

Natural Compounds as DPP-4 Inhibitors: 3D-Similarity Search, ADME Toxicity, and Molecular Docking Approaches


Abstract
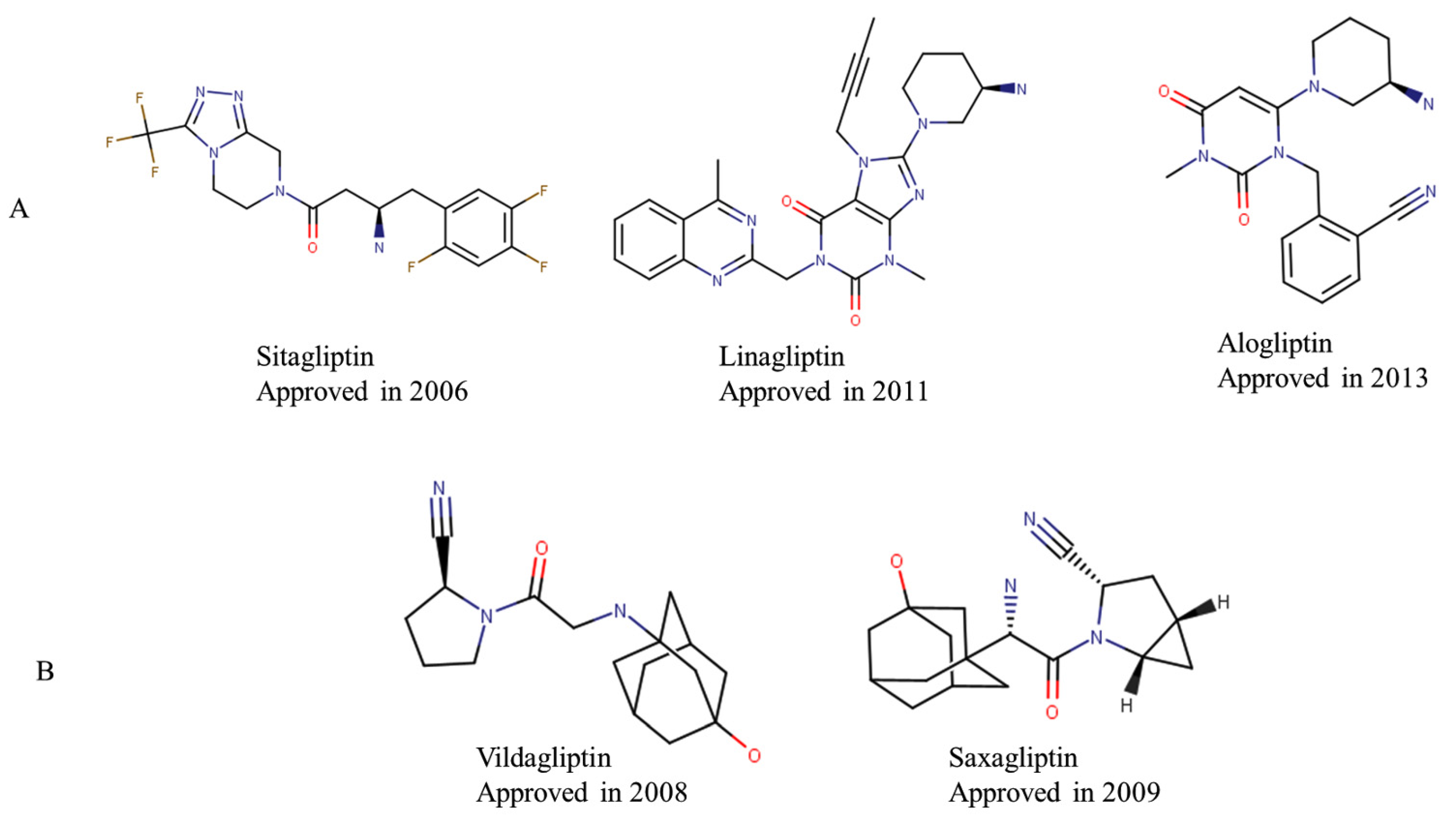
:1. Introduction
2. Materials and Methods
2.1. Data Set
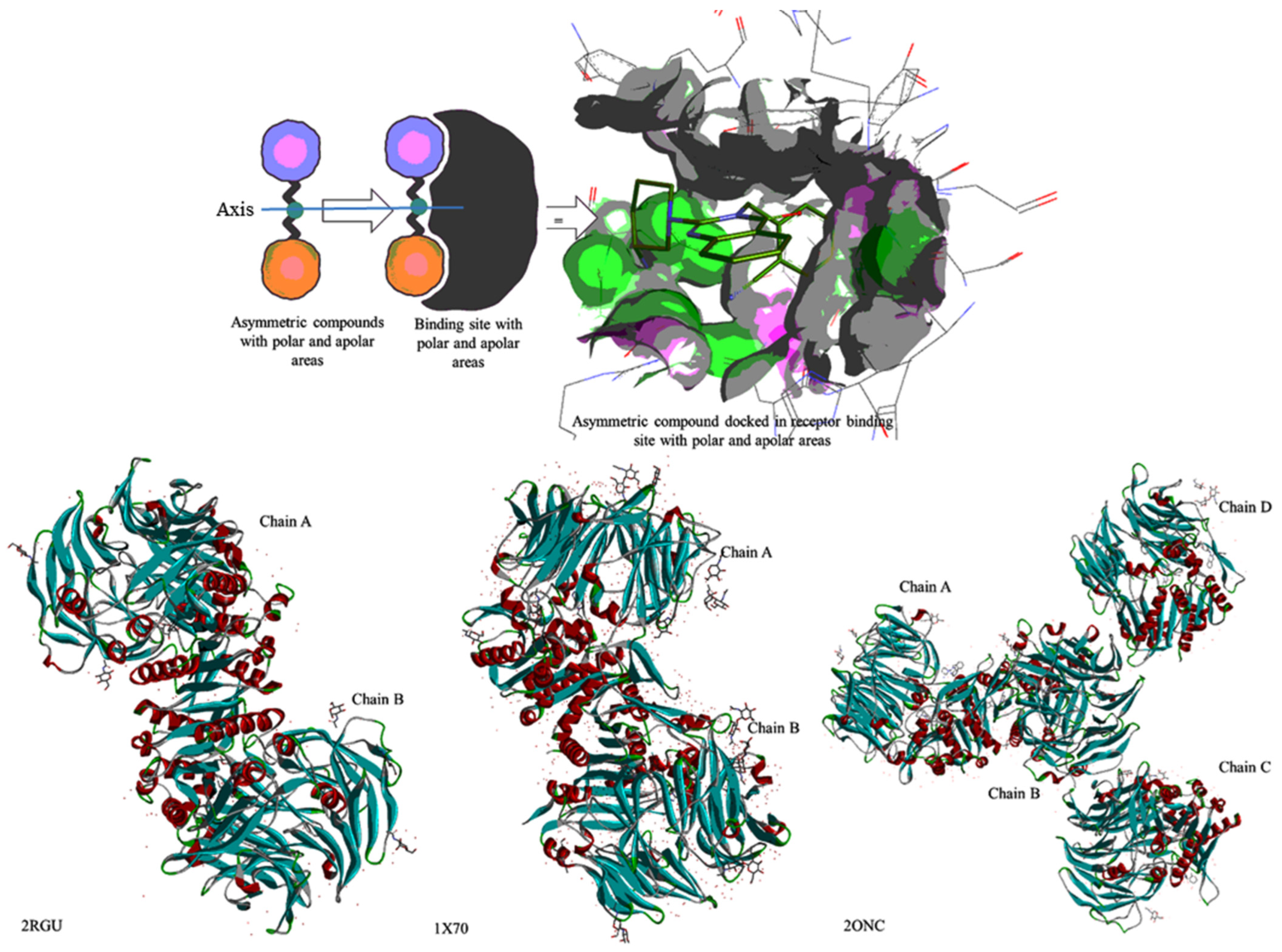
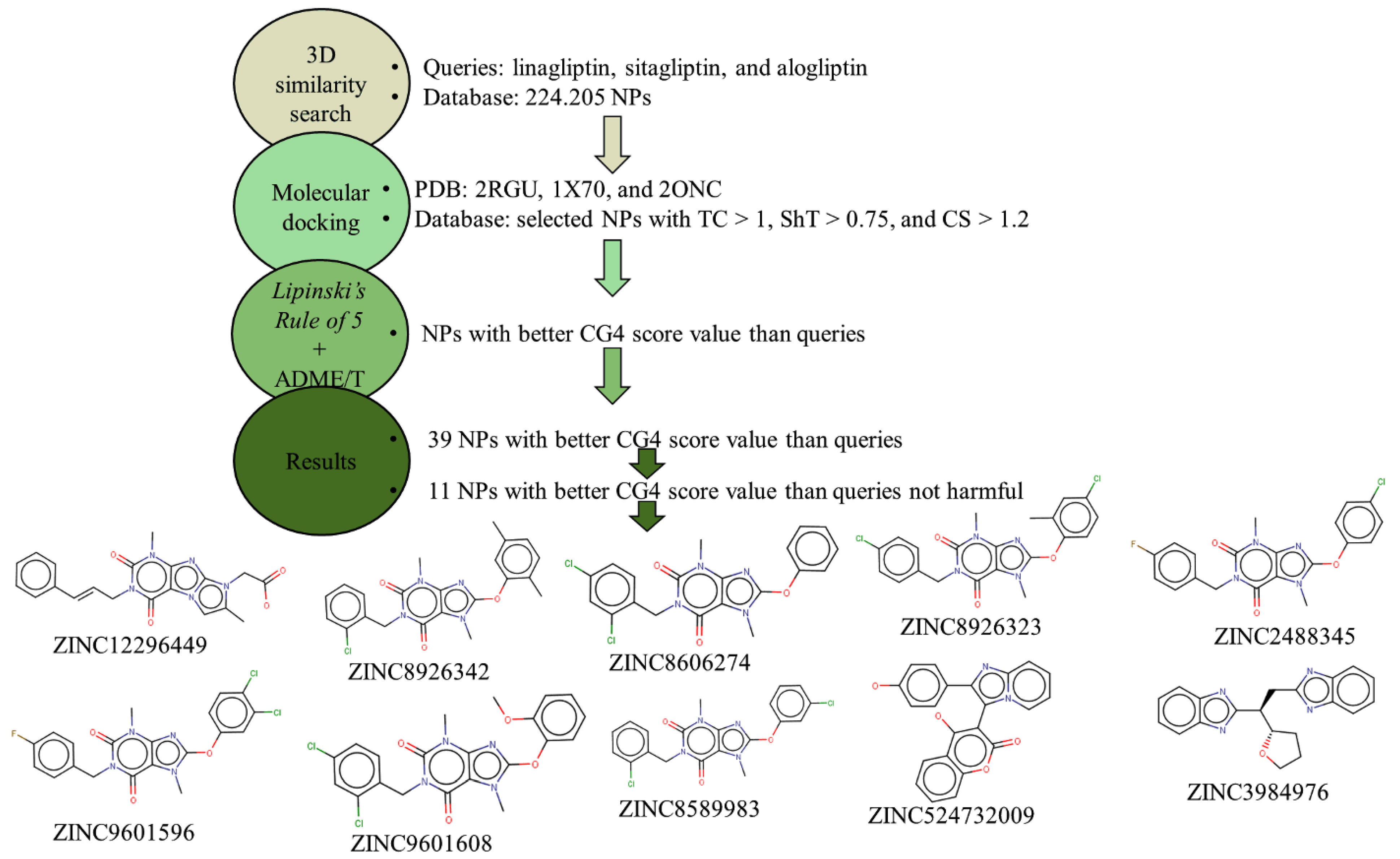
2.2. Shape-Based Similarity Search in a Chemical Database
- and are the self-overlap volumes of molecules and ;
- is the overlap volume between molecules and .
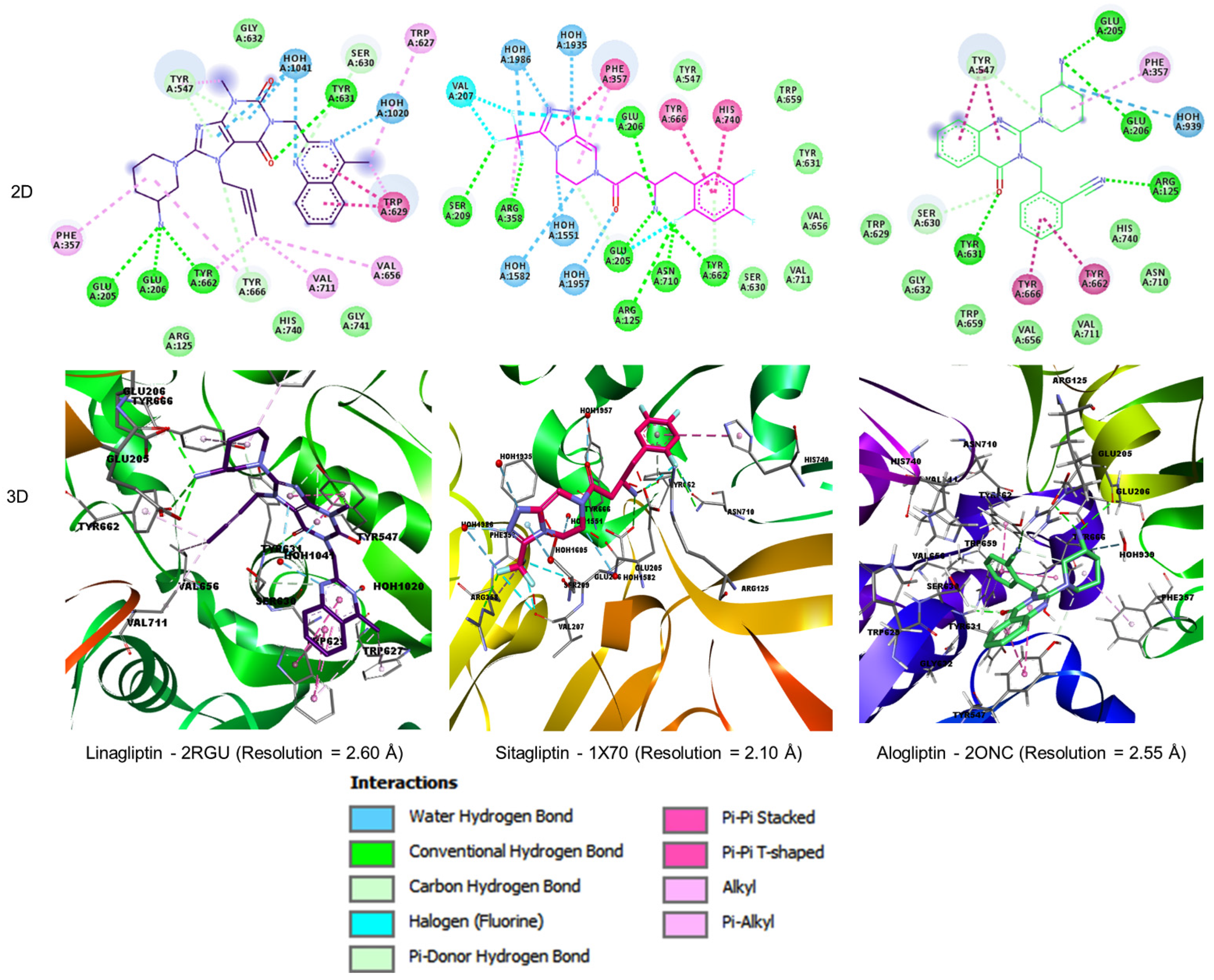
2.3. Docking Study
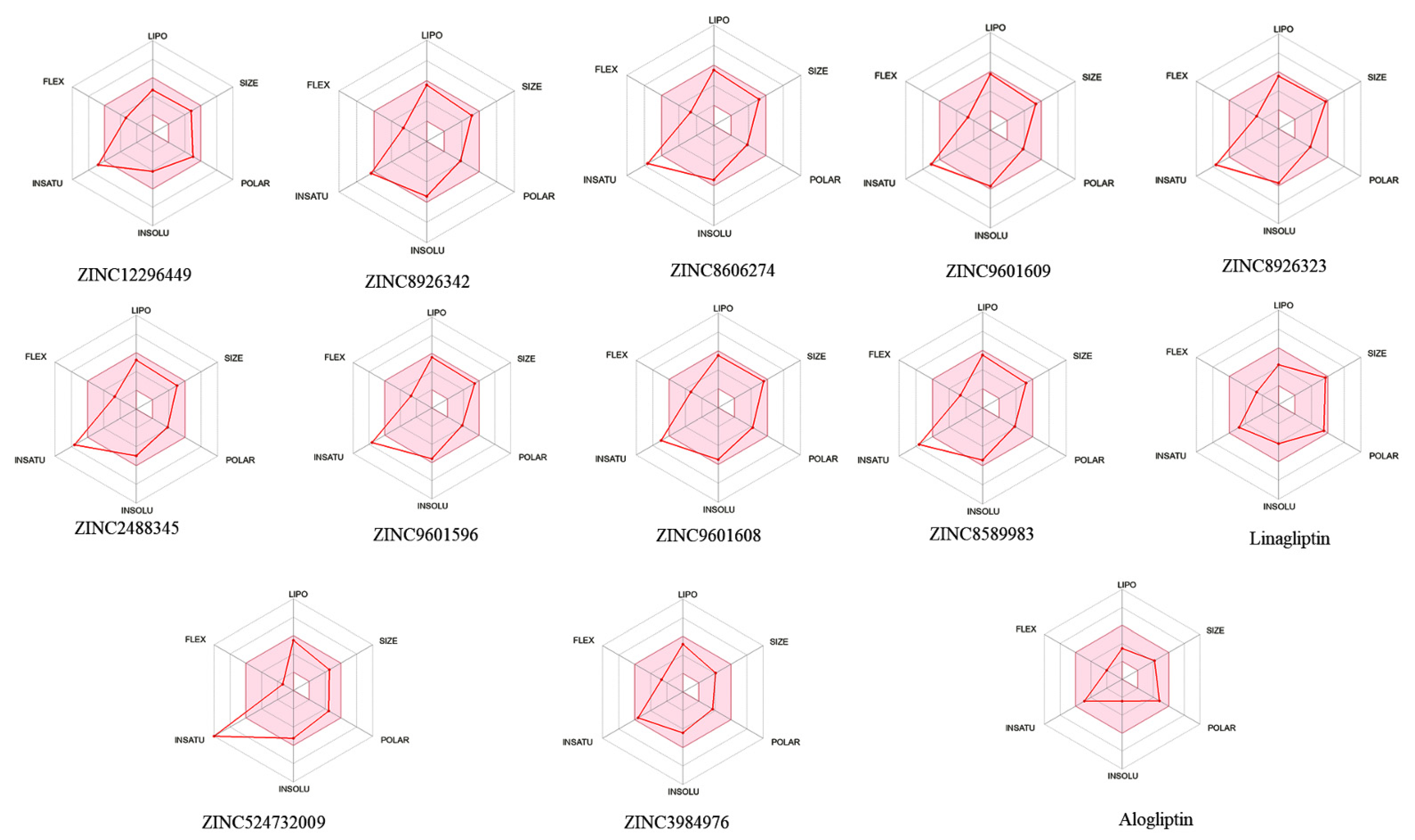
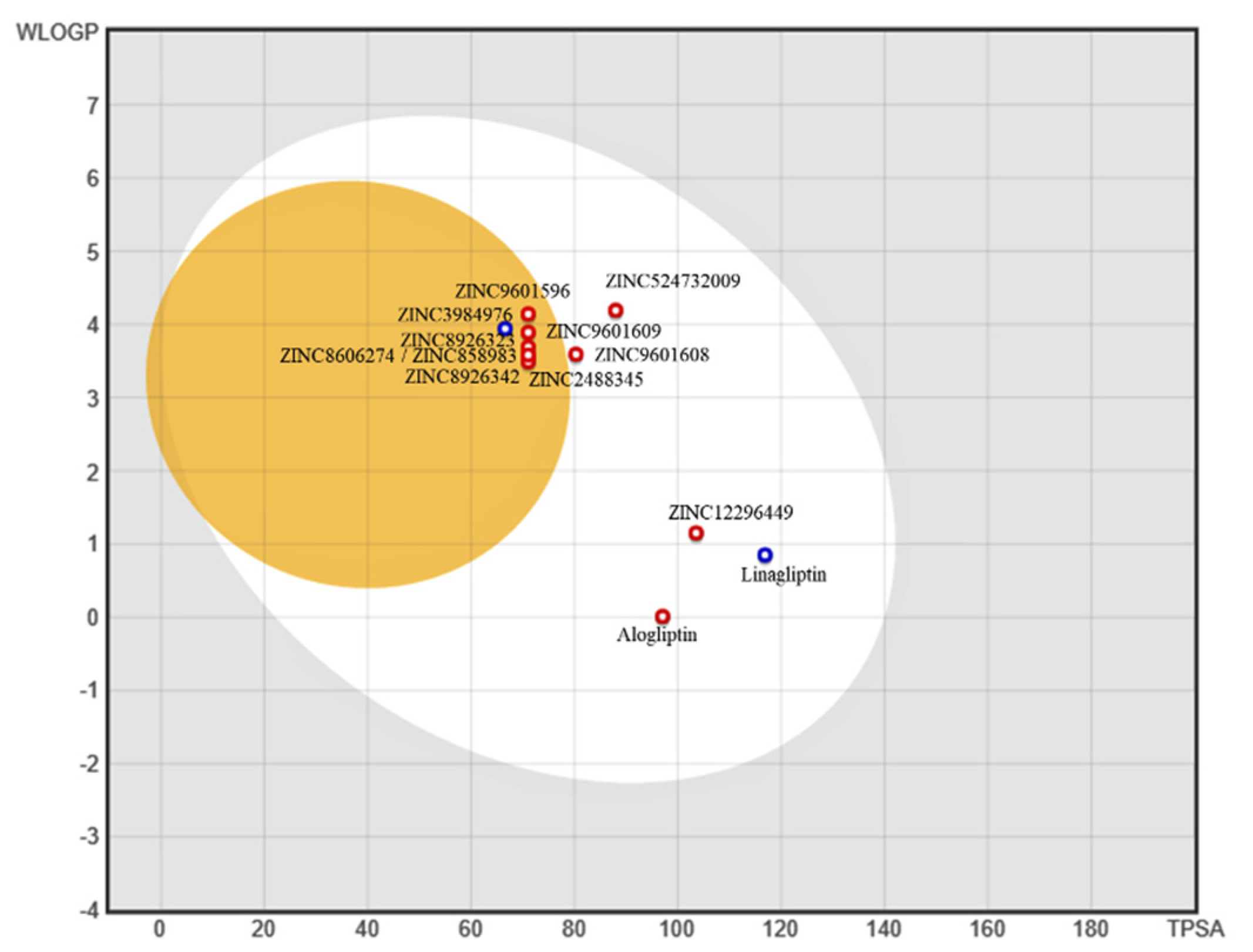
2.4. Bioavailability and In Silico ADME/T Screening for Drug-Likeness
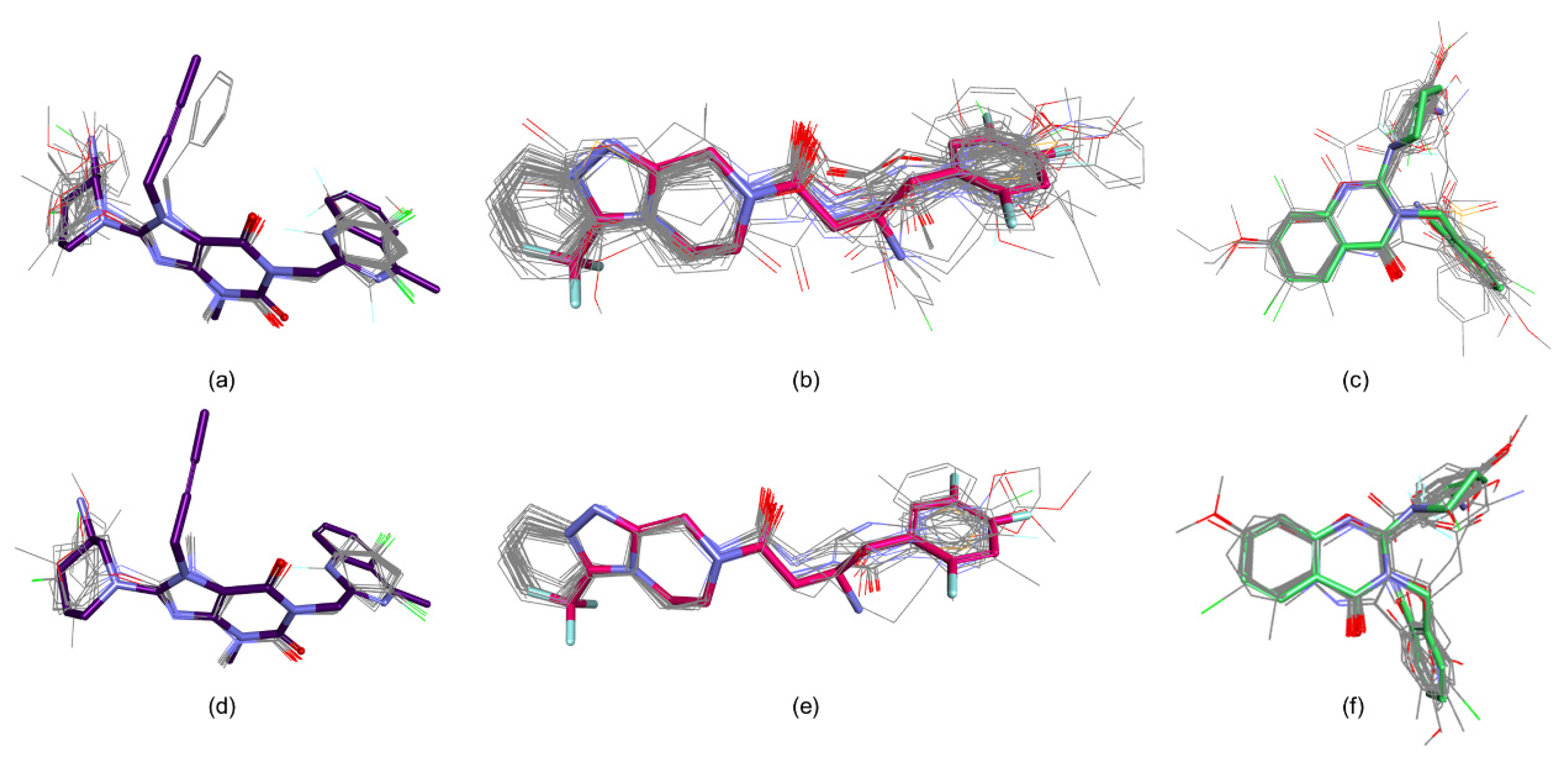
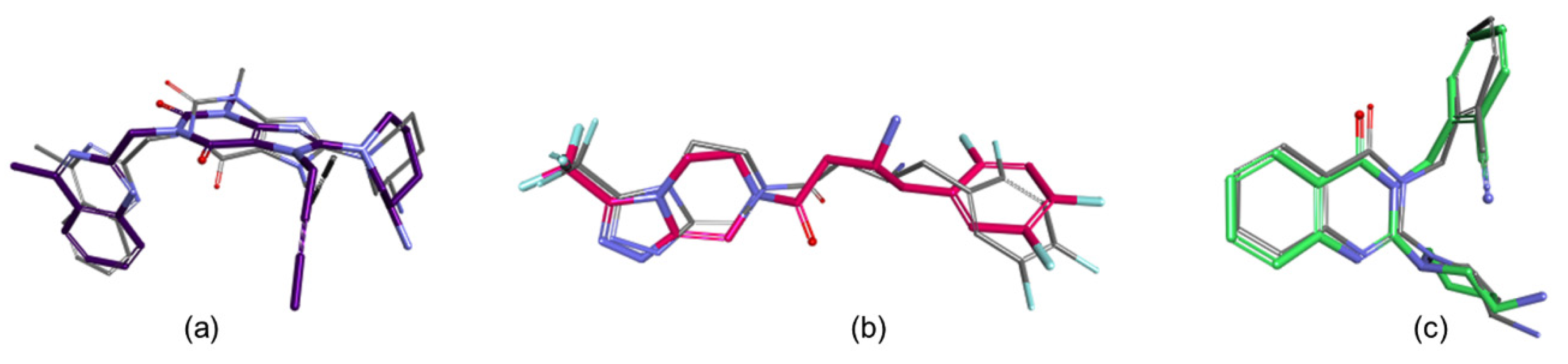
3. Results
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ADMETox | Absorption, distribution, metabolism, excretion, and toxicity; |
| BBB | Blood–brain barrier; |
| CS | Combo score; |
| DPP-4 | Dipeptidyl peptidase 4; |
| EMA | European Medicines Agency; |
| HIA | Human gastrointestinal absorption; |
| IDF | International Diabetes Federation; |
| NPs | Natural products; |
| PDB | RCSB Protein Data Bank; |
| ShT | Shape Tanimoto; |
| TC | Tanimoto Combo; |
| T2DM | Type 2 diabetes mellitus; |
| ZINC-NPs DB | ZINC15 natural products database; |
| WHO | World Health Organization. |
References
- World Health Organization. Available online: Tps://www.who.int/health-topics/diabetes (accessed on 8 April 2022).
- IDF Diabetes Atlas. Available online: http://www.idf.org/diabetesatlas (accessed on 8 April 2022).
- Souto, S.B.; Souto, E.B.; Braga, D.C.; Medina, J.L. Prevention and current onset delay approaches of type 2 diabetes mellitus (T2DM). Eur. J. Clin. Pharmacol. 2011, 67, 653–661. [Google Scholar] [CrossRef] [PubMed]
- Forbes, J.M.; Cooper, M.E. Mechanisms of diabetic complications. Physiology 2013, 93, 137–188. [Google Scholar] [CrossRef] [PubMed]
- Weyer, C.; Bogardus, C.; Mott, D.M.; Pratley, R.E. The natural history of insulin secretory dysfunction and insulin resistance in the pathogenesis of type 2 diabetes mellitus. J. Clin. Investig. 1999, 104, 787–794. [Google Scholar] [CrossRef] [PubMed]
- Crisan, L.; Pacureanu, L.; Avram, S.; Bora, A.; Avram, S.; Kurunczi, L. PLS and shape-based similarity analysis of maleimides–GSK-3 inhibitors. J. Enzyme Inhib. Med. Chem. 2014, 29, 599–610. [Google Scholar] [CrossRef]
