Optimisation of Radium Removal from Saline Produced Waters during Oil and Gas Extraction

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Radium Recovery by Barite Co-Precipitation
2.3. Kinetics of Radium Recovery by Barite Post-Precipitation
2.4. Mineralogical Analysis
3. Results
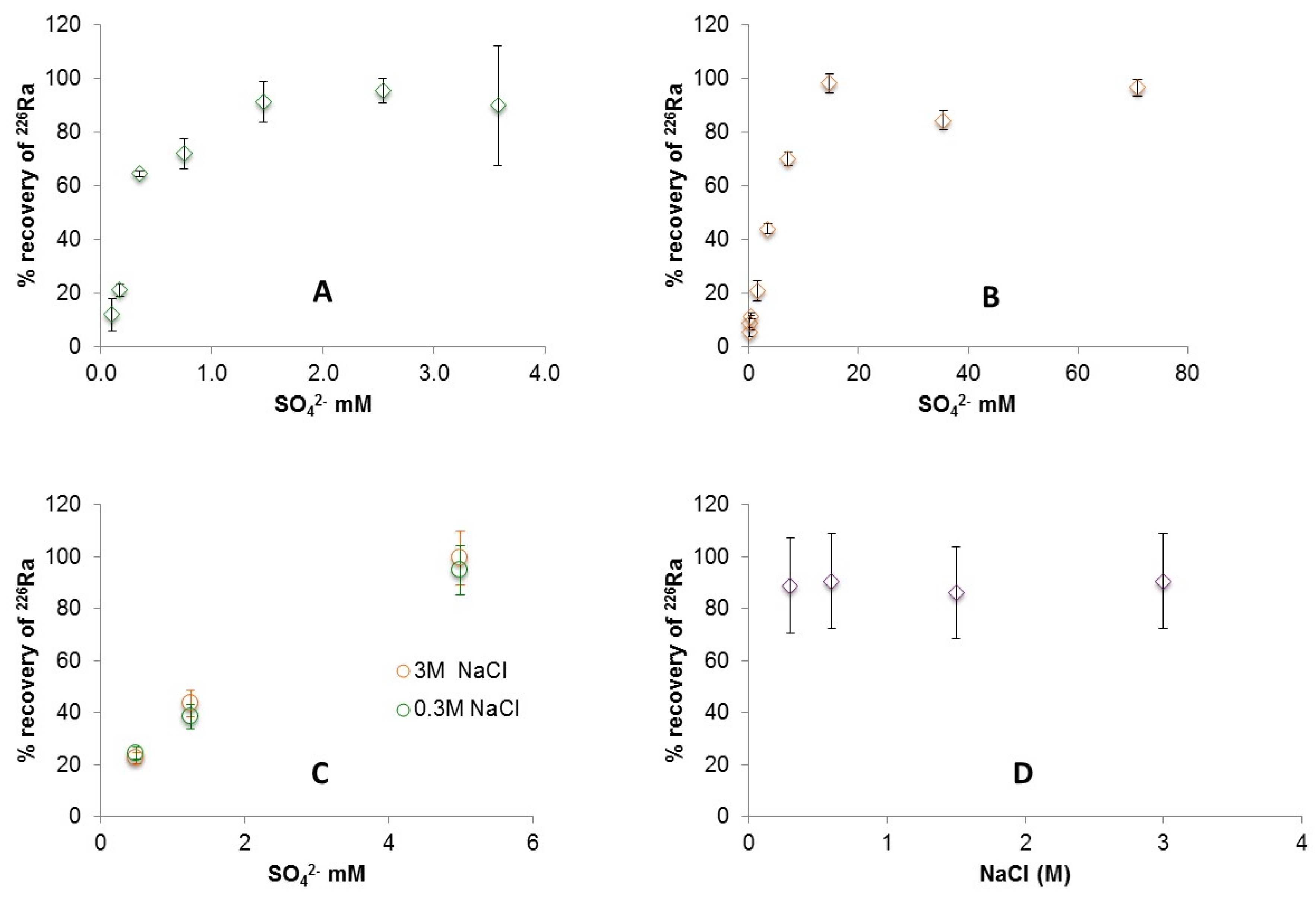
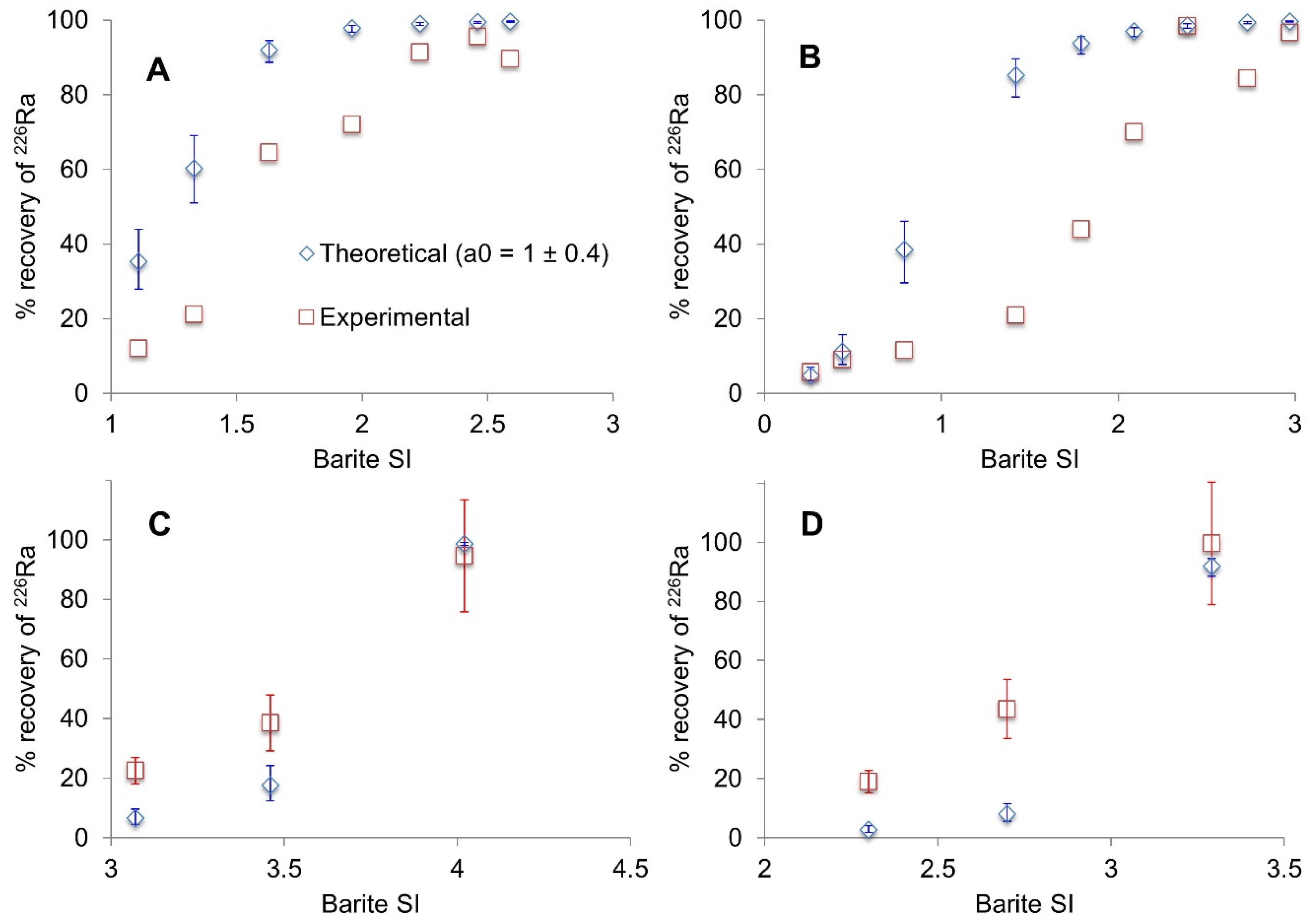
3.1. Radium Recovery by Barite Co-Precipitation
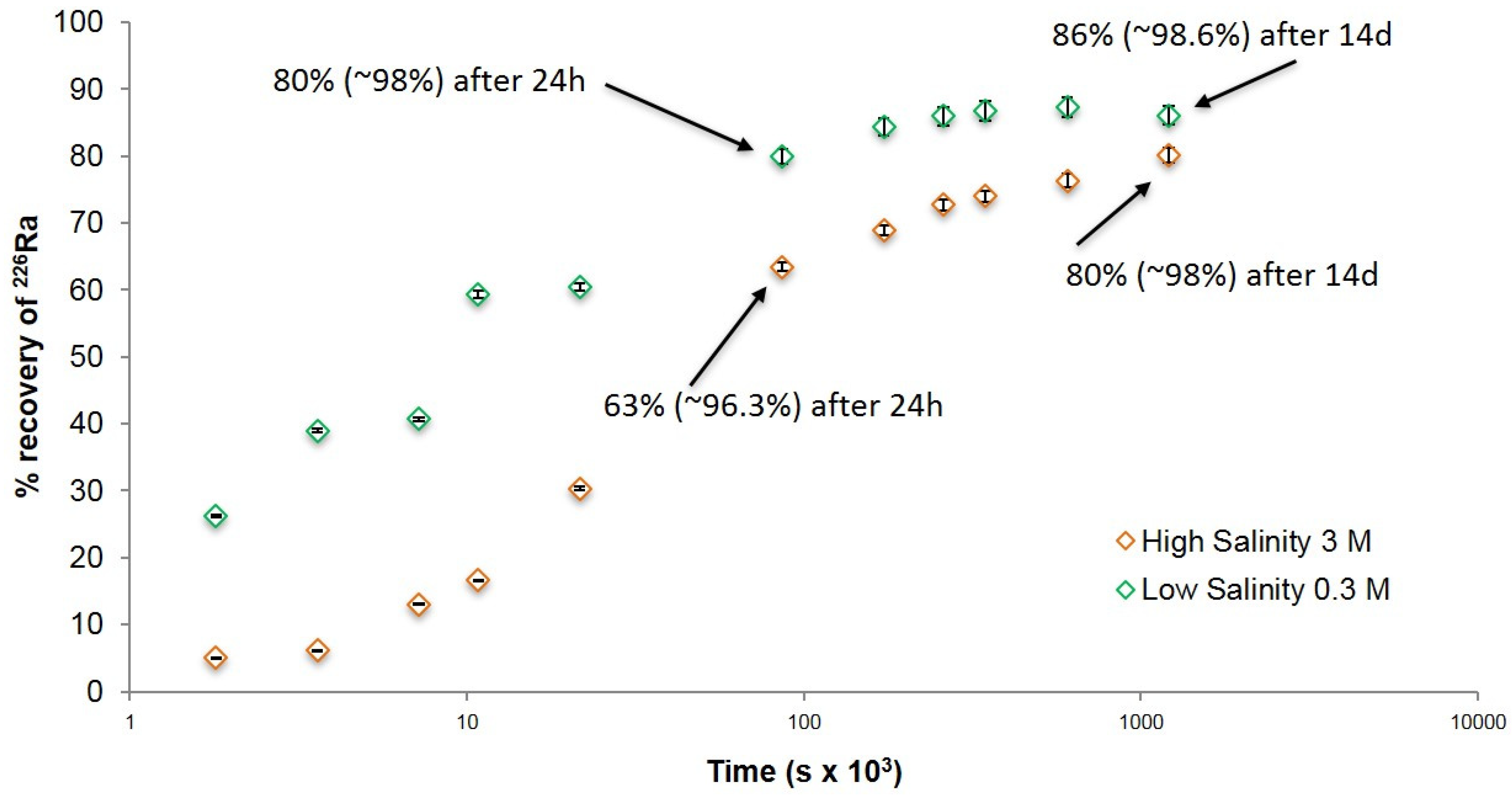
3.2. Kinetics of Radium Recovery by Barite Recrystallization Post-Precipitation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Elster, J.; Geitel, H. Über die radioaktive Substanz, deren Emanation in der Bodenluft und der Atmosphäre enthalten ist. Phys. Z. 1904, 5, 321–325. [Google Scholar]
- Schmidt, H.W.; Kurz, K. Natural radioactive substances in thermal brines. Phys. Z. 1906, 7, 213–224. [Google Scholar]
- Thompson, H. Fracking boom spurs environmental audit. Nature 2012, 485, 556–557. [Google Scholar] [CrossRef] [PubMed]
