Capacity of Chlorate to Oxidize Ferrous Iron: Implications for Iron Oxide Formation on Mars




Abstract
:1. Introduction
2. Materials and Methods
2.1. Kinetic Experiments
2.2. Mineral Precipitation Studies
3. Results
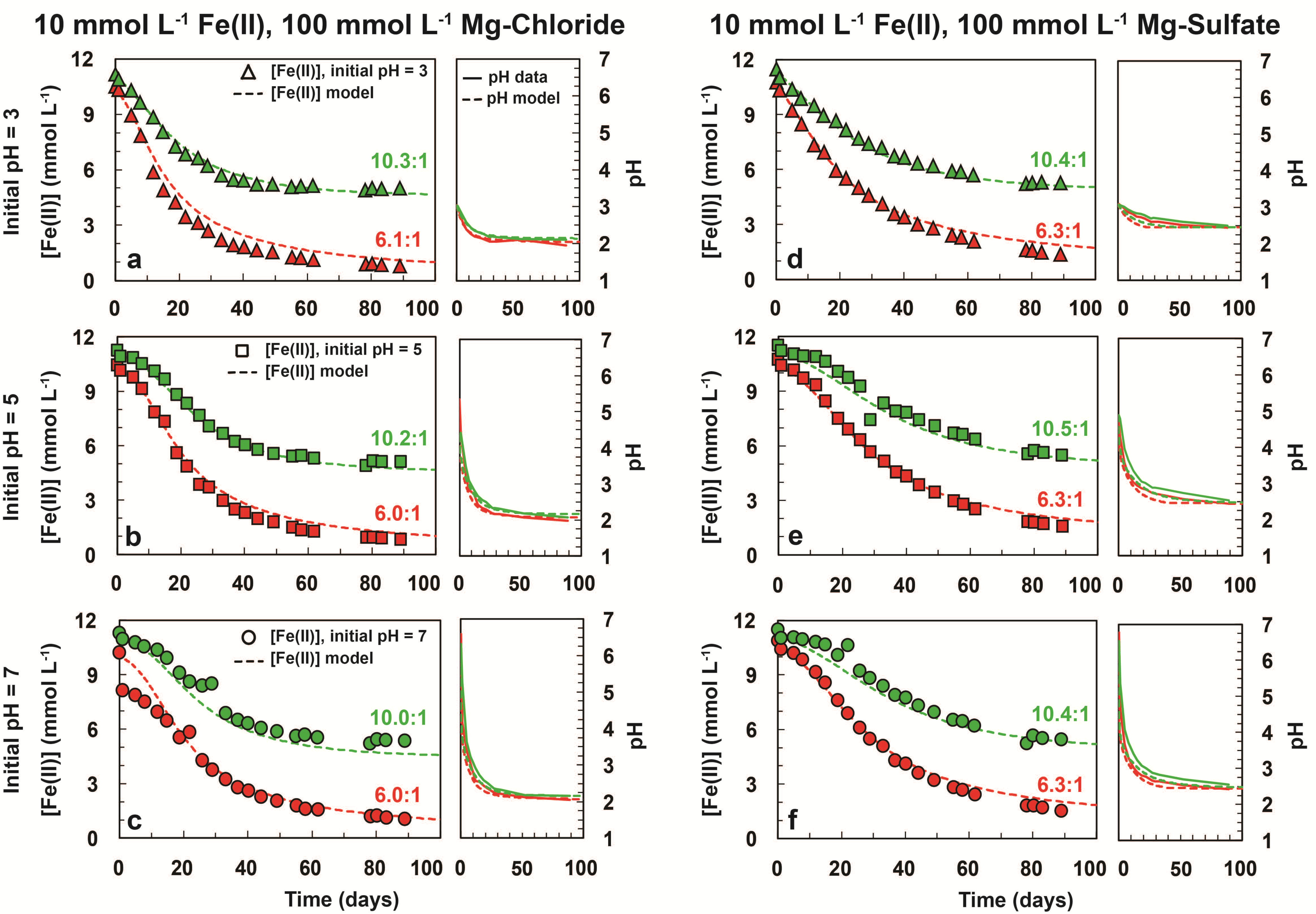
3.1. Iron Oxidation in Chlorate-Equivalent vs. Chlorate-Deficient Systems
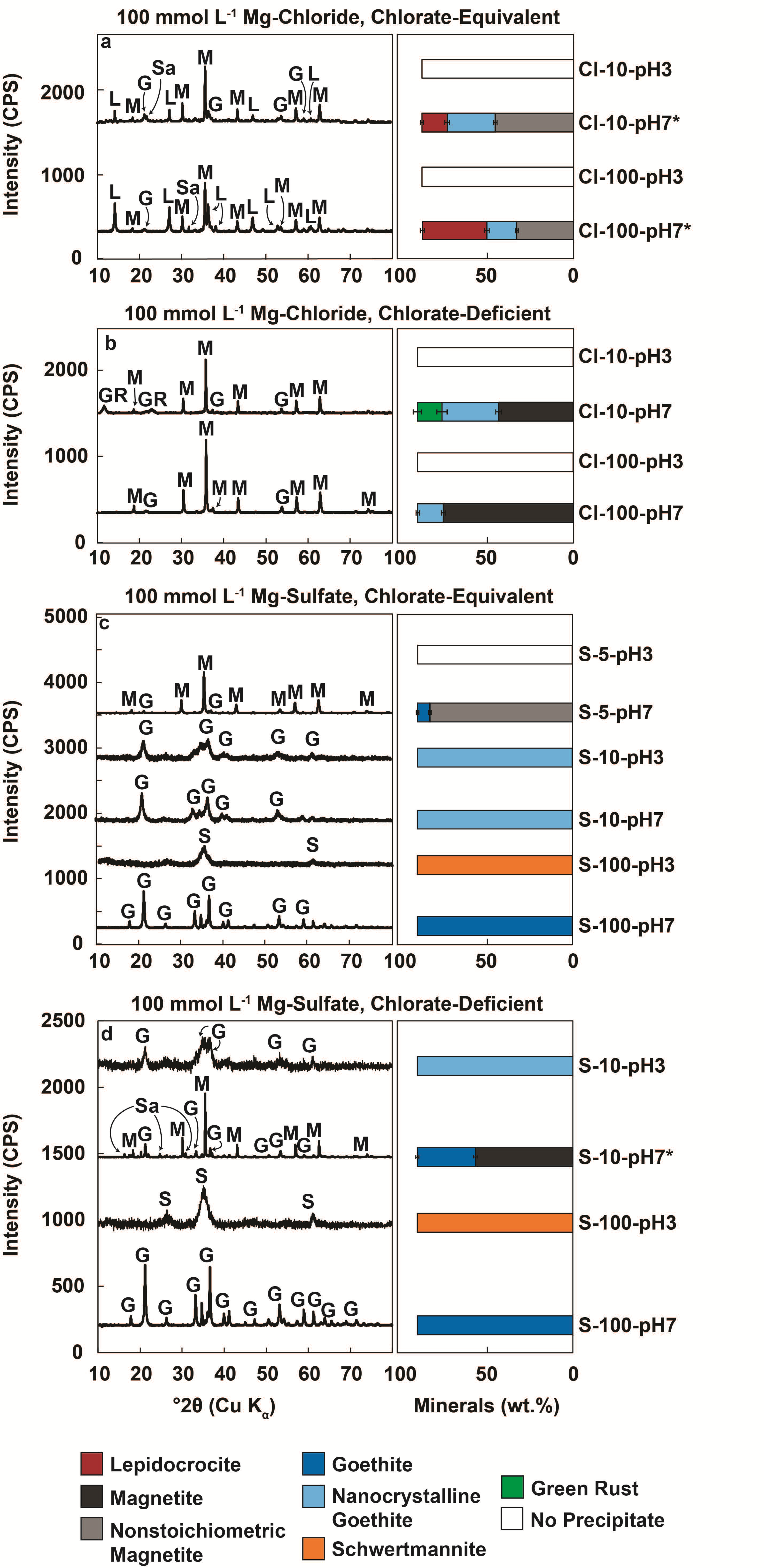
3.2. Mineral Products in Chlorate-Equivalent vs. Chlorate-Deficient Systems
3.2.1. Minerals Produced in Chloride-Rich Fluids
3.2.2. Minerals Produced in Sulfate-Rich Fluids
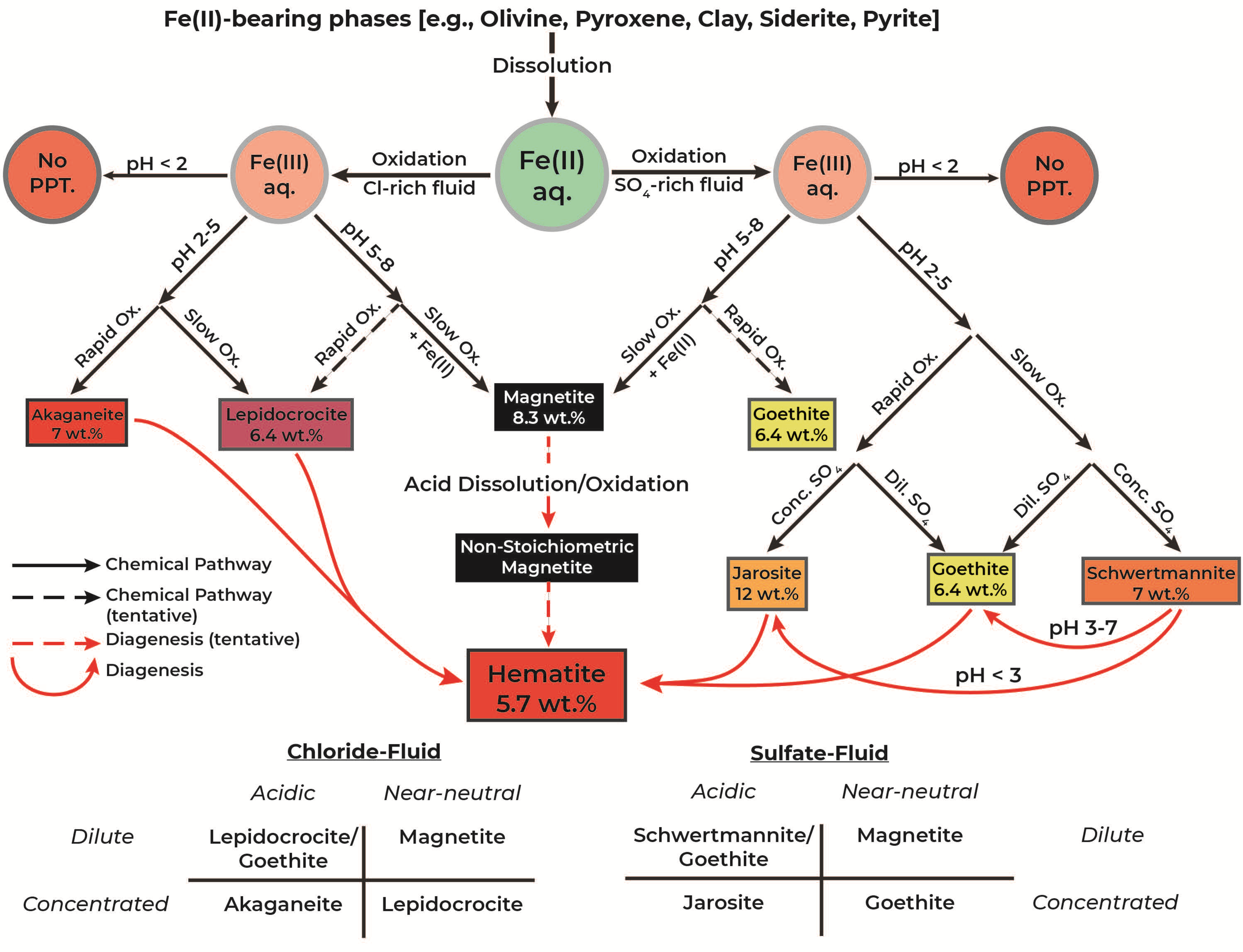
4. Discussion
4.1. Fe(II) Oxidation Capacity of Chlorate
4.2. Geochemical Parameters Determining Mineral Products
4.2.1. Fluid Composition
4.2.2. pH Evolution
4.2.3. Oxidation Rate
4.3. Quantitative Measure of Mineral Production
4.4. Implications for Gale Crater, Mars
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Christensen, P.R.; Bandfield, J.L.; Clark, R.N.; Edgett, K.S.; Hamilton, V.E.; Hoefen, T.; Kieffer, H.H.; Kuzmin, R.O.; Lane, M.D.; Malin, M.C.; et al. Detection of crystalline hematite mineralization on Mars by the Thermal Emission Spectrometer: Evidence for near-surface water. J. Geophys. Res. Planets 2000, 105, 9623–9642. [Google Scholar] [CrossRef]
- Lane, M.D.; Morris, R.V.; Mertzman, S.A.; Christensen, P.R. Evidence for platy hematite grains in Sinus Meridiani, Mars. J. Geophys. Res. Planets 2002, 107, 5126. [Google Scholar] [CrossRef]
- Christensen, P.R.; Wyatt, M.B.; Glotch, T.D.; Rogers, A.D.; Anwar, S.; Arvidson, R.E.; Bandfield, J.L.; Blaney, D.L.; Budney, C.; Calvin, W.M.; et al. Mineralogy at Meridiani Planum from the Mini-TES experiment on the Opportunity Rover. Science 2004, 306, 1733–1739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klingelhöfer, G.; Morris, R.V.; Bernhardt, B.; Schroder, C.; Rodionov, D.S.; de Souza, P.A.; Yen, A.; Gellert, R.; Evlanov, E.N.; Zubkov, B.; et al. Jarosite and hematite at Meridiani Planum from Opportunity’s Mossbauer spectrometer. Science 2004, 306, 1740–1745. [Google Scholar] [CrossRef] [PubMed]