- Crisan, L.; Avram, S.; Pacureanu, L. Pharmacophore-based screening and drug repurposing exemplified on glycogen synthase kinase-3 inhibitors. Mol. Divers. 2017, 21, 385–405. [Google Scholar] [CrossRef]
- Mulakayala, N.; Reddy, U.C.H.; Iqbal, J.; Pal, M. Synthesis of dipeptidyl peptidase-4 inhibitors: A brief overview. Tetrahedron 2010, 66, 4919–4938. [Google Scholar] [CrossRef]
- Augustyns, K.; Van der Veken, P.; Senten, K.; Haerners, A. Dipeptidyl peptidase IV inhibitors as new therapeutic agents for the treatment of Type 2 diabetes. Expert Opin. Ther. Patents 2003, 13, 499–510. [Google Scholar] [CrossRef]
- Wang, N.; Yang, T.; Li, J.; Zhang, X. Dipeptidyl peptidase-4 inhibitors as add-on therapy to insulin in patients with type 2 diabetes mellitus: A meta-analysis of randomized controlled trials. Diabetes Metab. Syndr. Obes. 2019, 12, 1513–1526. [Google Scholar] [CrossRef]
- Seshadri, K.G.; Kirubha, M.H.B. Gliptins: A new class of oral antidiabetic agents. Indian J. Pharm. Sci. 2009, 71, 608–614. [Google Scholar] [CrossRef]
- Gupta, V.; Kalra, S. Choosing a gliptin. Indian J. Endocrinol. Metab. 2011, 15, 298–308. [Google Scholar] [CrossRef]
- PLoSker, G.L. Sitagliptin: A review of its use in patients with type 2 diabetes mellitus. Drugs 2014, 74, 223–242. [Google Scholar] [CrossRef] [PubMed]
- Dhillon, S. Sitagliptin: A review of its use in the management of type 2 diabetes mellitus. Drugs 2010, 70, 489–512. [Google Scholar] [CrossRef]
- Keating, G.M. Vildagliptin: A review of its use in type 2 diabetes mellitus. Drugs 2010, 70, 2089–2112. [Google Scholar] [CrossRef] [PubMed]
- Dhillon, S. Saxagliptin: A Review in Type 2 Diabetes. Drugs 2015, 75, 1783–1796. [Google Scholar] [CrossRef]
- Lajara, R. Use of the dipeptidyl peptidase-4 inhibitor linagliptin in combination therapy for type 2 diabetes. Expert Opin. Pharmacother. 2012, 13, 2663–2671. [Google Scholar] [CrossRef] [PubMed]
- Keating, G.M. Alogliptin: A review of its use in patients with type 2 diabetes mellitus. Drugs 2015, 75, 777–796. [Google Scholar] [CrossRef] [PubMed]
- Deeks, E.D. Linagliptin: A review of its use in the management of type 2 diabetes mellitus. Drugs 2012, 72, 1793–1824. [Google Scholar] [CrossRef]
- Scott, L.J. Linagliptin in type 2 diabetes mellitus. Drugs 2011, 71, 611–624. [Google Scholar] [CrossRef]
