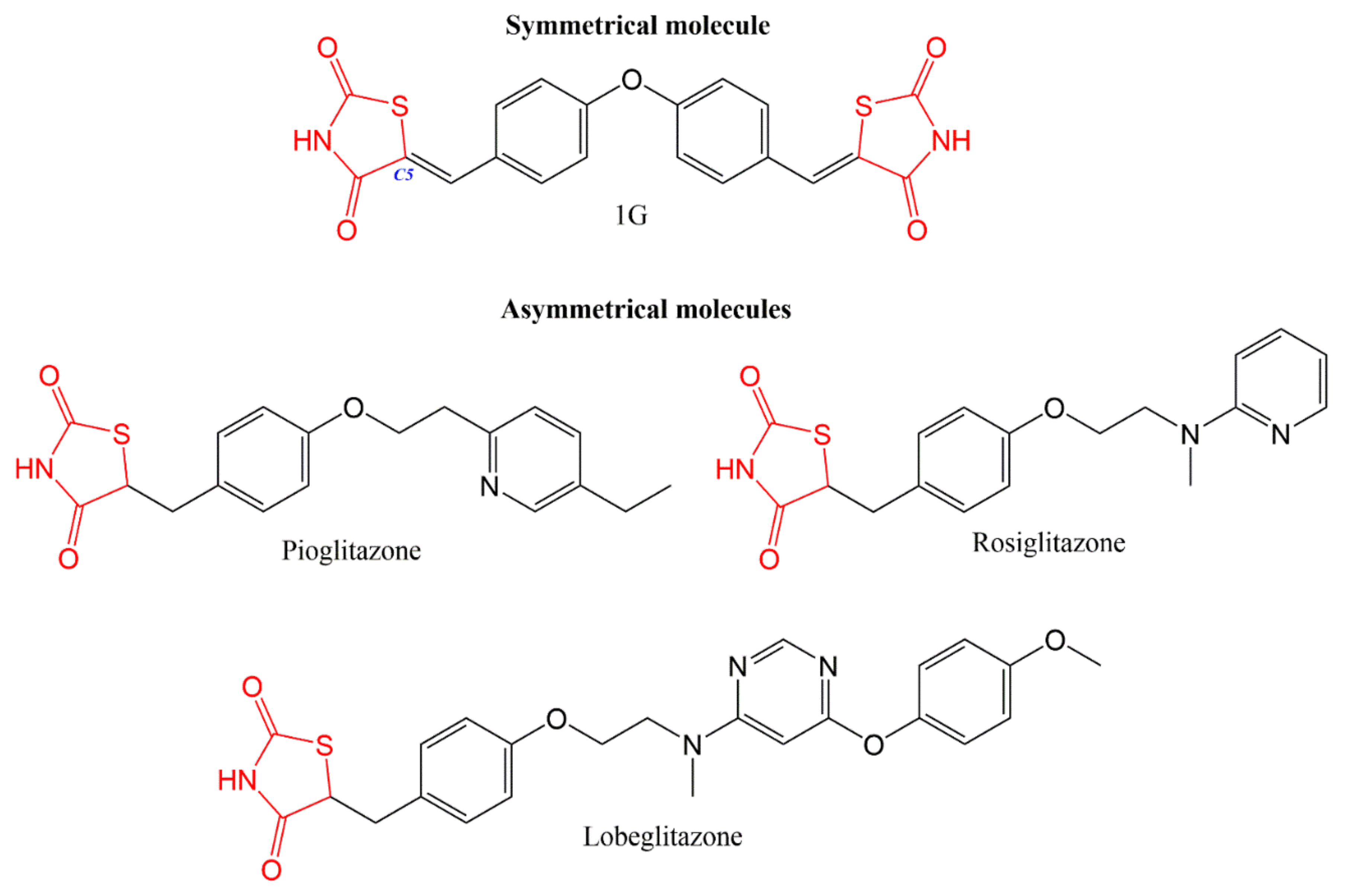
4.1. Design and Synthesis of Symmetrical and Asymmetrical Molecules
4.2. Structure-Related Physicochemical Properties and Pharmacokinetics
4.3. Prediction of Drug Targets
5. In Vitro Studies of Symmetrical and Asymmetrical Molecules
5.1. Activity of Thiazolidinediones on Different Cells
5.2. Effect of Biguanides on Different Cells
6. In Vivo Studies of Symmetrical and Asymmetrical Molecules
6.1. Symmetrical Compounds
6.2. Asymmetrical Compounds
6.3. Toxicity
6.4. Ex Vivo Studies of Symmetrical and Asymmetrical Molecules
7. Other Uses of Symmetrical and Asymmetrical Molecules
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Álvarez-Almazán, S.; Navarrete-Vázquez, G.; Padilla-Martínez, I.I.; Correa-Basurto, J.; Alemán-González-duhart, D.; Tamay-Cach, F.; Mendieta-Wejebe, J.E. A new symmetrical thiazolidinedione derivative: In silico design, synthesis, and in vivo evaluation on a streptozotocin-induced rat model of diabetes. Processes 2021, 9, 1294. [Google Scholar] [CrossRef]
- Alemán-González-Duhart, D.; Álvarez-Almazán, S.; Valdes, M.; Tamay-Cach, F.; Mendieta-Wejebe, J.E. In Vivo and Ex Vivo Evaluation of 1, 3-Thiazolidine-2, 4-Dione Derivatives as Euglycemic Agents. PPAR Res. 2021, 2021, 5100531. [Google Scholar] [CrossRef] [PubMed]
- Álvarez-Almazán, S.; Filisola-Villaseñor, J.G.; Alemán-González-Duhart, D.; Tamay-Cach, F.; Mendieta-Wejebe, J.E. Current molecular aspects in the development and treatment of diabetes. J. Physiol. Biochem. 2020, 76, 13–35. [Google Scholar] [CrossRef] [PubMed]
- Alemán-González-Duhart, D.; Tamay-Cach, F.; Álvarez-Almazán, S.; Mendieta-Wejebe, J.E. Current Advances in the Biochemical and Physiological Aspects of the Treatment of Type 2 Diabetes Mellitus with Thiazolidinediones. PPAR Res. 2016, 2016, 7614270. [Google Scholar] [CrossRef] [Green Version]
- Mansour, M. The roles of peroxisome proliferator-activated receptors in the metabolic syndrome. In Progress in Molecular Biology and Translational Science; Elsevier Inc.: Amsterdam, The Netherlands, 2014; Volume 121, pp. 217–266. ISBN 9780128001011. [Google Scholar]
- Zhou, J.; Massey, S.; Story, D.; Li, L. Metformin: An old drug with new applications. Int. J. Mol. Sci. 2018, 19, 2863. [Google Scholar] [CrossRef] [Green Version]
- Pérez-Mellor, A.F.; Spezia, R.; Zehnacker, A. How Symmetry Influences the Dissociation of Protonated Cyclic Peptides. Symmetry 2022, 14, 679. [Google Scholar] [CrossRef]
- Bai, W.J.; Wang, X. Appreciation of symmetry in natural product synthesis. Nat. Prod. Rep. 2017, 34, 1345–1358. [Google Scholar] [CrossRef]
- Nejo, A.A.; Kolawole, G.A.; Opoku, A.R.; Wolowska, J.; O’Brien, P. Synthesis, characterization and preliminary insulin-enhancing studies of symmetrical tetradentate Schiff base complexes of oxovanadium(IV). Inorganica Chim. Acta 2009, 362, 3993–4001. [Google Scholar] [CrossRef]
- Mollazadeh, S.; Hadizadeh, F.; Ferreira, R.J. Theoretical studies on 1,4-dihydropyridine derivatives as P-glycoprotein allosteric inhibitors: Insights on symmetry and stereochemistry. J. Biomol. Struct. Dyn. 2021, 39, 4752–4763. [Google Scholar] [CrossRef]
- Patujo, J.; Azeem, M.; Khan, M.; Muhammad, H.; Raheel, A.; Fatima, S.; Mirza, B.; Hussain, Z.; Badshah, A. Assessing the biological potential of new symmetrical ferrocene based bisthiourea analogues. Bioorg. Chem. 2021, 106, 104180. [Google Scholar] [CrossRef]
- Naz, S.; Zahoor, M.; Umar, M.N.; Ali, B.; Ullah, R.; Shahat, A.A.; Mahmood, H.M.; Sahibzada, M.U.K. Enzyme inhibitory, antioxidant and antibacterial potentials of synthetic symmetrical and unsymmetrical thioureas. Drug Des. Dev. Ther. 2019, 13, 3485–3495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lorieau, J.L. Partial alignment, residual dipolar couplings and molecular symmetry in solution NMR. J. Biomol. NMR 2019, 73, 477–491. [Google Scholar] [CrossRef]
- Crommelin, D.J.A.; Sindelar, R.D.; Meibohm, B. Pharmaceutical Biotechnology. In Fundamentals and Applications, 5th ed.; Springer: Cham, Switzerland, 2019; ISBN 9783030007096. [Google Scholar]
- Shen, J.; Cheng, F.; Xu, Y.; Li, W.; Tang, Y. Estimation of ADME properties with substructure pattern recognition. J. Chem. Inf. Model. 2010, 50, 1034–1041. [Google Scholar] [CrossRef]
- Álvarez-Almazán, S.; Bello, M.; Tamay-Cach, F.; Martínez-Archundia, M.; Alemán-González-Duhart, D.; Correa-Basurto, J.; Mendieta-Wejebe, J.E. Study of new interactions of glitazone’s stereoisomers and the endogenous ligand 15d-PGJ2 on six different PPAR gamma proteins. Biochem. Pharmacol. 2017, 142, 168–193. [Google Scholar] [CrossRef] [PubMed]
- Iyer, P.; Bolla, J.; Kumar, V.; Gill, M.S.; Sobhia, M.E. In silico identification of targets for a novel scaffold, 2-thiazolylimino-5-benzylidin-thiazolidin-4-one. Mol. Divers. 2015, 19, 855–870. [Google Scholar] [CrossRef]
- Jang, J.Y.; Bae, H.; Lee, Y.J.; Choi, Y.I.; Kim, H.J.; Park, S.B.; Suh, S.W.; Kim, S.W.; Han, B.W. Structural Basis for the Enhanced Anti-Diabetic Efficacy of Lobeglitazone on PPARγ. Sci. Rep. 2018, 8, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shahzad, D.; Saeed, A.; Larik, F.A.; Channar, P.A.; Abbas, Q.; Alajmi, M.F.; Ifzan Arshad, M.; Erben, M.F.; Hassan, M.; Raza, H.; et al. Novel C-2 symmetric molecules as α-glucosidase and α-amylase inhibitors: Design, synthesis, kinetic evaluation, molecular docking and pharmacokinetics. Molecules 2019, 24, 1511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geldenhuys, W.J.; Skolik, R.; Konkle, M.E.; Menze, M.A.; Long, T.E.; Robart, A.R. Binding of thiazolidinediones to the endoplasmic reticulum protein nutrient-deprivation autophagy factor-1. Bioorg. Med. Chem. Lett. 2019, 29, 901–904. [Google Scholar] [CrossRef]