- Morris, R.V.; Klingelhöfer, G.; Schroder, C.; Rodionov, D.S.; Yen, A.; Ming, D.W.; de Souza, P.A.; Fleischer, I.; Wdowiak, T.; Gellert, R.; et al. Mössbauer mineralogy of rock, soil, and dust at Gusev crater, Mars: Spirit’s journey through weakly altered olivine basalt on the plains and pervasively altered basalt in the Columbia Hills. J. Geophys. Res. Planets 2006, 111, E02S13. [Google Scholar] [CrossRef]
- Rampe, E.; Blake, D.; Bristow, T.; Ming, D.; Vaniman, D.; Morris, R.; Achilles, C.; Chipera, S.; Morrison, S.; Tu, V. Mineralogy and geochemistry of sedimentary rocks and eolian sediments in Gale crater, Mars: A review after six Earth years of exploration with Curiosity. Geochemistry 2020, 80, 125605. [Google Scholar] [CrossRef]
- Rampe, E.; Ming, D.; Blake, D.; Bristow, T.; Chipera, S.; Grotzinger, J.; Morris, R.; Morrison, S.; Vaniman, D.; Yen, A. Mineralogy of an ancient lacustrine mudstone succession from the Murray formation, Gale crater, Mars. Earth Planet. Sci. Lett. 2017, 471, 172–185. [Google Scholar] [CrossRef]
- Bridges, J.C.; Catling, D.; Saxton, J.; Swindle, T.; Lyon, I.; Grady, M. Alteration assemblages in Martian meteorites: Implications for near-surface processes. Space Sci. Rev. 2001, 96, 365–392. [Google Scholar] [CrossRef]
- Vaniman, D.T.; Bish, D.L.; Ming, D.W.; Bristow, T.F.; Morris, R.V.; Blake, D.F.; Chipera, S.J.; Morrison, S.M.; Treiman, A.H.; Rampe, E.B.; et al. Mineralogy of a mudstone at Yellowknife Bay, Gale Crater, Mars. Science 2014, 343, 1243480. [Google Scholar] [CrossRef]
- Carter, J.; Viviano-Beck, C.; Loizeau, D.; Bishop, J.; Le Deit, L. Orbital detection and implications of akaganeite on Mars. Icarus 2015, 253, 296–310. [Google Scholar] [CrossRef]
- Morris, R.V.; Golden, D.C.; Bell, J.F.; Shelfer, T.D.; Scheinost, A.C.; Hinman, N.W.; Furniss, G.; Mertzman, S.A.; Bishop, J.L.; Ming, D.W.; et al. Mineralogy, composition, and alteration of Mars Pathfinder rocks and soils: Evidence from multispectral, elemental, and magnetic data on terrestrial analogue, SNC meteorite, and Pathfinder samples. J. Geophys. Res. Planets 2000, 105, 1757–1817. [Google Scholar] [CrossRef]