- Marino, A.B.; Cole, S.W. Alogliptin: Safety, efficacy, and clinical implications. J. Pharm. Pract. 2015, 28, 99–106. [Google Scholar] [CrossRef]
- Saisho, Y. Alogliptin benzoate for management of type 2 diabetes. Vasc. Health Risk. Manag. 2015, 11, 229–243. [Google Scholar] [CrossRef] [Green Version]
- Haak, T. Combination of Linagliptin and Metformin for the Treatment of Patients with Type 2 Diabetes. Clin. Med. Insights Endocrinol. Diabetes 2015, 8, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Takeshita, Y.; Kita, Y.; Kato, K.-I.; Kanamori, T.; Misu, H.; Kaneko, S.; Takamura, T. Effects of metformin and alogliptin on body composition in people with type 2 diabetes. J. Diabetes. Investig. 2019, 10, 723–730. [Google Scholar] [CrossRef] [PubMed]
- Shinya, N.; Mariko, A.; Hiroyuki, I. Anagliptin in the treatment of type 2 diabetes: Safety, efficacy, and patient acceptability. Metab. Syndrome Obes. Targets Ther. 2015, 8, 163–171. [Google Scholar]
- Kim, S.H.; Lee, S.H.; Yim, H.J. Gemigliptin, a novel dipeptidyl peptidase 4 inhibitor: First new anti-diabetic drug in the history of Korean pharmaceutical industry. Arch. Pharm. Res. 2013, 36, 1185–1188. [Google Scholar] [CrossRef]
- Scott, L.J. Teneligliptin: A review in type 2 diabetes. Clin. Drug Invest. 2015, 35, 765–772. [Google Scholar] [CrossRef] [PubMed]
- Mccormack, P.L. Evogliptin: First Global Approval. Drugs 2015, 75, 2045–2049. [Google Scholar] [CrossRef] [PubMed]
- Biftu, T.; Sinha-Roy, R.; Chen, P.; Qian, X.; Feng, D.; Kuethe, J.T.; Scapin, G.; Gao, Y.D.; Yan, Y.; Krueger, D.; et al. Omarigliptin (MK-3102): A novel long-acting DPP-4 inhibitor for once-weekly treatment of type 2 diabetes. J. Med. Chem. 2014, 57, 3205–3212. [Google Scholar] [CrossRef]
- Kaku, K. First novel once-weekly DPP-4 inhibitor, trelagliptin, for the treatment of type 2 diabetes mellitus. Expert Opin. Pharmacother. 2015, 16, 2539–2547. [Google Scholar] [CrossRef]
- Sharma, R.; Sun, H.; Piotrowski, D.W.; Ryder, T.F.; Doran, S.D.; Dai, H.Q.; Prakash, C. Metabolism, excretion, and pharmacokinetics of (3,3-Diuoropyrrolidin1-yl)((2S,4S)-4-(4-(pyrimidin-2-yl)piperazin-1-yl)pyrrolidin-2-yl)methanone, a dipeptidyl peptidase inhibitor, in rat, dog and human. Drug Metab. Dispos. 2012, 40, 2143–2161. [Google Scholar] [CrossRef]