- Shih, J.-S.; Saiers, J.E.; Anisfeld, S.C.; Chu, Z.; Muehlenbachs, L.A.; Olmstead, S.M. Characterization and analysis of liquid waste from Marcellus Shale gas development. Environ. Sci. Technol. 2015, 49, 9557–9565. [Google Scholar] [CrossRef]
- Heaton, B.; Lambley, J. TENORM in the oil, gas and mineral mining industry. Appl. Radiat. Isot. 1995, 46, 577–581. [Google Scholar] [CrossRef]
- Worden, R.H.; Manning, D.A.C.; Lythgoe, P.R. The origin and production geochemistry of radioactive lead (210Pb) in NORM-contaminated formation waters. J. Geochem. Explor. 2000, 70, 695–699. [Google Scholar] [CrossRef]
- Garner, J.; Cairns, J.; Read, D. NORM in the East Midlands’ oil and gas producing region of the UK. J. Envrion. Radioact. 2015, 150, 49–56. [Google Scholar] [CrossRef]
- Cooke, C.E., Jr. Hydraulic Fracturing Method. U.S. Patent US3888311A, 10 June 1975. [Google Scholar]
- Environment Agency. Shale Gas North West-Monitoring of Flowback Water; Environment Agency: Bristol, UK, 2011.
- Her Majesty’s Stationery Office. The Environmental Permitting (England and Wales) Regulations; UK Statutory Instruments, No. 1154; Her Majesty’s Stationery Office: London, UK, 2016. [Google Scholar]
- U.S. Nuclear Regulatory Commission (NRC). Limits for Industrial Wastewater Discharge. Available online: https://www.nrc.gov/waste.html (accessed on 31 January 2020).
- Maxwell, S.L.; Culligan, B.K.; Warren, R.A.; McAlister, D.R. Rapid method for the determination of 226Ra in hydraulic fracturing wastewater samples. J. Radioanal. Nucl. Chem. 2016, 309, 1333–1340. [Google Scholar] [CrossRef]
- Haluszczak, L.O.; Rose, A.W.; Kump, L.R. Geochemical evaluation of flowback brine from Marcellus gas wells in Pennsylvania, USA. Appl. Geochem. 2013, 28, 55–61. [Google Scholar] [CrossRef]
- Elliott, E.G.; Ettinger, A.S.; Leaderer, B.P.; Bracken, M.B.; Deziel, N.C. A systematic evaluation of chemicals in hydraulic-fracturing fluids and wastewater for reproductive and developmental toxicity. J. Expo. Sci. Environ. Epidemiol. 2016, 27, 90–99. [Google Scholar] [CrossRef]
- Kondash, A.J.; Albright, E.; Vengosh, A. Quantity of flowback and produced waters from unconventional oil and gas exploration. Sci. Total Environ. 2017, 574, 314–321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lester, Y.; Ferrer, I.; Thurman, E.M.; Sitterley, K.A.; Korak, J.A.; Aiken, G.; Linden, K.G. Characterization of hydraulic fracturing flowback water in Colorado: Implications for water treatment. Sci. Total Environ. 2015, 512–513, 637–644. [Google Scholar] [CrossRef] [PubMed]
- Rowan, E.L.; Engle, M.A.; Kirby, C.S.; Kraemer, T.F. Radium Content of Oil- and Gas-Field Produced Waters in the Northern Appalachian Basin (USA)—Summary and Discussion of Data; U.S. Geological Survey Scientific Investigations Report 2011–5135; USGS: Reston, VA, USA, 2011; 31p.