- Milliken, R.E.; Swayze, G.A.; Arvidson, R.E.; Bishop, J.L.; Clark, R.N.; Ehlmann, B.L.; Green, R.O.; Grotzinger, J.P.; Morris, R.V.; Murchie, S.L.; et al. Opaline silica in young deposits on Mars. Geology 2008, 36, 847–850. [Google Scholar] [CrossRef]
- Farrand, W.H.; Glotch, T.D.; Rice, J.W.; Hurowitz, J.A.; Swayze, G.A. Discovery of jarosite within the Mawrth Vallis region of Mars: Implications for the geologic history of the region. Icarus 2009, 204, 478–488. [Google Scholar] [CrossRef]
- Thollot, P.; Mangold, N.; Ansan, V.; Le Mouelic, S.; Milliken, R.E.; Bishop, J.L.; Weitz, C.M.; Roach, L.H.; Mustard, J.F.; Murchie, S.L. Most Mars minerals in a nutshell: Various alteration phases formed in a single environment in Noctis Labyrinthus. J. Geophys. Res. Planets 2012, 117, E00J06. [Google Scholar] [CrossRef] [Green Version]
- Weitz, C.M.; Bishop, J.L.; Thollot, P.; Mangold, N.; Roach, L.H. Diverse mineralogies in two troughs of Noctis Labyrinthus, Mars. Geology 2011, 39, 899–902. [Google Scholar] [CrossRef]
- Ehlmann, B.L.; Mustard, J.F. An in-situ record of major environmental transitions on early Mars at Northeast Syrtis Major. Geophys. Res. Lett. 2012, 39, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Arvidson, R.E.; Wolff, M.J.; Mellon, M.T.; Catalano, J.G.; Wang, A.; Bishop, J.L. Lambert Albedo retrieval and analyses over Aram Chaos from OMEGA hyperspectral imaging data. J. Geophys. Res. Planets 2012, 117. [Google Scholar] [CrossRef] [Green Version]
- Morris, R.V.; Klingelhoefer, G.; Schröder, C.; Rodionov, D.S.; Yen, A.; Ming, D.W.; De Souza, P.; Wdowiak, T.; Fleischer, I.; Gellert, R. Mössbauer mineralogy of rock, soil, and dust at Meridiani Planum, Mars: Opportunity’s journey across sulfate-rich outcrop, basaltic sand and dust, and hematite lag deposits. J. Geophys. Res. Planets 2006, 111. [Google Scholar] [CrossRef] [Green Version]
- Bradley, J.P.; Harvey, R.P.; McSween, H.Y., Jr. Magnetite whiskers and platelets in the ALH84001 Martian meteorite: Evidence of vapor phase growth. Geochim. Cosmochim. Acta 1996, 60, 5149–5155. [Google Scholar] [CrossRef]