- Rotermund, C.; Machetanz, G.; Fitzgerald, J.C. The therapeutic potential of metformin in neurodegenerative diseases. Front. Endocrinol. 2018, 9, 400. [Google Scholar] [CrossRef]
- Kroker, A.J.; Bruning, J.B. Review of the structural and dynamic mechanisms of PPAR γ partial agonism. PPAR Res. 2015, 2015, 816856. [Google Scholar] [CrossRef] [Green Version]
- Fan, L.Y.; Zhu, D.; Yang, Y.; Huang, Y.; Zhang, S.N.; Yan, L.C.; Wang, S.; Zhao, Y.H. Comparison of modes of action among different trophic levels of aquatic organisms for pesticides and medications based on interspecies correlations and excess toxicity: Theoretical consideration. Ecotoxicol. Environ. Saf. 2019, 177, 25–31. [Google Scholar] [CrossRef]
- Gilbert, T.; Rodriguez-Boulan, E. Induction of vacuolar apical compartments in the Caco-2 intestinal epithelial cell line. J. Cell Sci. 1991, 100, 451–458. [Google Scholar] [CrossRef] [PubMed]
- Veber, D.F.; Johnson, S.R.; Cheng, H.Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef] [PubMed]
- Alemán-González-Duhart, D.; Tamay-Cach, F.; Correa-Basurto, J.; Padilla-Martínez, I.I.; Álvarez-Almazán, S.; Mendieta-Wejebe, J.E. In silico design, chemical synthesis and toxicological evaluation of 1,3-thiazolidine-2,4-dione derivatives as PPARγ agonists. Regul. Toxicol. Pharmacol. 2017, 86, 25–32. [Google Scholar] [CrossRef]
- Elhenawy, A.A.; Salama, A.A.A.; All, M.M.A.; Alomri, A.A.; Nassar, H.S. Synthesis, characterization and discovery novel anti-diabetic and anti-hyperlipidemic thiazolidinedione derivatives. Int. J. Pharm. Sci. Rev. Res. 2015, 31, 23–30. [Google Scholar]
- Kaserer, T.; Obermoser, V.; Weninger, A.; Gust, R.; Schuster, D. Evaluation of selected 3D virtual screening tools for the prospective identification of peroxisome proliferator-activated receptor (PPAR) γ partial agonists. Eur. J. Med. Chem. 2016, 124, 49–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gfeller, D.; Grosdidier, A.; Wirth, M.; Daina, A.; Michielin, O.; Zoete, V. SwissTargetPrediction: A web server for target prediction of bioactive small molecules. Nucleic Acids Res. 2014, 42, W32–W38. [Google Scholar] [CrossRef] [PubMed]
- Bakry, R.; Vallant, R.M.; Najam-ul-Haq, M.; Rainer, M.; Szabo, Z.; Huck, C.W.; Bonn, G.K. Medicinal applications of fullerenes. Int. J. Nanomed. 2007, 2, 639–649. [Google Scholar] [CrossRef]
- Hidalgo-Figueroa, S.; Ramírez-Espinosa, J.J.; Estrada-Soto, S.; Almanza-Pérez, J.C.; Román-Ramos, R.; Alarcón-Aguilar, F.J.; Hernández-Rosado, J.V.; Moreno-Díaz, H.; Díaz-Coutiño, D.; Navarrete-Vázquez, G. Discovery of Thiazolidine-2,4-Dione/Biphenylcarbonitrile Hybrid as Dual PPAR α/γ Modulator with Antidiabetic Effect: In Vitro, In Silico and In Vivo Approaches. Chem. Biol. Drug Des. 2013, 81, 474–483. [Google Scholar] [CrossRef] [PubMed]
- Navarrete-Vázquez, G.; Torres-Gómez, H.; Hidalgo-Figueroa, S.; Ramírez-Espinosa, J.J.; Estrada-Soto, S.; Medina-Franco, J.L.; León-Rivera, I.; Alarcón-Aguilar, F.J.; Almanza-Pérez, J.C. Synthesis, in vitro and in silico studies of a PPARγ and GLUT-4 modulator with hypoglycemic effect. Bioorg. Med. Chem. Lett. 2014, 24, 4575–4579. [Google Scholar] [CrossRef]
- Meyer, F.B.; Marx, C.; Spangel, S.B.; Thierbach, R. Butyrate and Metformin Affect Energy Metabolism Independently of the Metabolic Phenotype in the Tumor Therapy Model. Biomolecules 2021, 11, 1831. [Google Scholar] [CrossRef] [PubMed]
- Kumar, N.; Dey, C.S. Metformin enhances insulin signalling in insulin-dependent and -independent pathways in insulin resistant muscle cells. Br. J. Pharmacol. 2002, 137, 329–336. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, L.G.; Santos, R.N.; Oliva, G.; Andricopulo, A.D. Molecular Docking and Structure-Based Drug Design Strategies. Molecules 2015, 20, 13384–13421. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.-Y.; Zhang, H.-X.; Mezel, M.; Cui, M. Molecular Docking: A powerful approach for structure-based drug discovery. Curr. Comput. Aided Drug Des. 2011, 7, 146–157. [Google Scholar] [CrossRef]
- Mahapatra, M.K.; Kumar, R.; Kumar, M. Synthesis, biological evaluation and in silico studies of 5-(3-methoxybenzylidene)thiazolidine-2,4-dione analogues as PTP1B inhibitors. Bioorg. Chem. 2017, 71, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Hidalgo-Figueroa, S.; Estrada-Soto, S.; Ramírez-Espinosa, J.J.; Paoli, P.; Lori, G.; León-Rivera, I.; Navarrete-Vázquez, G. Synthesis and evaluation of thiazolidine-2,4-dione/benzazole derivatives as inhibitors of protein tyrosine phosphatase 1B (PTP-1B): Antihyperglycemic activity with molecular docking study. Biomed. Pharmacother. 2018, 107, 1302–1310. [Google Scholar] [CrossRef] [PubMed]
- Elkamhawy, A.; Youn Kim, N.; Hassan, A.H.E.; Park, J.-E.; Yang, J.E.; Elsherbeny, M.H.; Paik, S.; Oh, K.S.; Lee, B.H.; Lee, M.Y.; et al. Optimization study towards more potent thiazolidine-2,4-dione IKK-β modulator: Synthesis, biological evaluation and in silico docking simulation. Bioorg. Chem. 2019, 92, 103261. [Google Scholar] [CrossRef]
- Sameeh, M.Y.; Khowdiary, M.M.; Nassar, H.S.; Abdelall, M.M.; Alderhami, S.A.; Elhenawy, A.A. Discovery potent of thiazolidinedione derivatives as antioxidant, α-amylase inhibitor, and antidiabetic agent. Biomedicines 2022, 10, 24. [Google Scholar] [CrossRef]
- Hussain, F.; Khan, Z.; Jan, M.S.; Ahmad, S.; Ahmad, A.; Rashid, U.; Ullah, F.; Ayaz, M.; Sadiq, A. Synthesis, in-vitro α-glucosidase inhibition, antioxidant, in-vivo antidiabetic and molecular docking studies of pyrrolidine-2,5-dione and thiazolidine-2,4-dione derivatives. Bioorg. Chem. 2019, 91, 103128. [Google Scholar] [CrossRef]