- Crisan, L.; Pacureanu, L.; Bora, A.; Avram, S.; Kurunczi, L.; Simon, Z. QSAR study and molecular docking on indirubin inhibitors of Glycogen Synthase Kinase-3. Cent. Eur. J. Chem. 2013, 11, 63–67. [Google Scholar] [CrossRef]
- Ivan, D.; Crisan, L.; Funar-Timofei, S.; Mracec, M. A quantitative structure–activity relationships study for the anti-HIV-1 activities of 1-[(2-hydroxyethoxy)methyl]-6-(phenylthio)thymine derivatives using the multiple linear regression and partial least squares methodologies. J. Serb. Chem. Soc. 2013, 78, 495–506. [Google Scholar] [CrossRef]
- Petric, M.; Crisan, L.; Crisan, M.; Micle, A.; Maranescu, B.; Ilia, G. Synthesis and QSRR Study for a Series of Phosphoramidic Acid Derivatives. Heteroat. Chem. 2013, 24, 138–145. [Google Scholar] [CrossRef]
- Rogic, S.; Nukic, M.; Gagic, Z. Quantitative Structure-Activity Relationship Study of DPP-4 Enzyme Inhibitors as Drugs in Therapy of Type 2 Diabetes Mellitus. International Conference on Medical and Biological Engineering, CMBEBIH 2021. In CMBEBIH 2021; Badnjevic, A., Gurbeta Pokvić, L., Eds.; Springer: Berlin/Heidelberg, Germany; Volume 84, pp. 481–488.
- Upadhyay, J.; Gajjar, A.; Suhagia, B.N. Combined Ligand-Based and Structure-Based Virtual Screening Approach for Identification of New Dipeptidyl Peptidase 4 Inhibitors. Curr. Drug Discov. Technol. 2019, 16, 426–436. [Google Scholar] [CrossRef] [PubMed]
- Rakesh, K.P.; Virendra, N.; Vipin, K. Structure based virtual screening of natural compounds and molecular dynamics simulation: Butirosin as Dipeptidyl peptidase (DPP-IV) inhibitor. Biocat. Agricl. Biotech. 2021, 35, 102042. [Google Scholar]
- Wang, W.; Tian, Y.; Wan, Y.; Gu, S.; Ju, X.; Luo, X.; Liu, G. In silico studies of diarylpyridine derivatives as novel HIV-1 NNRTIs using docking-based 3D-QSAR, molecular dynamics, and pharmacophore modeling approaches. RSC Adv. 2018, 8, 40529–40543. [Google Scholar]
- Gajjar, K.A.; Gajjar, A.K. Combiphore (Structure and Ligand Based Pharmacophore)—Approach for the Design of GPR40 Modulators in the Management of Diabetes. Curr. Drug Discov. Technol. 2020, 17, 233–247. [Google Scholar] [CrossRef]
- Swilam, N.; Nawwar, M.A.M.; Radwan, R.A.; Mostaf, E.S. Antidiabetic Activity and In Silico Molecular Docking of Polyphenols from Ammannia baccifera L. subsp. Aegyptiaca (Willd.) Koehne Waste: Structure Elucidation of Undescribed Acylated Flavonol Diglucoside. Plants 2022, 11, 452. [Google Scholar] [CrossRef]