- Morris, R.V.; Klingelhoefer, G.; Bernhardt, B.; Schröder, C.; Rodionov, D.S.; De Souza, P.; Yen, A.; Gellert, R.; Evlanov, E.; Foh, J. Mineralogy at Gusev Crater from the Mössbauer spectrometer on the Spirit Rover. Science 2004, 305, 833–836. [Google Scholar] [CrossRef]
- Treiman, A.H.; Barrett, R.A.; Gooding, J.L. Preterrestrial aqueous alteration of the Lafayette (SNC) meteorite. Meteoritics 1993, 28, 86–97. [Google Scholar] [CrossRef]
- Gooding, J.L. Chemical weathering on Mars-thermodynamic stabilities of primary minerals (and their alteration products) from mafic igneous rocks. Icarus 1978, 33, 483–513. [Google Scholar] [CrossRef]
- Riveros, P.; Dutrizac, J. The precipitation of hematite from ferric chloride media. Hydrometallurgy 1997, 46, 85–104. [Google Scholar] [CrossRef]
- Cornell, R.M.; Schwertmann, U. The Iron Oxides: Structure, Properties, Reactions, Occurrences and Uses, 2nd ed.; John Wiley & Sons: Weinheim, Germany, 2003. [Google Scholar]
- Glotch, T.D.; Kraft, M. Thermal transformations of akaganeite and lepidocrocite to hematite: Assessment of possible precursors to Martian crystalline hematite. Phys. Chem. Miner. 2008, 35, 569–581. [Google Scholar] [CrossRef]
- Goss, C.J. The kinetics and reaction-mechanism of the goethite to hematite transformation. Mineral. Mag. 1987, 51, 437–451. [Google Scholar] [CrossRef] [Green Version]
- Glotch, T.D.; Morris, R.V.; Christensen, P.R.; Sharp, T.G. Effect of precursor mineralogy on the thermal infrared emission spectra of hematite: Application to Martian hematite mineralization. J. Geophys. Res. Planets 2004, 109, E07003. [Google Scholar] [CrossRef]
- Gehring, A.; Hofmeister, A. The transformation of lepidocrocite during heating: A magnetic and spectroscopic study. Clays Clay Miner. 1994, 42, 409–415. [Google Scholar] [CrossRef]
- Gómez-Villacieros, R.; Hernán, L.; Morales, J.; Tirado, J. Textural evolution of synthetic γ-FeOOH during thermal treatment by differential scanning calorimetry. J. Colloid Interface Sci. 1984, 101, 392–400. [Google Scholar] [CrossRef]
- Naono, H.; Fujiwara, R.; Sugioka, H.; Sumiya, K.; Yanazawa, H. Micropore formation due to thermal decomposition of acicular microcrystals of β-FeOOH. J. Colloid Interface Sci. 1982, 87, 317–332. [Google Scholar] [CrossRef]