- Bhutani, R.; Pathak, D.P.; Kapoor, G.; Husain, A.; Iqbal, M.A. Novel hybrids of benzothiazole-1,3,4-oxadiazole-4-thiazolidinone: Synthesis, in silico ADME study, molecular docking and in vivo anti-diabetic assessment. Bioorg. Chem. 2019, 83, 6–19. [Google Scholar] [CrossRef]
- Mahapatra, M.K.; Bera, K.; Singh, D.V.; Kumar, R.; Kumar, M. In silico modelling and molecular dynamics simulation studies of thiazolidine based PTP1B inhibitors. J. Biomol. Struct. Dyn. 2018, 36, 1195–1211. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.D.; Pasha, T.Y.; Lunagariya, P.; Shah, U.; Bhambharoliya, T.; Tripathi, R.K.P. A Library of Thiazolidin-4-one Derivatives as Protein Tyrosine Phosphatase 1B (PTP1B) Inhibitors: An Attempt To Discover Novel Antidiabetic Agents. ChemMedChem 2020, 15, 1229–1242. [Google Scholar] [CrossRef]
- Sawant, R.L.; Wadekar, J.B.; Kharat, S.B.; Makasare, H.S. Targeting PPAR-γ to design and synthesize antidiabetic thiazolidines. EXCLI J. 2018, 17, 598–607. [Google Scholar] [CrossRef] [PubMed]
- Sameeh, M.Y.; Khowdiary, M.M.; Nassar, H.S.; Abdelall, M.M.; Amer, H.H.; Hamed, A.; Elhenawy, A.A. Thiazolidinedione Derivatives: In Silico, In Vitro, In Vivo, Antioxidant and Anti-Diabetic Evaluation. Molecules 2022, 27, 830. [Google Scholar] [CrossRef]
- Benet, L.Z.; Hosey, C.M.; Ursu, O.; Oprea, T.I. BDDCS, the Rule of 5 and Drugability. Adv. Drug Deliv. Rev. 2016, 101, 89–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butler, A.; Helliwell, M.V.; Zhang, Y.; Hancox, J.C.; Dempsey, C.E. An update on the structure of HERG. Front. Pharmacol. 2020, 10, 1572. [Google Scholar] [CrossRef] [PubMed]
- Rachek, L.I.; Yuzefovych, L.V.; LeDoux, S.P.; Julie, N.L.; Wilson, G.L. Troglitazone, but not rosiglitazone, damages mitochondrial DNA and induces mitochondrial dysfunction and cell death in human hepatocytes. Toxicol. Appl. Pharmacol. 2009, 240, 348–354. [Google Scholar] [CrossRef] [Green Version]
- Eggleton, J.S.; Jialal, I. Thiazolidinediones. Available online: https://www.ncbi.nlm.nih.gov/books/NBK551656/ (accessed on 7 April 2022).
- Nazreen, S.; Alam, M.M.S.; Hamid, H.; Yar, M.S.; Dhulap, A.; Alam, P.; Pasha, M.A.Q.; Bano, S.; Alam, M.M.S.; Haider, S.; et al. Design, Synthesis, and Biological Evaluation of Thiazolidine-2,4-dione Conjugates as PPAR-g Agonists. Arch. Parm. Chem. Life Sci. 2015, 348, 421–432. [Google Scholar] [CrossRef]
- Naim, J.M.; Alam, O.; Alam, M.J.; Shaquiquzzaman, M.; Alam, M.M.; Naidu, V.G.M. Synthesis, docking, in vitro and in vivo antidiabetic activity of pyrazole-based 2, 4-thiazolidinedione derivatives as PPAR-γ modulators. Arch. Parm. Chem. Life Sci. 2018, 2018, e1700223. [Google Scholar] [CrossRef]
- Naim, J.M.; Alam, J.; Nawaz, F.; Naidu, V.G.M.; Aaghaz, S.; Sahu, M.; Siddiqui, N.; Alam, O. Synthesis, molecular docking and anti-diabetic evaluation of 2, 4-thiazolidinedione based amide derivatives. Bioorg. Chem. 2017, 73, 24–36. [Google Scholar] [CrossRef]
- Szychowski, K.A.; Skóra, B.; Kryshchyshyn-dylevych, A.; Kaminskyy, D.; Tobiasz, J.; Lesyk, R.B.; Gminski, J. 4-Thiazolidinone-based derivatives do not affect differentiation of mouse embryo fibroblasts ( 3T3-L1 cell line ) into adipocytes. Chem. Biol. Interact. 2021, 345, 109538. [Google Scholar] [CrossRef] [PubMed]
- Moreno-navarrete, J.M.; Ortega, F.; Moreno, M.; Xifra, G.; Ricart, W.; Fernández-real, J.M. PRDM16 sustains white fat gene expression profile in human adipocytes in direct relation with insulin action. Mol. Cell. Endocrinol. 2015, 405, 84–93. [Google Scholar] [CrossRef]
- Hidalgo-Figueroa, S.; Rodríguez-Luévano, A.; Almanza-Pérez, J.C.; Giacoman-Martínez, A.; Ortiz-Andrade, R.; León-Rivera, I.; Navarrete-Vázquez, G. Synthesis, molecular docking, dynamic simulation and pharmacological characterization of potent multifunctional agent ( dual GPR40-PPAR γ agonist ) for the treatment of experimental type 2 diabetes. Eur. J. Pharmacol. 2021, 907, 174244. [Google Scholar] [CrossRef]
- Martin, G.; Schoonjans, K.; Lefebvre, A.-M.; Staels, B.; Auwerx, J. Coordinate Regulation of the Expression of the Fatty Acid Transport Protein and Acyl-CoA Synthetase Genes by PPARα and PPARγ Activators. J. Biol. Chem. J. 1997, 272, 28210–28217. [Google Scholar] [CrossRef] [Green Version]
- Gong, L.; Jin, H.; Li, Y.; Quan, Y.; Yang, J.; Tang, Q.; Zou, Z. Rosiglitazone ameliorates skeletal muscle insulin resistance by decreasing free fatty acids release from adipocytes. Biochem. Biophys. Res. Commun. 2020, 533, 1122–1128. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Xu, C.; Kuang, J.; Liu, Q.; Jiang, H.; Mo, L.; Geng, B.; Xu, G. Thiazolidinediones attenuate lipolysis and ameliorate dexamethasone-induced insulin resistance. Metabolism 2015, 64, 826–836. [Google Scholar] [CrossRef]
- Kwon, M.J.; Lee, Y.J.; Jung, H.S.; Shin, H.M.; Kim, T.N.; Lee, S.H.; Rhee, B.D.; Kim, M.; Park, J.H. The direct effect of lobeglitazone, a new thiazolidinedione, on pancreatic beta cells: A comparison with other thiazolidinediones. Diabetes Res. Clin. Pract. 2019, 151, 209–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karunakaran, U.; Elumalai, S.; Moon, J.S.; Won, K.C. Pioglitazone-induced AMPK-Glutaminase-1 prevents high glucose-induced pancreatic β -cell dysfunction by glutathione antioxidant system. Redox Biol. 2021, 45, 102029. [Google Scholar] [CrossRef]
- Shannon, C.E.; Ragavan, M.; Palavicini, J.P.; Fourcaudot, M.; Bakewell, T.M.; Valdez, I.A.; Ayala, I.; Jin, E.S.; Madesh, M.; Han, X.; et al. Insulin resistance is mechanistically linked to hepatic mitochondrial remodeling in non-alcoholic fatty liver disease. Mol. Metab. 2021, 45, 101154. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhang, X.; Meng, L.; Gong, M.; Li, J.; Shi, W.; Qiu, J.; Yang, Y.; Zhao, J.; Suo, Y.; et al. Pioglitazone Inhibits Diabetes-Induced Atrial Mitochondrial Oxidative Stress and Improves Mitochondrial Biogenesis, Dynamics, and Function Through the PPAR-γ/ PGC-1α Signaling Pathway. Front. Pharmacol. 2021, 12, 658362. [Google Scholar] [CrossRef]