- Pangajavalli, S.; Ranjithkumar, R.; Ramaswamy, S. Structural, Hirshfeld, spectroscopic, quantum chemical and molecular docking studies on 6b′, 7′, 8′, 9′-Tetrahydro-2H,6′H-spiro[acenaphthylene-1,11′-chromeno [3,4-a]pyrrolizine]-2,6′(6a′H,11a′H)-dione. J. Mol. Struc. 2020, 1209, 127921. [Google Scholar] [CrossRef]
- Singh, P.; Singh, V.K.; Singh, A.K. Molecular docking analysis of candidate compounds derived from medicinal plants with type 2 diabetes mellitus targets. Bioinformation 2019, 15, 179–188. [Google Scholar] [CrossRef]
- Crisan, L.; Borota, A.; Bora, A.; Pacureanu, L. Diarylthiazole and diarylimidazole selective COX-1 inhibitor analysis through pharmacophore modeling, virtual screening, and DFT-based approaches. Struct. Chem. 2019, 30, 2311–2326. [Google Scholar] [CrossRef]
- Visa, A.; Maranescu, B.; Lupa, L.; Crisan, L.; Borota, A. New Efficient Adsorbent Materials for the Removal of Cd(II) from Aqueous Solutions. Nanomaterials 2020, 10, 899. [Google Scholar] [CrossRef]
- Jabeen, E.; Janjua, N.K.; Ahmed, S.; Murtaza, I.; Ali, T.; Masood, N.; Rizvi, A.S.; Murtaza, G. DFT predictions, synthesis, stoichiometric structures and anti-diabetic activity of Cu (II) and Fe (III) complexes of quercetin, morin, and primuletin. J. Mol. Struc. 2017, 1150, 459–468. [Google Scholar] [CrossRef]
- Visa, A.; Mracec, M.; Maranescu, B.; Maranescu, V.; Ilia, G.; Popa, A.; Mracec, M. Structure simulation into a lamellar supramolecular network and calculation of the metal ions/ligands ratio. Chem. Cent. J. 2012, 6, 91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meduru, H.; Wang, Y.-T.; Tsai, J.J.P.; Chen, Y.-C. Finding a Potential Dipeptidyl Peptidase-4 (DPP-4) Inhibitor for Type-2 Diabetes Treatment Based on Molecular Docking, Pharmacophore Generation, and Molecular Dynamics Simulation. Int. J. Mol. Sci. 2016, 17, 920. [Google Scholar] [CrossRef]
- Hidalgo-Figueroa, S.; Rodríguez-Luévano, A.; Almanza-Pérez, J.C.; Giacoman-Martínez, A.; Ortiz-Andrade, R.; León-Rivera, I.; Navarrete-Vázquez, G. Synthesis, molecular docking, dynamic simulation and pharmacological characterization of potent multifunctional agent (dual GPR40-PPARγ agonist) for the treatment of experimental type 2 diabetes. Eur. J. Pharmacol. 2021, 907, 174244. [Google Scholar] [CrossRef] [PubMed]
- ROCS, v. 3.5.0.1; OpenEye Scientific Software Inc.: Santa Fe, NM, USA, 2021.