- Rowan, E.L.; Engle, M.A.; Kraemer, T.F.; Schroeder, K.T.; Hammack, R.W.; Doughten, M.W. Geochemical and isotopic evolution of water produced from Middle Devonian Marcellus shale gas wells, Appalachian basin, Pennsylvania. AAPG Bull. 2015, 99, 181–206. [Google Scholar] [CrossRef]
- Blondes, M.S.; Gans, K.D.; Engle, M.A.; Kharaka, Y.K.; Reidy, M.E.; Saraswathula, V.; Thordsen, J.J.; Rowan, E.L.; Morrissey, E.A. U.S. Geological Survey National Produced Waters Geochemical Database; ver. 2.3; U.S. Geological Survey Data Release; USGS: Reston, VA, USA, 2018. [CrossRef]
- Zhang, T.; Gregory, K.; Hammack, R.W.; Vidic, R.D. Co-precipitation of radium with barium and strontium sulfate and its impact on the fate of radium during treatment of produced water from unconventional gas extraction. Environ. Sci. Technol. 2014, 48, 4596–4603. [Google Scholar] [CrossRef] [PubMed]
- Doerner, H.A.; Hoskins, W.M. Coprecipitation of radium with barium sulfates. J. Am. Chem. Soc. 1925, 47, 662–675. [Google Scholar] [CrossRef]
- Gordon, L.; Rowley, K. Coprecipitation of radium with barium sulfate. Anal. Chem. 1957, 29, 34–37. [Google Scholar] [CrossRef]
- Langmuir, D.; Riese, A.C. The thermodynamic properties of radium. Geochim. Cosmochim. Acta 1985, 49, 1573–1601. [Google Scholar] [CrossRef]
- Choppin, G.; Liljenzin, J.-O.; Rydberg, J.; Ekberg, C. Radiochemistry and Nuclear Chemistry, 4th ed.; Academic Press: Cambridge, MA, USA, 2013. [Google Scholar]
- Anderson, G.M.; Crerar, D.A. Thermodynamics of Geochemistry: The Equilibrium Model, 1st ed.; Oxford Univesity Press: Oxford, UK, 1993. [Google Scholar]
- Risthaus, P.; Bosbach, D.; Becker, U.; Putnis, A. Barite scale formation and dissolution at high ionic strength studied with atomic force microscopy. Colloids Surf. 2001, 191, 201–214. [Google Scholar] [CrossRef]
- Fernandez-Diaz, L.; Putnis, A.; Cumberbatch, J. Barite nucleation kinetics and the effects of additives. Eur. J. Mineral. 1990, 2, 495–501. [Google Scholar] [CrossRef]
- Nielsen, A.E.; Toft, J.M. Electrolyte crystal growth kinetics. J. Cryst. Growth 1984, 67, 278–288. [Google Scholar] [CrossRef]
- He, S.; Oddo, J.E.; Tomson, M.B. The nucleation kinetics of barium sulfate in NaCl solutions up to 6 M and 90 °C. J. Colloid Interface Sci. 1995, 174, 319–326. [Google Scholar] [CrossRef]
- Bosbach, D.; Böttle, M.; Metz, V. Experimental Study on Ra2+ Uptake by Barite (BaSO4); SKB Technical Report TR-10-43; Waste Management: Houston, TX, USA, 2010. [Google Scholar]
- Curti, E.; Fujiwara, K.; Iijima, K.; Tits, J.; Cuesta, C.; Kitamura, A.; Glaus, M.A.; Müller, W. Radium uptake during barite recrystallization at 23 ± 2 °C as a function of solution composition: An experimental 133Ba and 226Ra tracer study. Geochim. Cosmochim. Acta 2010, 74, 3553–3570. [Google Scholar] [CrossRef]
- Gilmore, G. Practical Gamma-Ray Spectrometry, 2nd ed.; Wiley: Hoboken, NJ, USA, 2011. [Google Scholar]
- International Atomic Energy Agency (IAEA). Reference material IAEA 434: Naturally Occurring Radionuclides in Phosphogypsum; Analytical Quality in Nuclear Applications. Series No. 17; IAEA: Vienna, Austria, 2010. [Google Scholar]
- Monnin, C. A thermodynamic model for the solubility of barite and celestite in electrolyte solutions and seawater to 200 °C and to 1 kbar. Chem. Geol. 1999, 153, 187–209. [Google Scholar] [CrossRef]
- Templeton, C.C. Solubility of barium sulfate in sodium chloride solutions from 25 to 95 °C. J. Chem. Eng. Data 1960, 5, 514–516. [Google Scholar] [CrossRef]
- Ceccarello, S.; Black, S.; Read, D.; Hodson, M.E. Industrial radioactive barite scale: Suppression of radium uptake by introduction of competing ions. Miner. Eng. 2004, 17, 323–330. [Google Scholar] [CrossRef]
- Maguire-Boyle, S.J.; Barron, A.R. Organic compounds in produced waters from shale gas wells. Environ. Sci. Process. Impacts 2014, 16, 2237–2248. [Google Scholar] [CrossRef]
- Vinograd, V.L.; Brandt, F.; Rozov, K.; Klinkenberg, M.; Refson, K.; Winkler, B.; Bosbach, D. Solid–aqueous equilibrium in the BaSO4–RaSO4–H2O system: First-principles calculations and a thermodynamic assessment. Geochim. Cosmochim. Acta 2013, 122, 398–417. [Google Scholar] [CrossRef]
- Parkhurst, D.L.; Appelo, C.A.J. User’s Guide to PHREEQC (version 2)—A Computer Program for Speciation, Batch-Reaction, One-Dimensional Transport and Inverse Geochemical Calculations; U.S. Geological Survey, Water-Resources Investigations Report; USGS: Reston, VA, USA, 1999.