- Oleksandr, G.; Myrgorodska, I.; Rocchi, S.; Ronco, C.; Benhida, R. Biguanides drugs: Past success stories and promising future for drug discover. Eur. J. Med. Chem. 2021, 224, 113726. [Google Scholar] [CrossRef]
- Jacobs, D.B.; Hayes, G.R.; Truglia, J.A.; Lockwood, D.H. Effects of metformin on insulin receptor tyrosine kinase activity in rat adipocytes. Diabetologia 1986, 29, 798–801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sasson, S.; Gorowits, N.; Joost, G.; King, G.L.; Cerasi, E.; Kaiser, N. Regulation by metformin of the hexose transport system in vascular endothelial and smooth muscle cells. Br. J. Pharmacol. 1996, 117, 1318–1324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grisouard, J.; Timper, K.; Radimerski, T.M.; Frey, D.M.; Peterli, R.; Kola, B.; Korbonits, M.; Herrmann, P.; Krähenbühl, S.; Zulewski, H.; et al. Mechanisms of metformin action on glucose transport and metabolism in human adipocytes. Biochem. Pharmacol. 2010, 80, 1736–1745. [Google Scholar] [CrossRef] [PubMed]
- Gunton, J.E.; Delhanty, P.J.D.; Takahashi, S.; Baxter, R.C. Metformin Rapidly Increases Insulin Receptor Activation in Human Liver and Signals Preferentially through Insulin-Receptor Substrate-2. J. Clin. Endocrinol. Metab. 2015, 88, 1323–1332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, N.; Kaul, C.L.; Ishrath, A.; Dey, C.S. Combination of metformin and thiazolidindiones restore insulin signalling in insulin-resistant cultured myotubes. Life Sci. 2004, 74, 1877–1888. [Google Scholar] [CrossRef]
- Meuillet, E.J.; Wiernsperger, N.; Mania-Farnell, B.; Hubert, P.; Cremel, G. Metformin modulates insulin receptor signaling in normal and cholesterol-treated human hepatoma cells (HepG2). Eur. J. Pharmacol. 1999, 377, 241–252. [Google Scholar] [CrossRef]
- Yuan, L.; Ziegler, R.; Hamann, A. Metformin modulates insulin post-receptor signaling transduction in chronically insulin-treated Hep G2 cells. Acta Pharm. Sin. B 2003, 24, 55–60. [Google Scholar]
- Hundal, H.S.; Ramlal, T.; Reyes, R.; Leiter, L.A.; Klip, A. Cellular Mechanism of Metformin Action Involves Glucosa Transporter Translocation from an Intracellular Pool to the Plasma Membrane in L6 Muscle Cells. Endocrinology 1992, 131, 1165–1173. [Google Scholar] [CrossRef]
- Pryor, P.; Liu, S.; Clark, A.; Yang, J.; Holman, G.; Tosh, D. Chronic insulin effects on insulin signalling and GLUT4 endocytosis are reversed by metformin. Biochem. J. 2000, 348, 83–91. [Google Scholar] [CrossRef]
- Melin, B.; Cherqui, G.; Blivet, M.J.; Caron, M.; Lascols, O.; Capeau, J.; Picard, J. Dual effect of metformin in cultured rat hepatocytes: Potentiation of insulin action and prevention of insulin-induced resistance. Metabolism 1990, 39, 1089–1095. [Google Scholar] [CrossRef]
- Wollen, N.; Bailey, C.J. Inhibition of hepatic gluconeogenesis by metformin. Synergism with insulin. Biochem. Pharmacol. 1988, 37, 4353–4358. [Google Scholar] [CrossRef]
- Argaud, D.; Roth, H.; Wiernsperger, N.; Leverve, X.M. Metformin decreases gluconeogenesis by enhancing the pyruvate kinase flux in isolated rat hepatocytes. Eur. J. Biochem. 1993, 213, 1341–1348. [Google Scholar] [CrossRef] [PubMed]
- Yu, B.; Pugazhenthi, S.; Khandelwal, R.L. Effects of Metformin on Glucose and Glucagon Regulated Gluconeogenesis in Cultured Normal and Diabetic Hepatocytes. Biochem. Biophys. Res. Commun. 1994, 48, 949–954. [Google Scholar]
- Fulgencio, J.P.; Kohl, C.; Girard, J.; Pégorier, J.P. Effect of metformin on fatty acid and glucose metabolism in freshly isolated hepatocytes and on specific gene expression in cultured hepatocytes. Biochem. Pharmacol. 2001, 62, 439–446. [Google Scholar] [CrossRef]
- Otto, M.; Breinholt, J.; Westergaard, N. Metformin inhibits glycogen synthesis and gluconeogenesis in cultured rat hepatocytes. Diabetes Obes. Metab. 2003, 5, 189–194. [Google Scholar] [CrossRef] [PubMed]
- Owen, M.R.; Doran, E.; Halestrap, A.P. Evidence that metformin exerts its anti-diabetic effects through inhibition of complex 1 of the mitochondrial respiratory chain. Biochem. J. 2000, 348, 607–614. [Google Scholar] [CrossRef]
- LaMoia, T.E.; Butrico, G.M.; Kalpage, H.A.; Goedeke, L.; Hubbard, B.T.; Vatner, D.F.; Gaspar, R.C.; Zhang, X.-M.; Cline, G.W.; Nakahara, K.; et al. Metformin, phenformin, and galegine inhibit complex IV activity and reduce glycerol-derived gluconeogenesis. Proc. Natl. Acad. Sci. USA 2022, 119, e2122287119. [Google Scholar] [CrossRef] [PubMed]
- Yuan, L.; Ziegler, R.; Hamann, A. Inhibition of phosphoenolpyruvate carboxykinase gene expression by metformin in cultured hepatocytes. Chin. Med. J. 2002, 115, 1843–1848. [Google Scholar]
- Morioka, K.; Nakatani, K.; Matsumoto, K.; Urakawa, H.; Kitagawa, N.; Katsuki, A.; Hori, Y.; Gabazza, E.C.; Yano, Y.; Nishioka, J.; et al. Metformin-induced suppression of glucose-6-phosphatase expression is independent of insulin signaling in rat hepatoma cells. Int. J. Mol. Med. 2005, 15, 449–452. [Google Scholar] [PubMed]
- Ji, X.; Wang, S.; Tang, H.; Zhang, Y.; Zhou, F.; Zhang, L.; Zhu, Q.; Zhu, K.; Liu, Q.; Liu, Y.; et al. PPP1R3C mediates metformin-inhibited hepatic gluconeogenesis. Metabolism 2019, 98, 62–75. [Google Scholar] [CrossRef]
- Cao, J.; Meng, S.; Chang, E.; Beckwith-Fickas, K.; Xiong, L.; Cole, R.N.; Radovick, S.; Wondisford, F.E.; He, L. Low concentrations of metformin suppress glucose production in hepatocytes through AMP-activated protein kinase (AMPK). J. Biol. Chem. 2014, 289, 20435–20446. [Google Scholar] [CrossRef] [Green Version]