- Bigham, J.; Nordstrom, D.K. Iron and aluminum hydroxysulfates from acid sulfate waters. Rev. Mineral. Geochem. 2000, 40, 351–403. [Google Scholar] [CrossRef]
- Stanjek, H.; Weidler, P. The effect of dry heating on the chemistry, surface area, and oxalate solubility of synthetic 2-line and 6-line ferrihydrites. Clay Miner. 1992, 27, 397–411. [Google Scholar] [CrossRef]
- Zolotov, M.Y.; Shock, E.L. Formation of jarosite-bearing deposits through aqueous oxidation of pyrite at Meridiani Planum, Mars. Geophys. Res. Lett. 2005, 32, L21203. [Google Scholar] [CrossRef] [Green Version]
- Christensen, P.R.; Ruff, S.W.; Fergason, R.L.; Knudson, A.T.; Anwar, S.; Arvidson, R.E.; Bandfield, J.L.; Blaney, D.L.; Budney, C.; Calvin, W.M.; et al. Initial results from the Mini-TES experiment in Gusev crater from the Spirit rover. Science 2004, 305, 837–842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atkinson, R.; Posner, A.; Quirk, J. Crystal nucleation and growth in hydrolysing iron (III) chloride solutions. Clays Clay Miner. 1977, 25, 49–56. [Google Scholar] [CrossRef]
- Hamada, S.; Matijevic, E. Ferric hydrous oxide sols IV. Preparation of uniform cubic hematite particles by hydrolysis of ferric-chloride in alcohol-water solutions. J. Colloid Interface Sci. 1981, 84, 274–277. [Google Scholar] [CrossRef]
- Hamada, S.; Matijevic, E. Formation of monodispersed colloidal cubic hematite particles in ethanol + water solutions. J. Chem. Soc. Faraday Trans. 1982, 78, 2147–2156. [Google Scholar] [CrossRef]
- Golden, D.C.; Ming, D.W.; Morris, R.V.; Graff, T.G. Hydrothermal synthesis of hematite spherules and jarosite: Implications for diagenesis and hematite spherule formation in sulfate outcrops at Meridiani Planum, Mars. Am. Miner. 2008, 93, 1201–1214. [Google Scholar] [CrossRef]
- Beitler, B.; Parry, W.T.; Chan, M.A. Fingerprints of fluid flow: Chemical diagenetic history of the Jurassic Navajo sandstone, Southern Utah, U.S.A. J. Sediment. Res. 2005, 75, 547–561. [Google Scholar] [CrossRef]