- FRED, v.4.1.2.1; OpenEye Scientific Software Inc.: Santa Fe, NM, USA, 2021.
- BIOVIA. Discovery Studio Visualizer v. 20.1.0; Accelrys Software Inc.: San Diego, CA, USA, 2020. [Google Scholar]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef]
- Pires, D.E.V.; Blundell, T.L.; Ascher, D.B. pkCSM: Predicting Small-Molecule Pharmacokinetic and Toxicity Properties Using Graph-Based Signatures. J. Med. Chem. 2015, 58, 4066–4072. [Google Scholar] [CrossRef]
- Sterling, T.; Irwin, J.J. ZINC 15—Ligand Discovery for Everyone. J. Chem. Inf. Model. 2015, 55, 2324–2337. [Google Scholar] [CrossRef]
- Crisan, L.; Bora, A. Small Molecules of Natural Origin as Potential Anti-HIV Agents: A Computational Approach. Life 2021, 11, 722. [Google Scholar] [CrossRef]
- Schrödinger Release 2021-4: LigPrep (2021) Schrödinger; LLC: New York, NY, USA, 2021.
- Hawkins, P.C.D.; Skillman, A.G.; Warren, G.L.; Ellingson, B.A.; Stahl, M.T. Conformer Generation with OMEGA: Algorithm and Validation Using High Quality Structures from the Protein Databank and Cambridge Structural Database. J. Chem. Inf. Model. 2010, 50, 572–584. [Google Scholar] [CrossRef]
- Thom, R.; Löffler, B.; Stihle, M.; Huber, W.; Ruf, A.; Hennig, M. Structural Basis of Proline-Specific Exopeptidase Activity as Observed in Human Dipeptidyl Peptidase-IV. Structure 2003, 11, 947–959. [Google Scholar] [CrossRef]
- Brown, J.H. Breaking symmetry in protein dimers: Designs and functions. Protein Sci. 2006, 15, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Wang, L.; Beconi, M.; Eiermann, G.J.; Fisher, M.H.; He, H.; Hickey, G.J.; Kowalchick, J.E.; Leiting, B.; Lyons, K.; et al. (2R)-4-Oxo-4-[3-(Trifluoromethyl)-5,6-dihydro[1,2,4]triazolo[4,3-a]pyrazin-7(8H)-yl]-1-(2,4,5-trifluorophenyl)butan-2-amine: A Potent, Orally Active Dipeptidyl Peptidase IV Inhibitor for the Treatment of Type 2 Diabetes. J. Med. Chem. 2005, 48, 141–151. [Google Scholar] [CrossRef]
- Eckhardt, M.; Langkopf, E.; Mark, M.; Tadayyon, M.; Thomas, L.; Nar, H.; Pfrengle, W.; Guth, B.; Lotz, R.; Sieger, P.; et al. 8-(3-(R)-Aminopiperidin-1-yl)-7-but-2-ynyl-3-methyl-1-(4-methyl-quinazolin-2-ylmethyl)-3,7-dihydropurine-2,6-dione (BI 1356), a Highly Potent, Selective, Long-Acting, and Orally Bioavailable DPP-4 Inhibitor for the Treatment of Type 2 Diabetes. J. Med. Chem. 2007, 50, 6450–6453. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Zhang, Z.; Wallace, M.B.; Stafford, J.A.; Kaldor, S.W.; Kassel, D.B.; Navre, M.; Shi, L.; Skene, R.J.; Asakawa, T.; et al. Discovery of Alogliptin: A Potent, Selective, Bioavailable, and Efficacious Inhibitor of Dipeptidyl Peptidase IV. J. Med. Chem. 2007, 50, 2297–2300. [Google Scholar] [CrossRef]
- MakeReceptor, v. 3.5.0.4; OpenEye Scientific Software Inc.: Santa Fe, NM, USA, 2020.
- Schrödinger Release 2021-4: Maestro, v.13.0.135 (2021) Schrödinger; LLC: New York, NY, USA, 2021.
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 1997, 23, 3–25. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Duffy, E.M. Prediction of drug solubility from structure. Adv. Drug Deliv. Rev. 2002, 54, 355–366. [Google Scholar] [CrossRef]
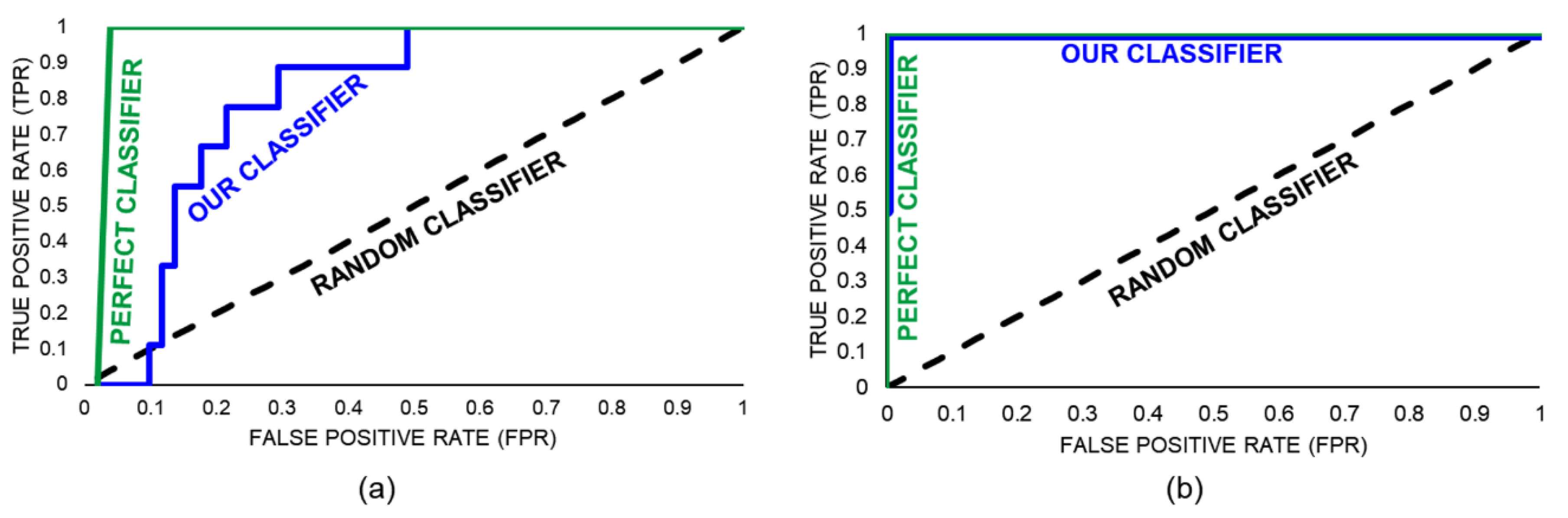
- Hanley, J.A.; McNeil, B.J. The meaning and use of the area under a receiver operating characteristic (ROC) curve. Radiology 1982, 143, 29–36. [Google Scholar] [CrossRef]
- Avram, S.I.; Pacureanu, L.M.; Bora, A.; Crisan, L.; Avram, S.; Kurunczi, L. ColBioSFlavRC: A Collection of Bioselective Flavonoids and Related Compounds Filtered from HighThroughput Screening Outcomes. J. Chem. Inf. Model. 2014, 54, 2360–2370. [Google Scholar] [CrossRef]
- Mysinger, M.M.; Carchia, M.; Irwin, J.J.; Shoichet, B.K. Directory of useful decoys, enhanced (DUD-E): Better ligands and decoys for better benchmarking. J. Med. Chem. 2012, 26, 6582–6594. [Google Scholar] [CrossRef]