- Aatsinki, S.M.; Buler, M.; Salomäki, H.; Koulu, M.; Pavek, P.; Hakkola, J. Metformin induces PGC-1α expression and selectively affects hepatic PGC-1α functions. Br. J. Pharmacol. 2014, 171, 2351–2363. [Google Scholar] [CrossRef] [Green Version]
- Adeghate, E.; Adem, A.; Hasan, M.Y.; Tekes, K.; Kalasz, H. Medicinal Chemistry and Actions of Dual and Pan PPAR Modulators. Open Med. Chem. J. 2011, 5, 93–98. [Google Scholar] [CrossRef]
- Abdelhamid, A.M.; Saber, S.; Youssef, M.E.; Gaafar, A.G.A.; Eissa, H.; Abd-Eldayem, M.A.; Alqarni, M.; Batiha, G.E.S.; Obaidullah, A.J.; Shahien, M.A.; et al. Empagliflozin adjunct with metformin for the inhibition of hepatocellular carcinoma progression: Emerging approach for new application. Biomed. Pharmacother. 2022, 145, 112455. [Google Scholar] [CrossRef] [PubMed]
- Boulanger, H.; Mansouri, R.; Gautier, J.F.; Glotz, D. Are peroxisome proliferator-activated receptors new therapeutic targets in diabetic and non-diabetic nephropathies? Nephrol. Dial. Transplant. 2006, 21, 2696–2702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, K.; Li, Y.; Guo, Z.; Zeng, Y.; Zhang, W.; Wang, H. Metformin: Current clinical applications in nondiabetic patients with cancer. Aging 2020, 12, 3993–4009. [Google Scholar] [CrossRef]
- Cho, M.C.; Lee, D.H.; Kim, E.J.; Lee, J.Y.; Kang, J.W.; Song, J.H.; Chong, Y.; Kim, Y.; Hong, J.T.; Yoon, D.Y. Novel PPARγ partial agonists with weak activity and no cytotoxicity; Identified by a simple PPARγ ligand screening system. Mol. Cell. Biochem. 2011, 358, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Derosa, G.; Cicero, A.F.G.; Gaddi, A.; Ragonesi, P.D.; Fogari, E.; Bertone, G.; Ciccarelli, L.; Piccinni, M.N. Metabolic effects of pioglitazone and rosiglitazone in patients with diabetes and metabolic syndrome treated with glimepiride: A twelve-month, multicenter, double-blind, randomized, controlled, parallel-group trial. Clin. Ther. 2004, 26, 744–754. [Google Scholar] [CrossRef]
- Spence, J.D.; Viscoli, C.; Kernan, W.N.; Young, L.H.; Furie, K.; DeFronzo, R.; Abdul-Ghani, M.; Dandona, P.; Inzucchi, S.E. Efficacy of lower doses of pioglitazone after stroke or transient ischaemic attack in patients with insulin resistance. Diabetes Obes. Metab. 2022, 24, 1150–1158. [Google Scholar] [CrossRef]
- Nesti, L.; Tricò, D.; Mengozzi, A.; Natali, A. Rethinking pioglitazone as a cardioprotective agent: A new perspective on an overlooked drug. Cardiovasc. Diabetol. 2021, 20, 109. [Google Scholar] [CrossRef] [PubMed]
- Bahare, R.S.; Ganguly, S.; Agrawal, R.; Dikshit, S.N. Thiazolidine: A Potent Candidate for Central Nervous Systems Diseases. Cent. Nerv. Syst. Agents Med. Chem. 2017, 17, 26–29. [Google Scholar] [CrossRef]
- Sharma, R.K.; Younis, Y.; Mugumbate, G.; Njoroge, M.; Gut, J.; Rosenthal, P.J.; Chibale, K. Synthesis and structure-activity-relationship studies of thiazolidinediones as antiplasmodial inhibitors of the Plasmodium falciparum cysteine protease falcipain-2. Eur. J. Med. Chem. 2015, 90, 507–518. [Google Scholar] [CrossRef] [PubMed]
- Asati, V.; Mahapatra, D.K.; Bharti, S.K. Thiazolidine-2,4-diones as multi-targeted scaffold in medicinal chemistry: Potential anticancer agents. Eur. J. Med. Chem. 2014, 87, 814–833. [Google Scholar] [CrossRef] [PubMed]
- Begum, A.B.; Begum, M.; Ranganatha, V.L.; Prashanth, T.; Zameer, F.; Hegdekatte, R.; Khanum, S.A. Synthesis, antioxidant, and xanthine oxidase inhibitory activities of 5-[4-[2-(5-ethyl-2-pyridinyl)ethoxy]phenyl]methyl]-2,4-thiazolidinedione derivatives. Arch. Pharm. 2014, 347, 247–255. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, P.S.; Karale, S.N.; Khandebharad, A.U.; Agrawal, B.R.; Sarda, S.R. Design, Synthesis, and Biological Evaluation of Newer Arylidene Incorporated 4-Thiazolidinones Derivatives as Potential Antimicrobial Agents. Polycycl. Aromat. Compd. 2020, 1–14. [Google Scholar] [CrossRef]
- Sucheta; Tahlan, S.; Verma, P.K. Biological potential of thiazolidinedione derivatives of synthetic origin. Chem. Cent. J. 2017, 11, 130. [Google Scholar] [CrossRef]
- Saraei, P.; Asadi, I.; Kakar, M.A.; Moradi-Kor, N. The beneficial effects of metformin on cancer prevention and therapy: A comprehensive review of recent advances. Cancer Manag. Res. 2019, 11, 3295–3313. [Google Scholar] [CrossRef] [Green Version]
- Song, A.; Zhang, C.; Meng, X. Mechanism and application of metformin in kidney diseases: An update. Biomed. Pharmacother. 2021, 138, 111454. [Google Scholar] [CrossRef]
- Nestler, J.E. Metformin for the Treatment of the Polycystic Ovary Syndrome. N. Engl. J. Med. 2008, 358, 47–54. [Google Scholar] [CrossRef] [Green Version]
- Lashen, H. Role of metformin in the management of polycystic ovary syndrome. Ther. Adv. Endocrinol. Metab. 2010, 1, 117–128. [Google Scholar] [CrossRef] [PubMed]
- Mohammed, I.; Hollenberg, M.D.; Ding, H.; Triggle, C.R. A Critical Review of the Evidence That Metformin Is a Putative Anti-Aging Drug That Enhances Healthspan and Extends Lifespan. Front. Endocrinol. 2021, 12, 718942. [Google Scholar] [CrossRef]
- Gettings, S.D.; Reeve, J.E.; King, L.J. Possible role of intracellular Ca2+ in the toxicity of phenformin. Biochem. Pharmacol. 1988, 37, 281–289. [Google Scholar] [CrossRef]
- Dykens, J.A.; Jamieson, J.; Marroquin, L.; Nadanaciva, S.; Billis, P.A.; Will, Y. Biguanide-induced mitochondrial dysfunction yields increased lactate production and cytotoxicity of aerobically-poised HepG2 cells and human hepatocytes in vitro. Toxicol. Appl. Pharmacol. 2008, 233, 203–210. [Google Scholar] [CrossRef] [PubMed]
- National Center for Biotechnology Information Proguanil|C11H16ClN5-PubChem. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/chloroguanide (accessed on 12 April 2022).