- Chan, M.A.; Bowen, B.B.; Parry, W.; Ormö, J. Red rock and red planet diagenesis. GSA Today 2005, 15, 4–10. [Google Scholar] [CrossRef]
- Chan, M.A.; Parry, W.; Bowman, J. Diagenetic hematite and manganese oxides and fault-related fluid flow in Jurassic sandstones, southeastern Utah. AAPG Bull. 2000, 84, 1281–1310. [Google Scholar] [CrossRef]
- Reiners, P.W.; Chan, M.A.; Evenson, N.S. (U-Th)/He geochronology and chemical compositions of diagenetic cement, concretions, and fracture-filling oxide minerals in Mesozoic sandstones of the Colorado Plateau. Geol. Soc. Am. Bull. 2014, 126, 1363–1383. [Google Scholar] [CrossRef]
- Ormö, J.; Komatsu, G.; Chan, M.A.; Beitler, B.; Parry, W.T. Geological features indicative of processes related to the hematite formation in Meridiani Planum and Aram Chaos, Mars: A comparison with diagenetic hematite deposits in southern Utah, USA. Icarus 2004, 171, 295–316. [Google Scholar] [CrossRef]
- Potter, S.L.; Chan, M.A.; Petersen, E.U.; Dyar, M.D.; Sklute, E. Characterization of Navajo Sandstone concretions: Mars comparison and criteria for distinguishing diagenetic origins. Earth Planet. Sci. Lett. 2011, 301, 444–456. [Google Scholar] [CrossRef]
- Singer, P.C.; Stumm, W. Acidic mine drainage: The rate-determining step. Science 1970, 167, 1121–1123. [Google Scholar] [CrossRef] [PubMed]
- Hurowitz, J.A.; Fischer, W.; Tosca, N.J.; Milliken, R.E. Origin of acidic surface waters and the evolution of atmospheric chemistry on early Mars. Nat. Geosci. 2010, 3, 323–326. [Google Scholar] [CrossRef]
- Nie, N.X.; Dauphas, N.; Greenwood, R.C. Iron and oxygen isotope fractionation during iron UV photo-oxidation: Implications for early Earth and Mars. Earth Planet. Sci. Lett. 2017, 458, 179–191. [Google Scholar] [CrossRef]
- Lasne, J.; Noblet, A.; Szopa, C.; Navarro-Gonzalez, R.; Cabane, M.; Poch, O.; Stalport, F.; Francois, P.; Atreya, S.K.; Coll, P. Oxidants at the surface of Mars: A review in light of recent exploration results. Astrobiology 2016, 16, 977–996. [Google Scholar] [CrossRef] [Green Version]