- Giangreco, I.; Mukhopadhyay, A.; Cole, J.C. Validation of a Field-Based Ligand Screener Using a Novel Benchmarking Data Set for Assessing 3D-Based Virtual Screening Methods. J. Chem. Inf. Model. 2021, 61, 5841–5852. [Google Scholar] [CrossRef] [PubMed]
- Jain, A.N.; Nicholls, A. Recommendations for evaluation of computational methods. J. Comput. Aided. Mol. Des. 2008, 22, 133–139. [Google Scholar] [CrossRef] [PubMed]
- Filisola-Villaseñor, J.G.; Aranda-Barradas, M.E.; Miranda-Castro, S.P.; Mendieta-Wejebe, J.E.; Valdez Guerrero, A.S.; Guillen Castro, S.A.; Martínez Castillo, M.; Tamay-Cach, F.; Álvarez-Almazán, S. Impact of Molecular Symmetry/Asymmetry on Insulin-Sensitizing Treatments for Type 2 Diabetes. Symmetry 2022, 14, 1240. [Google Scholar] [CrossRef]
- Sever, B.; Soybir, H.; Görgülü, Ş.; Cantürk, Z.; Altıntop, M.D. Pyrazole Incorporated New Thiosemicarbazones: Design, Synthesis and Investigation of DPP-4 Inhibitory Effects. Molecules 2020, 25, 5003. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, T.; Akahoshi, F.; Sakashita, H.; Kitajima, H.; Nakamura, M.; Sonda, S.; Takeuchi, M.; Tanaka, Y.; Ueda, N.; Sekiguchi, S.; et al. Discovery and preclinical profile of teneligliptin (3-[(2S,4S)-4-[4-(3-methyl1-phenyl-1H-pyrazol-5-yl)piperazin-1-yl]pyrrolidin-2- ylcarbonyl]thiazolidine): A highly potent, selective, long-lasting and orally active dipeptidyl peptidase IV inhibitor for the treatment of type 2 diabetes. Bioorg. Med. Chem. 2012, 20, 5705–5719. [Google Scholar] [PubMed]
- Pantaleão, S.Q.; Maltarollo, V.G.; Araujo, S.C.; Gertrudes, J.C.; Honorio, K.M. Molecular docking studies and 2D analyses of DPP-4 inhibitors as candidates in the treatment of diabetes. Mol. Biosyst. 2015, 11, 3188–3193. [Google Scholar] [CrossRef]
- Pan, J.; Zhang, Q.; Zhang, C.; Yang, W.; Liu, H.; Lv, Z.; Liu, J.; Jiao, Z. Inhibition of Dipeptidyl Peptidase-4 by Flavonoids: Structure-Activity Relationship, Kinetics and Interaction Mechanism. Front Nutr. 2022, 12, 892426. [Google Scholar] [CrossRef] [PubMed]
- Alon, G.; Tuvi-Arad, I. Improved algorithms for symmetry analysis: Structure preserving permutations. J. Math. Chem. 2018, 56, 193–212. [Google Scholar] [CrossRef]
- Zabrodsky, H.; Peleg, S.; Avnir, D. Continuous symmetry measures. J. Am. Chem. Soc. 1992, 114, 7843–7851. [Google Scholar] [CrossRef]
- Bochevarov, A.D.; Harder, E.; Hughes, T.F.; Greenwood, J.R.; Braden, D.A.; Philipp, D.M.; Rinaldo, D.; Halls, M.D.; Zhang, J.; Friesner, R.A. Jaguar: A high-performance quantum chemistry software program with strengths in life and materials sciences. Int. J. Quantum Chem. 2013, 113, 2110–2142. [Google Scholar] [CrossRef]
- Li, D.; Wang, B.; Long, X.; Xu, W.; Xia, Y.; Yang, D.; Yao, X. Controlled Asymmetric Charge Distribution of Active Centers in Conjugated Polymers for Oxygen Reduction. Angew. Chem. 2021, 60, 26483–26488. [Google Scholar] [CrossRef] [PubMed]
- Tripathy, T.; De, B.R. Making sense about Dipole Moments. J. Phys. Sci. 2008, 12, 155–172. [Google Scholar]
- Álvarez-Almazán, S.; Filisola-Villaseñor, J.G.; Alemán-González-Duhart, D.; Tamay-Cach, F.; Mendieta-Wejebe, J.E. Current molecular aspects in the development and treatment of diabetes. J. Physiol. Biochem. 2020, 76, 13–35. [Google Scholar] [CrossRef] [PubMed]
- Morimoto, J.; Miyamoto, K.; Ichikawa, Y.; Uchiyama, M.; Makishima, M.; Hashimoto, Y.; Ishikawa, M. Improvement in aqueous solubility of achiral symmetric cyclofenil by modification to a chiral asymmetric analog. Sci. Rep. 2021, 11, 12697. [Google Scholar] [CrossRef] [PubMed]
- Veber, D.F.; Johnson, S.R.; Cheng, H.Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef] [PubMed]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| NPs ID | CG4 | MW | HBA | HBD | RBN | TPSA | LogP | Lipinski #Violations |
|---|---|---|---|---|---|---|---|---|
| ZINC84153787 | −8.931 | 478.40 | 12 | 7 | 5 | 199.51 | 2.11 | 2 |