- Guggenheim, E.A.; Turgeon, J.C. Specific interaction of ions. Trans. Faraday Soc. 1955, 51, 747–761. [Google Scholar] [CrossRef]
- Ciavatta, L. The specific interaction theory in the evaluating ionic equilibria. Ann. Chim. 1980, 70, 551–562. [Google Scholar]
- Zhu, C. Coprecipitation in the barite isostructural family: 1. binary mixing properties. Geochim. Cosmochim. Acta 2004, 68, 3327–3337. [Google Scholar] [CrossRef]
- Kowacz, M.; Putnis, C.V.; Putnis, A. The effect of cation:anion ratio in solution on the mechanism of barite growth at constant supersaturation: Role of the desolvation process on the growth kinetics. Geochim. Cosmochim. Acta 2007, 71, 5168–5179. [Google Scholar] [CrossRef]
- Piana, S.; Jones, F.; Gale, J.D. Assisted desolvation as a key kinetic step for crystal growth. J. Am. Chem. Soc. 2006, 128, 13568–13574. [Google Scholar] [CrossRef] [PubMed]
- Her Majesty’s Stationery Office. The Water Supply (Water Quality) Regulations; UK Statutory Instruments No. 614; Her Majesty’s Stationery Office: London, UK, 2016. [Google Scholar]
- European Council. Laying down Requirements for the Protection of the Health of the General Public with Regard to Radioactive Substances in Water Intended for Human Consumption; Council Directive 2013/51/Euratom; CEC: Brussels, Belgium, 2013. [Google Scholar]
- Olsson, O.; Weichgrebe, D.; Rosenwinkel, K.H. Hydraulic fracturing wastewater in Germany: Composition, treatment, concerns. Environ. Earth Sci. 2013, 70, 3895–3906. [Google Scholar] [CrossRef]
- Haghshenas, A.; Nasr-El-Din, H.A. Effect of dissolved solids on reuse of produced water at high temperature in hydraulic fracturing jobs. J. Nat. Gas Sci. Eng. 2014, 21, 316–325. [Google Scholar] [CrossRef]
- Zhang, T.; Hammack, R.W.; Vidic, R.D. Fate of radium in Marcellus Shale flowback water impoundments and assessment of associated health risks. Environ. Sci. Technol. 2015, 49, 9347–9354. [Google Scholar] [CrossRef]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Garner, J.; Read, D. Optimisation of Radium Removal from Saline Produced Waters during Oil and Gas Extraction. Minerals 2020, 10, 278. https://doi.org/10.3390/min10030278
Garner J, Read D. Optimisation of Radium Removal from Saline Produced Waters during Oil and Gas Extraction. Minerals. 2020; 10(3):278. https://doi.org/10.3390/min10030278
Chicago/Turabian StyleGarner, Joel, and David Read. 2020. "Optimisation of Radium Removal from Saline Produced Waters during Oil and Gas Extraction" Minerals 10, no. 3: 278. https://doi.org/10.3390/min10030278
APA StyleGarner, J., & Read, D. (2020). Optimisation of Radium Removal from Saline Produced Waters during Oil and Gas Extraction. Minerals, 10(3), 278. https://doi.org/10.3390/min10030278