- Abbasi, E.; Aval, S.F.; Akbarzadeh, A.; Milani, M.; Nasrabadi, H.T.; Joo, S.W.; Hanifehpour, Y.; Nejati-Koshki, K.; Pashaei-Asl, R. Dendrimers: Synthesis, applications, and properties. Nanoscale Res. Lett. 2014, 9, 247. [Google Scholar] [CrossRef] [Green Version]
- Fang, X.; Meng, Q.; Zhang, H.; Liang, B.; Zhu, S.; Wang, J.; Zhang, C.; Huang, L.S.; Zhang, X.; Schooley, R.T.; et al. Design, synthesis, and biological characterization of a new class of symmetrical polyamine-based small molecule CXCR4 antagonists. Eur. J. Med. Chem. 2020, 200, 112410. [Google Scholar] [CrossRef]
- Vrancken, J.P.M.; Tame, J.R.H.; Voet, A.R.D. Development and applications of artificial symmetrical proteins. Comput. Struct. Biotechnol. J. 2020, 18, 3959–3968. [Google Scholar] [CrossRef] [PubMed]
- Paquin, A.; Reyes-Moreno, C.; Bérubé, G. Recent advances in the use of the dimerization strategy as a means to increase the biological potential of natural or synthetic molecules. Molecules 2021, 26, 2340. [Google Scholar] [CrossRef]
- Fayed, M.A.A.; El-Behairy, M.F.; Abdallah, I.A.; Abdel-Bar, H.M.; Elimam, H.; Mostafa, A.; Moatasim, Y.; Abouzid, K.A.M.; Elshaier, Y.A.M.M. Structure- and Ligand-Based in silico Studies towards the Repurposing of Marine Bioactive Compounds to Target SARS-CoV-2. Arab. J. Chem. 2021, 14, 103092. [Google Scholar] [CrossRef]
- National Center for Biotechnology Information Sceptrin|C22H24Br2N10O2-PubChem. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/157394#section=Literature (accessed on 13 April 2022).
- National Center for Biotechnology Information Complanadine A|C32H42N4-PubChem. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/11443064#section=2D-Structure (accessed on 13 April 2022).



{kind=link}
{kind=link}
| Molecule Name | Solubility (mg/mL) | Reference | |||||
|---|---|---|---|---|---|---|---|
| DMSO | Ethanol | dH2O | Methanol | Acetone | Ethyl Acetate | ||
| Symmetrical compound | |||||||
| 1G | 18.0 | 3.6 | 2.7 | NR | NR | NR | [1] |
| Asymmetrical compound | |||||||
| C40 | Good | NR | NR | Good | Good | Good | [26] |
| Molecule | Receptors Involved in Docking | Docking Program/Hydrogen Bonds | Physico- Chemical Properties | Target Predictions | Toxico- Logical Predictions | Pharmaco- Kinetic Predictions | Reference |
|---|---|---|---|---|---|---|---|
| Symmetrical compound | |||||||
| 1G | PPARγ (2PRG) | AutoDock 4.2/5 H-bonds | Molinspiration and Osiris Property Explorer | SwissTarget- Prediction | ACD/Tox Suite and Osiris Property Explorer | NR | [1] |
| Asymmetrical compounds | |||||||
| 1 | PPARα (1I7G) and PPARγ (1I7I) | AutoDock 4.2/3 H-bonds for PPARα and 4 for PPARγ | NR | NR | ACD/Tox Suite | NR | [31] |
| 5 | PPARγ (2PRG) | Molecular Operating Environment (MOE)/3 H-bonds | MOE and ADME-T | NR | ADME-T | ADME-T | [27] |
| ChEMBL:259883, 1563849, 1599789, 1523092, 259883, 405972, and 1599789 | PPARγ (3K8S) and ALR2 (3RX3) | Glide Maestro 9.0 Schrödinger Suite and GOLD/5 H- bonds for PPARγ and 3 for ALR2 | QikProp 3.2 | TarFisDock, DRAR-CPI, and Pharm- Mapper, BindingDB, ChEMBL, and Specs | DEREK | QikProp 3.2 | [17] |
| Lobeglitazone | PPARγ (1PRG) | AutoDock 4.0/ 4 H-bonds | NR | NR | NR | NR | [18] |
| 24 and 26 | PPARγ (4A4W and 2XKW) | GOLD 5.2/5 H-bonds for 24 and 26 | NR | SEA, PASS, and Pharm- Mapper | NR | NR | [28] |
| 13 and 16 | PTP1B (2NT7) | Glide 5.8 Schrödinger 2012/6 H- bonds for 13 and 16 | QikProp 3.5 | PASS | NR | QikProp 3.5 | [37] |
| C40 | PPARγ (2PRG) | AutoDock 4.0/ 5 H-bonds | Molinspiration and Osiris Property Explorer | NR | Osiris Property Explorer | NR | [26] |
| 46 | PTP1B (2NT7) | Glide 5.8, Schrödinger 2012/5 H- bonds | QikProp 3.5 | NR | NR | QikProp 3.5 | [43] |
| 4b | PPARγ (P37231) | VLife MDS 4.3/2 H-bonds | NR | NR | NR | NR | [45] |
| 1 | PTP1B (1c83) | AutoDock 4.2/4 H-bonds | admetSAR | NR | admetSAR | admetSAR | [38] |
| Tz21, Tz7, and Tz10 | PPARγ (1FM9) and α- glucosidase (2QMJ) | Maestro 9.0 Schrödinger suite /Regarding PPARγ, 4 H-bonds for Tz21 and 3 for Tz17 and Tz10 | Molins- piration | NR | NR | NR | [42] |
| 11n, 11o, and 22a | α-glucosidase (homology modeling) | MOE 2016.0208/ 2 H-bonds for 11o and 4 for 22a | NR | NR | NR | NR | [41] |
| 7m | IKK-β (3QA8) | Glide Maestro Schrödinger suite/ 3 H-bonds | NR | NR | NR | NR | [39] |
| 17 | PTP1B (2QB5) | AutoDock 4.2/ 6 H-bonds | pkCSM | NR | ProTox | pkCSM | [44] |
| 5 and 9 | PPARγ (2PRG) and α-amy-lase (2QV4) | MOE 2019/ Regarding PPARγ, 1 H-bond for 5 and 9; Regarding α- amylase, 1 H-bond for 5 and 4 for 9 | admetSAR | NR | admetSAR ADME-Tox | admetSAR ADME-Tox | [40] |
| 4 and 5 | PPARγ (2PRG) and α-amylase (2QV4) | MOE 2019/ Regarding α- amylase, 1 H-bond for 4 and 2 for 5 | NR | NR | NR | NR | [46] |
| Structure | Number of Reactions | Total Synthesis Time | Yield (%) | Reference |
|---|---|---|---|---|
| Symmetrical compound | ||||
 | 1 | 3 h | 83.0 | [1] |
| Asymmetrical compounds | ||||
 | 2 | 3–4 h | 80.0 | [31] |
 | 5 | 6 h | 70.0 | [27] |
 | 3 | 48.2 h | 58.4 for 13 and 68.4 for 16 | [37] |
 | 1 | 2 h | 90.6 | [26] |
 | 4 | 15.45 h | 67.6 | [45] |
 | 5 | 104.15 h | 56.0 | [38] |
 | 5 | 58.5 h | 69.0 for Tz21 and Tz7, and 67.0 for Tz10 | [42] |
 | 4 | 24 h | 59.9 | [41] |
 | 7 | 50.5 | 52.0 | [39] |
 | 3 | 25 h | 45.0 | [44] |
 | 5 | 8 h | 71.0 for 6 and 73.0 for 11 | [40] |
   | 5 | 9 h | 74.0 for 4, 65.0 for 5, 61.0 for 6, and 58.0 for 7 | [46] |
| Compound | Duration of Treatment | Cells or Assays | Control Treatment | Aim | Effectiveness | Reference |
|---|---|---|---|---|---|---|
| 1 | 24 h | 3T3-L1 fibroblasts | Cells without treatment | Relative expression of mRNA | mRNA of PPARγ (5-fold), PPARα (6-fold), and LUT-4 (3-fold) | [31] |
| Lobeglitazone | Assay | Kinase assay | ROSI | Comparison of blocking phosphorylation | Better inhibition of phosphorylation. | [18] |
| 1 | Assay | Inhibition assay | NR | Inhibition of PTP1B | 85% inhibition at 20 µM | [38] |
| Tz21 | Assay | Inhibition assay | Acarbose | Inhibition of α-glucosidase | 0.21 µM | [42] |
| 5 | Assay | DPPH assay | Ascorbic acid | Antioxidant activity | ∼10% decrease | [27] |
| 13 and 16 | Assay | Inhibition assay | Suramin at 9.76 µM | Inhibition of PTP1B | 7.31 and 8.13 µM | [37] |
| Molecule Name/Dosage | Duration of Treatment | Model/Dosage | Control Treatment/ Dosage | Higher Effectiveness? | Reference |
|---|---|---|---|---|---|
| Symmetrical compound | |||||
| 1G at 35.7 mg/kg/day | 2 w | STZ rat model at 45 mg/kg | PIO (Agopar®) at 30 mg/kg/day | Yes | [1] |
| Asymmetrical compounds | |||||
| 1 at 50 mg/kg/ single dose | - | STZ (at 65 mg/kg) and NIC (at 110 mg/kg) rat model | Glibenclamide at 5 mg/kg | Yes | [31] |
| Tz21 at 36 mg/kg | 4 h | STZ (60 mg/kg) rat model | PIO at 36 mg/kg | Yes | [42] |
| C40 and C81 at 18 and 21 mg/kg/day, respectively | 3 w | STZ (at 45 mg/kg) rat model | PIO (Agopar®) at 30 mg/kg/day | Yes | [2] |
| 5 at 50 mg/kg | 4 d | Alloxan (100 mg/kg) rat model | Metformin at 500 mg/kg | Yes | [27] |
| 4b (NR) | 8 d | Alloxan (120 mg/kg) rat model | PIO at 40 mg/kg | Similar | [45] |
| 16 (NR) | 1 w | Alloxan (185 mg/kg) albino mouse model | PIO (NR) | Similar | [37] |
| Study | Molecule Name/Control Treatment | Effectiveness | Reference |
|---|---|---|---|
| Symmetrical compound | |||