- Mitra, K.; Catalano, J.G. Chlorate as a potential oxidant on Mars: Rates and products of dissolved Fe(II) oxidation. J. Geophys. Res. Planets 2019, 124, 2893–2916. [Google Scholar] [CrossRef]
- Sutter, B.; Quinn, R.C.; Archer, P.D.; Glavin, D.P.; Glotch, T.D.; Kounaves, S.P.; Osterloo, M.M.; Rampe, E.B.; Ming, D.W. Measurements of oxychlorine species on Mars. Int. J. Astrobiol. 2017, 16, 203–217. [Google Scholar] [CrossRef] [Green Version]
- Ming, D.W.; Archer, P.D.; Glavin, D.P.; Eigenbrode, J.L.; Franz, H.B.; Sutter, B.; Brunner, A.E.; Stern, J.C.; Freissinet, C.; McAdam, A.C.; et al. Volatile and organic compositions of sedimentary rocks in Yellowknife Bay, Gale Crater, Mars. Science 2014, 343, 9. [Google Scholar] [CrossRef]
- Leshin, L.A.; Mahaffy, P.R.; Webster, C.R.; Cabane, M.; Coll, P.; Conrad, P.G.; Archer, P.D.; Atreya, S.K.; Brunner, A.E.; Buch, A.; et al. Volatile, isotope, and organic analysis of Martian fines with the Mars Curiosity Rover. Science 2013, 341, 1238937. [Google Scholar] [CrossRef]
- Glavin, D.P.; Freissinet, C.; Miller, K.E.; Eigenbrode, J.L.; Brunner, A.E.; Buch, A.; Sutter, B.; Archer, P.D.; Atreya, S.K.; Brinckerhoff, W.B.; et al. Evidence for perchlorates and the origin of chlorinated hydrocarbons detected by SAM at the Rocknest aeolian deposit in Gale Crater. J. Geophys. Res. Planets 2013, 118, 1955–1973. [Google Scholar] [CrossRef]
- Sutter, B.; McAdam, A.C.; Mahaffy, P.R.; Ming, D.W.; Edgett, K.S.; Rampe, E.B.; Eigenbrode, J.L.; Franz, H.B.; Freissinet, C.; Grotzinger, J.P.; et al. Evolved gas analyses of sedimentary rocks and eolian sediment in Gale Crater, Mars: Results of the curiosity rover’s sample analysis at Mars instrument from Yellowknife Bay to the Namib Dune. J. Geophys. Res. Planets 2017, 122, 2574–2609. [Google Scholar] [CrossRef] [Green Version]
- Hogancamp, J.; Sutter, B.; Morris, R.; Archer, P.D.; Ming, D.; Rampe, E.; Mahaffy, P.; Navarro-Gonzalez, R. Chlorate/Fe-bearing phase mixtures as a possible source of oxygen and chlorine detected by the Sample Analysis at Mars instrument in Gale Crater, Mars. J. Geophys. Res. Planets 2018, 123, 2920–2938. [Google Scholar] [CrossRef]