| ZINC33833997 | −8.354 | 432.38 | 10 | 6 | 4 | 170.05 | 1.71 | 1 |
| ZINC33833796 | −8.209 | 446.36 | 11 | 6 | 4 | 187.12 | 1.15 | 2 |
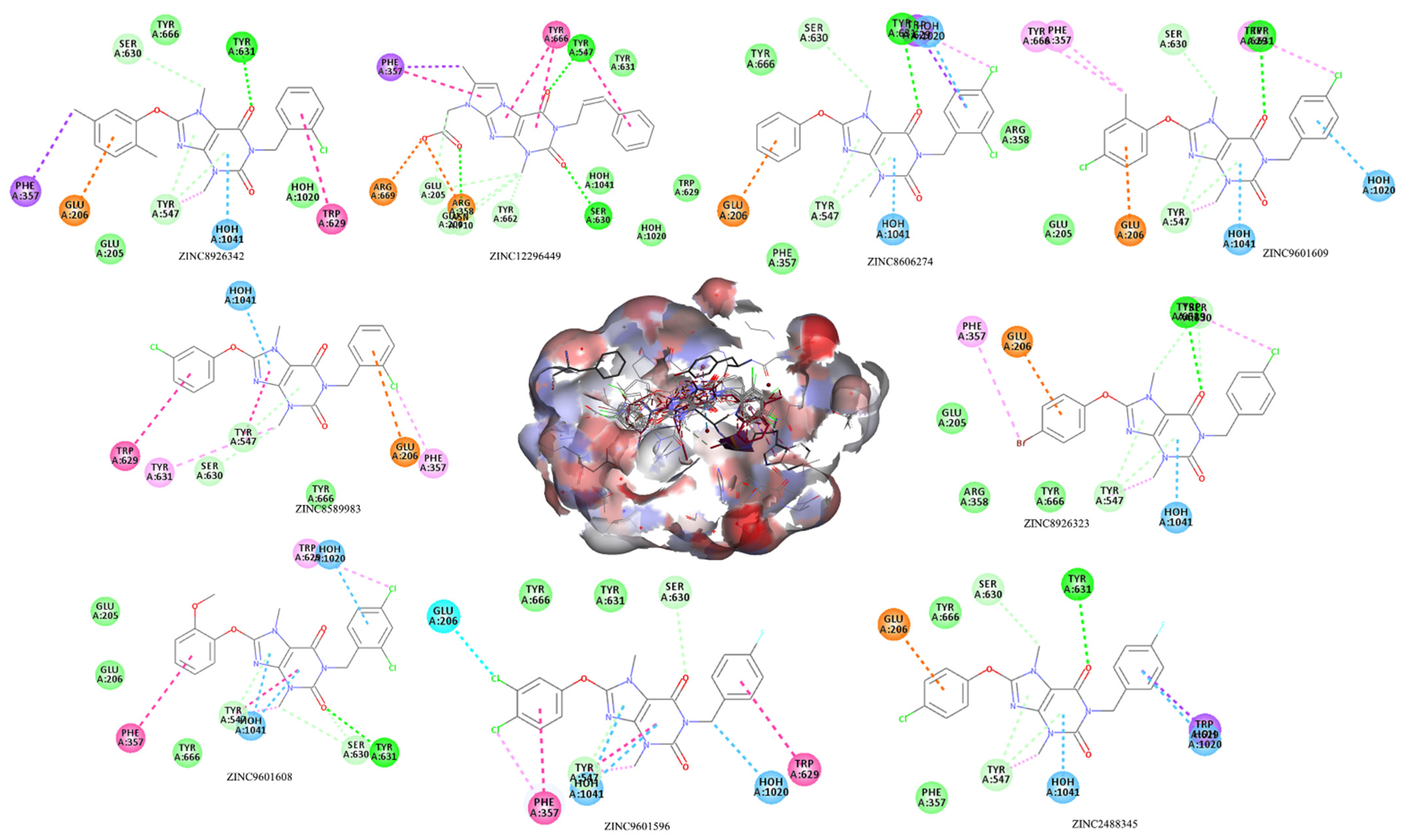
| ZINC8926342 | −6.555 | 424.88 | 4 | 0 | 4 | 71.05 | 3.94 | 0 |
| ZINC12296449 | −6.354 | 393.40 | 5 | 1 | 5 | 103.53 | 2.98 | 0 |
| ZINC8606274 | −6.350 | 431.27 | 4 | 0 | 4 | 71.05 | 3.94 | 0 |
| ZINC9601609 | −6.208 | 445.30 | 4 | 0 | 4 | 71.05 | 4.24 | 0 |
| ZINC8926323 | −6.177 | 475.72 | 4 | 0 | 4 | 71.05 | 3.97 | 0 |
| ZINC2488345 | −6.005 | 414.82 | 5 | 0 | 4 | 71.05 | 3.75 | 0 |
| ZINC9601596 | −5.765 | 449.26 | 5 | 0 | 4 | 71.05 | 3.98 | 0 |
| ZINC1094329 | −5.719 | 436.33 | 3 | 0 | 3 | 65.06 | 4.07 | 0 |
| ZINC8916385 | −5.618 | 422.31 | 3 | 0 | 3 | 65.06 | 3.87 | 0 |
| ZINC640985 | −5.496 | 377.83 | 4 | 1 | 5 | 85.29 | 3.29 | 0 |
| ZINC832612 | −5.467 | 432.31 | 3 | 0 | 3 | 65.06 | 3.77 | 0 |
| ZINC2334127 | −5.462 | 446.34 | 3 | 0 | 3 | 65.06 | 3.88 | 0 |
| ZINC838204 | −5.433 | 377.83 | 4 | 2 | 5 | 94.08 | 2.98 | 0 |
| ZINC838200 | −5.433 | 377.83 | 4 | 2 | 5 | 94.08 | 3.00 | 0 |
| ZINC6732655 | −5.428 | 410.30 | 3 | 1 | 6 | 73.85 | 3.83 | 0 |
| ZINC9601608 | −5.385 | 461.30 | 5 | 0 | 5 | 80.28 | 4.09 | 0 |
| ZINC838142 | −5.353 | 385.44 | 4 | 0 | 3 | 65.06 | 3.70 | 0 |
| ZINC1108640 | −5.265 | 410.30 | 3 | 1 | 5 | 73.85 | 3.73 | 0 |
| ZINC2488364 | −5.058 | 377.83 | 4 | 1 | 6 | 83.08 | 3.40 | 0 |
| ZINC9601615 | −5.012 | 436.30 | 4 | 1 | 7 | 83.08 | 3.66 | 0 |
| ZINC4147162 | −4.938 | 420.30 | 3 | 1 | 6 | 73.85 | 3.73 | 0 |
| ZINC1094334 | −4.890 | 389.84 | 4 | 0 | 3 | 74.29 | 3.49 | 0 |
| ZINC2488384 | −4.883 | 422.28 | 4 | 1 | 6 | 83.08 | 3.58 | 0 |
| ZINC789645 | −4.880 | 371.41 | 4 | 0 | 3 | 65.06 | 3.62 | 0 |
| ZINC8589983 | −4.865 | 431.27 | 4 | 0 | 4 | 71.05 | 3.76 | 0 |
| ZINC13512682 | −4.767 | 412.27 | 4 | 1 | 6 | 83.08 | 3.60 | 0 |
| ZINC13142224 | −4.745 | 387.43 | 5 | 1 | 8 | 92.31 | 3.58 | 0 |
| ZINC6565526 | −4.550 | 407.83 | 5 | 0 | 3 | 74.29 | 3.27 | 0 |
| Linagliptin | −4.460 | 472.54 | 6 | 1 | 4 | 116.86 | 3.73 | 0 |
| ZINC2508009 | −10.551 | 290.36 | 2 | 2 | 4 | 55.12 | 2.44 | 0 |
| ZINC85879571 | −9.829 | 321.38 | 3 | 2 | 4 | 73.91 | 1.92 | 0 |
| ZINC11692316 | −9.682 | 363.41 | 3 | 3 | 4 | 85.43 | 1.81 | 0 |
| ZINC96112842 | −9.297 | 307.37 | 5 | 1 | 5 | 81.60 | 1.52 | 0 |
| ZINC604405970 | −9.149 | 335.44 | 1 | 1 | 6 | 41.03 | 3.10 | 0 |
| ZINC12296782 | −9.138 | 326.39 | 2 | 2 | 2 | 68.44 | 2.72 | 0 |
| Sitagliptin | −8.859 | 407.31 | 10 | 1 | 4 | 77.04 | 2.35 | 0 |
| ZINC15674091 | −11.613 | 360.41 | 5 | 2 | 4 | 78.18 | 2.58 | 0 |
| ZINC524732009 | −11.498 | 370.36 | 5 | 2 | 2 | 87.97 | 2.50 | 0 |
| ZINC15830055 | −11.158 | 365.47 | 5 | 3 | 3 | 68.09 | 3.47 | 0 |
| ZINC3984976 | −11.003 | 332.40 | 3 | 2 | 4 | 66.59 | 2.60 | 0 |
| ZINC85509222 | −10.850 | 450.39 | 11 | 7 | 3 | 186.37 | 0.95 | 2 |
| Alogliptin | −10.635 | 339.39 | 4 | 1 | 3 | 97.05 | 2.38 | 0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Istrate, D.; Crisan, L. Natural Compounds as DPP-4 Inhibitors: 3D-Similarity Search, ADME Toxicity, and Molecular Docking Approaches. Symmetry 2022, 14, 1842. https://doi.org/10.3390/sym14091842
Istrate D, Crisan L. Natural Compounds as DPP-4 Inhibitors: 3D-Similarity Search, ADME Toxicity, and Molecular Docking Approaches. Symmetry. 2022; 14(9):1842. https://doi.org/10.3390/sym14091842
Chicago/Turabian StyleIstrate, Daniela, and Luminita Crisan. 2022. "Natural Compounds as DPP-4 Inhibitors: 3D-Similarity Search, ADME Toxicity, and Molecular Docking Approaches" Symmetry 14, no. 9: 1842. https://doi.org/10.3390/sym14091842
APA StyleIstrate, D., & Crisan, L. (2022). Natural Compounds as DPP-4 Inhibitors: 3D-Similarity Search, ADME Toxicity, and Molecular Docking Approaches. Symmetry, 14(9), 1842. https://doi.org/10.3390/sym14091842