| Blood glucose and triacylglyceride levels | 1G/PIO (Agopar®) | Similar effect | [1] |
| Asymmetrical compounds | |||
| Blood glucose level | 1/glibenclamide | Similar effect | [31] |
| Blood glucose level | 5/metformin | Lesser effect | [27] |
| Serum glucose, cholesterol, TAG, LDL level | 4b/PIO | Similar effects (only a minor effect for TAG) | [45] |
| Levels of blood glucose, TAG, cholesterol, and antioxidant molecules (SOD and GSH) | C40/PIO (Agopar®) | Greater effect | [2] |
| Application | Findings | Model | Reference |
|---|---|---|---|
| Thiazolidinediones | |||
| Insulin sensitizer | A dose of <45 mg per day lowered the level of edema as well as the rate of weight gain and heart failure. However, it was not possible to reduce the risk of fractures. | Insulin resistance intervention after a stroke (IRIS) trial in humans | [93] |
| Cardioprotective agent PIO | The cardioprotective activity of PIO may owe itself to the depleted level of collagenase III in plasma. | Clinical trials | [94] |
| Anticancer, antiaging | PIO and ROSI presented good affinity for NAF-1 and could be linked to anticancer and anti-aging activity. Additionally, ROSI moderately inhibit complex I of the mitochondrial chain. | Human hepatocellular carcinoma (HepG2) cells overexpressing NAF-1 and complex I. | [20] |
| Antihyperglycemic, α-amylase inhibitors, antioxidants, and antihyperlipidemic agents | Some TZD derivatives were able to inhibit α-amylase more effectively than acarbose. They showed a great capacity for scavenging free radicals (better than vitamin C), leading to a decrease in blood glucose and an antihyperlipidemic effect. | Alloxan-induced diabetes in male Wistar rats. The inhibition of α-amylase was measured in vitro. The DPPH assay revealed antioxidant capacity. | [40] |
| Anti-inflammatory agents, anticonvulsants and antidepressants | TZDs reduced the expression of microglial and inflammatory cytokines and chemokines in the brain. Likewise, they lowered the level of proinflammatory transcription factors in the CNS. TZDs were capable of inhibiting COX-2, an essential enzyme in the inflammatory cascade. They also activated PPARγ, causing a decline in the amount of TNFα and iNOS. This diminished inflammatory damage and improved the cognitive abilities of patients with Alzheimer’s. The antidepressant and anticonvulsant effects were better than the standard drug. | Parkinson’s produced by 1-methyl-4-phenyl-1,2,3,6 tetrahydropyridine (MPTP) in mice, in other animal models, and in cells. | [95] |
| Antimalarial | TZDs displayed moderate activity against the growth of P. falciparum and weakly inhibited FP-2. Although addition of halogen or electron-withdrawing groups significantly increased the inhibition of the FP-2 enzyme, there was no decrease in whole-cell activity. Hence, the compounds were evaluated with liver microsomes, resulting in rapid degradation, which suggests their metabolic instability. | In vitro inhibition of cysteine protease falcipain-2 (FP-2), whole cells of Plasmodium falciparum and hepatic microsomes from human, rat, and mouse liver. | [96] |
| Anticancer | In some cell lines, TZDs induced apoptosis by inter-nucleosomal DNA fragmentation. Similarly, they inhibited the growth of some adenocarcinomas. TZDs also lowered the level of endotrophin, a vital substance in cancer cells. Additionally, cytotoxic and cytostatic effects have been detected, perhaps due to the repression of human telomerase reverse transcriptase (hTERT). | HL-60 and U937 human myeloid leukemia cells; human alveolar basal epithelial adenocarcinoma A549; human chronic myelogenous leukemia K562; MCF-7 human breast adenocarcinoma; human acute lymphoblastic leukemia MOLT-4; and H1299 cells. | [97] |
| Antioxidant and antigout | TZDs inhibited xanthine oxidase, a metallo-flavoprotein overexpressed in gout, and produced greater levels of reactive oxygen species. | For in vitro tests, the enzyme xanthine oxidase was obtained from rat liver. The antioxidant capacity was measured by the DPPH radical assay. | [98] |
| Antimicrobial | TZDs with methoxy, fluoro, chloro, and bromo groups helped improve antimicrobial activity by increasing specificity, evidenced by the lack of cytotoxicity for cell lines. | The minimum inhibitory concentration was quantified in vitro with gram-positive bacteria (Staphylococcus aureus, Bacillus cereus, Bacillus subtilis, Listeria monocytogenes, and Micrococcus luteus) and Gram-negative bacteria (Pseudomonas fluorescens, aeruginosa, Escherichia coli, Salmonella typhi, and Flavobacterium devorans). Cytotoxicity was assessed in HeLa and MCF-7 cells. | [99,100] |
| Biguanides | |||
| Several types of cancer treatment | Activation of LKB1 and AMPK and inhibition of mTOR activity, inhibition of protein synthesis, cell cycle arrest, triggering of apoptosis and autophagy by p53 and p21, respectively, lowering of blood insulin levels, inhibition of UPR, activation of the immune system, destruction of cancer stem cells, prevention of angiogenesis, and decreased hyperlipidemia. | Clinical trials in non-diabetic patients | [90,101] |
| Neurodegenerative diseases | AMPK activation via metformin was neuroprotective against Aβ. According to other in vitro studies, metformin reduced phosphorylation through signaling by mTOR/PP2A (protein phosphates 2A) and produced a lesser degree of molecular pathologies associated with Alzheimer’s disease. In rodent Parkinson’s disease models, dietary metformin diminished oxidative phosphorylation by inhibiting complex I in mitochondria and by inhibiting gluconeogenesis, which further aided neurons to decrease their oxidative burden by minimizing the utilization of NADH. The mTOR pathway links several biological pathways underlying neurodegenerative diseases, and metformin inhibited this signaling cascade. | In vitro studies, mouse models, and clinical trials with diabetic and non-diabetic patients | [21] |
| Acute kidney diseases | Metformin protected renal tubular cells from inflammation, apoptosis, ROS, endoplasmic reticulum stress, and epithelial mesenchymal transition via AMPK activation. Additionally, it inhibited cystic fibrosis transmembrane conductance regulator (CFTR)-mediated fluid secretion and the mTOR-induced cyst formation negatively regulated by AMPK in autosomal dominant polycystic kidney disease. For diabetic patients with kidney diseases, however, clinical investigations have shown an insignificant, even detrimental, effect of metformin. | In vitro studies, animal models, and clinical trials | [102] |
| Obesity-induced inflammation | Short-term metformin treatment led to greater cytokine levels in hepatocytes, including IL-1 , TNF-α, IL-6, MCP-1, and IFN-α, as well as a higher concentration of IL-1 and IL-6 in a hepatocyte culture medium. Metformin decreased the phosphorylation of c-JNK-1 and the level of fat deposition. In hepatocytes, it diminished the level of pro-inflammatory cytokines, increased AMPK phosphorylation, and reduced fat deposition after long-term treatment. | Obese mice | [6] |
| Non-alcoholic fatty liver disease (NAFLD) | Metformin lowered hepatocyte triglyceride accumulation triggered by hyperglycemia and hyperinsulinemia. It reduced ApoA5 expression. Metformin-induced down-regulation of ApoA5 was associated with enhanced phosphorylation of cellular AMPK, a metabolite-sensing protein kinase, and LXR. Metformin also decreased the expression of stearyl-coenzyme A desaturase 1 (SCD1), which participates in lipid de novo synthesis and catalyzes saturated fatty acids to form monounsaturated fatty acids. Animal and in vitro models were used. | HepG2 cell line and hepatocytes from obese mice | [6] |
| Polycystic ovary syndrome (PCOS) | Metformin elicited ovulation. It diminished hyperandrogenism through its effect on both the ovary and adrenal gland by suppressing androgen production. This in turn lowered the level of the pituitary luteinizing hormone and increased the generation of sex hormones and their binding with globulin in the liver. There was a decline in ovarian cytochrome P450c17-α activity. | Non-diabetic and diabetic patients, both obese and lean | [103,104] |
| Dyslipidemia | Metformin decreased the mRNA expression of sterol regulatory element-binding protein 1, ACC1, and ApoA-IV (involved in the secretion of chylomicrons). | Diabetic patients | [6] |
| Modulation of gut microbiota | A 30-day treatment with metformin significantly modified the expression of 46 gut microbes. After the diversity of the gut microbiota was significantly reduced in mice with diet-induced obesity, and Akkermansia spp. was introduced into their gut, glucose homeostasis improved. | Healthy and obese mice | [6] |
| Antihypertensive effects | Metformin inhibited angiotensin II-induced ER stress by means of AMPK activation. | Diabetic patients | [6] |
| Cardiovascular Protective Effects | Metformin protected against cardiac ischemia reperfusion injury by activating AMPK, which promoted glycolysis and protected myocyte viability through the closure of the mitochondrial permeability transition pore (PTP), preventing it from opening and rupturing. This effect was mediated by greater phosphorylation of eNOS, resulting in nitric oxide production. Metformin has also been observed to reduce post-ischemia myocardial injury by restoring depleted PGC-1 levels and enhancing in mitochondrial biogenesis. | Clinical trials | [6] |
| Anti-aging | Metformin is involved in the activation of AMPK and the inhibition of signaling through the mTOR pathway. Signaling via mTOR is associated with accelerated aging. AMPK is a key regulator of many cellular pathways linked to both health and lifespan, including the beneficial effects of calorie restriction. | In vitro studies, animal models, and clinical trials | [105] |
| Molecule | Structure | Application | Findings | Reference |
|---|---|---|---|---|
| Proguanil |  | Prophylactic antimalarial drug | Both drugs inhibit dihydrofolate reductase, an enzyme involved in the reproduction of the malaria parasites Plasmodium falciparum and Plasmodium vivax in red blood cells. | [108] |
| Chlorpro- guanil |  | Clinical trials for the treatment of malaria |
| Molecule | Structure | Origin | Findings | Reference |
|---|---|---|---|---|
| Dendrimers (Polypropylene amine, PAMAM, pseudorotaxane, ethylene diamine, etc.) |   | Synthetic | Analogous to proteins, enzymes and viruses. Delivers anticancer drugs. Delivers genes. Forms part of contrast agents in magnetic resonance imaging. Serves as a sensor. Enhances solubility. Participates in photodynamic therapy. Dendrimers and other molecules can either be attached to the periphery or encapsulated in their interior voids. Modern medicine uses a variety of these molecules as artificial blood substitutes (e.g., PAMAM dendrimers). Drug–dendrimer conjugates show high solubility, reduced systemic toxicity, and selective accumulation in solid tumors. | [109] |
| Polyamines synthesized as potential small molecule CXCR4 antagonists | Fragment-¡-Linker-¡- Fragment Fragment =  Linker Linker | Synthetic | Antagonist that blocks the entry of human immunodeficiency virus type 1 (HIV-1). | [110] |
| Thioureas |  | Synthetic | Antioxidant activity that scavenges ABTS, and antibacterial activity against Agrobacterium tumefaction. | [12] |
| Proteins |  | Synthetic | The redesign of existing proteins may result in enhanced functions. Energy minimization is achieved by symmetrical assemblies. | [111] |
| Steroid dimers |  | Synthetic | Improvement of biological potential leads to antiproliferative activity in human cell lines of cervical cancer (HeLa), breast cancers (MDA-MB-453 and MDA-MB-361), and leukemia (K562), with values ranging from 14.9 to 27.1 μM (values for cisplatin ranged from 2.1 to 17.1 μM). Dimeric compounds exhibited antifungal activity against Saccharomyces cerevisiae. | [112] |
| Sceptrin |  | Natural product | Antibacterial, antiviral, antihistaminic, and antimuscarinic agent, and possibly beneficial in treating coronavirus disease (COVID). | [8,113,114] |
| Complanadine A |  | Natural product | Treatment for Alzheimer’s disease or spinal cord injury. | [8,115] |
| G3F |  | Synthetic | Antifungal and antidiabetic activity. Docking results show that the lowest energy value is for α-amylase and α-glucosidase. In vitro studies with these two enzymes yielded IC50 values of 22.8 and 21 µg/mL, respectively. | [11] |
| 5g |  | Synthetic | α-Glucosidase and α-amylase inhibitors. Electron attracting substituents on the aromatic ring favor inhibition. | [19] |
| Dendro fullerenes |  | Synthetic | Antiviral. Fullerene is able to fit inside the hydrophobic cavity of HIV proteases, inhibiting the access of substrates to the catalytic site of the enzyme. If exposed to light, fullerene produces singlet oxygen with high quantum yields. This activity, together with direct electron transfer from the excited state of fullerene and DNA bases, can be used to cleave DNA. | [30] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Filisola-Villaseñor, J.G.; Aranda-Barradas, M.E.; Miranda-Castro, S.P.; Mendieta-Wejebe, J.E.; Valdez Guerrero, A.S.; Guillen Castro, S.A.; Martínez Castillo, M.; Tamay-Cach, F.; Álvarez-Almazán, S. Impact of Molecular Symmetry/Asymmetry on Insulin-Sensitizing Treatments for Type 2 Diabetes. Symmetry 2022, 14, 1240. https://doi.org/10.3390/sym14061240
Filisola-Villaseñor JG, Aranda-Barradas ME, Miranda-Castro SP, Mendieta-Wejebe JE, Valdez Guerrero AS, Guillen Castro SA, Martínez Castillo M, Tamay-Cach F, Álvarez-Almazán S. Impact of Molecular Symmetry/Asymmetry on Insulin-Sensitizing Treatments for Type 2 Diabetes. Symmetry. 2022; 14(6):1240. https://doi.org/10.3390/sym14061240
Chicago/Turabian StyleFilisola-Villaseñor, Jessica Georgina, María E. Aranda-Barradas, Susana Patricia Miranda-Castro, Jessica Elena Mendieta-Wejebe, Amaranta Sarai Valdez Guerrero, Selene Amasis Guillen Castro, Macario Martínez Castillo, Feliciano Tamay-Cach, and Samuel Álvarez-Almazán. 2022. "Impact of Molecular Symmetry/Asymmetry on Insulin-Sensitizing Treatments for Type 2 Diabetes" Symmetry 14, no. 6: 1240. https://doi.org/10.3390/sym14061240
APA StyleFilisola-Villaseñor, J. G., Aranda-Barradas, M. E., Miranda-Castro, S. P., Mendieta-Wejebe, J. E., Valdez Guerrero, A. S., Guillen Castro, S. A., Martínez Castillo, M., Tamay-Cach, F., & Álvarez-Almazán, S. (2022). Impact of Molecular Symmetry/Asymmetry on Insulin-Sensitizing Treatments for Type 2 Diabetes. Symmetry, 14(6), 1240. https://doi.org/10.3390/sym14061240