- Hecht, M.H.; Kounaves, S.P.; Quinn, R.C.; West, S.J.; Young, S.M.M.; Ming, D.W.; Catling, D.C.; Clark, B.C.; Boynton, W.V.; Hoffman, J.; et al. Detection of perchlorate and the soluble chemistry of Martian soil at the Phoenix Lander site. Science 2009, 325, 64–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanley, J.; Chevrier, V.F.; Berget, D.J.; Adams, R.D. Chlorate salts and solutions on Mars. Geophys. Res. Lett. 2012, 39, L08201. [Google Scholar] [CrossRef]
- Kounaves, S.P.; Carrier, B.L.; O’Neil, G.D.; Stroble, S.T.; Claire, M.W. Evidence of martian perchlorate, chlorate, and nitrate in Mars meteorite EETA79001: Implications for oxidants and organics. Icarus 2014, 229, 206–213. [Google Scholar] [CrossRef]
- Rao, B.; Anderson, T.A.; Redder, A.; Jackson, W.A. Perchlorate formation by ozone oxidation of aqueous chlorine/oxy-chlorine Species: Role of ClxOy radicals. Environ. Sci. Technol. 2010, 44, 2961–2967. [Google Scholar] [CrossRef]
- Jackson, W.A.; Bohlke, J.K.; Andraski, B.J.; Fahlquist, L.; Bexfield, L.; Eckardt, F.D.; Gates, J.B.; Davila, A.F.; McKay, C.P.; Rao, B.; et al. Global patterns and environmental controls of perchlorate and nitrate co-occurrence in arid and semi-arid environments. Geochim. Cosmochim. Acta 2015, 164, 502–522. [Google Scholar] [CrossRef] [Green Version]
- Brown, G.M.; Gu, B. The chemistry of perchlorate in the environment. In Perchlorate; Springer: Boston, MA, USA, 2006; pp. 17–47. [Google Scholar] [CrossRef]
- Urbansky, E.T. Perchlorate as an environmental contaminant. Environ. Sci. Pollut. Res. 2002, 9, 187–192. [Google Scholar] [CrossRef]
- Madlo, K. Kinetics of oxidation of bivalent iron by chlorate. Collect. Czech. Chem. Commun. 1979, 44, 2760–2768. [Google Scholar] [CrossRef]
- Ang, K.P.; Creak, G.A.; Kwik, W.L. Effect of ionic-strength on kinetics of oxidation of ferrous ion by chlorate ion. J. Chem. Soc. Dalton Trans. 1972, 2560–2562. [Google Scholar] [CrossRef]
- Mitzner, R.; Fischer, G.; Leupold, P. Kinetics of oxidation of iron(II) by means of chlorate in perchloric-acid solutions. Z. Phys. Chem. 1973, 253, 81–95. [Google Scholar]
- Higginson, W.C.; Simpson, M.E. Formation of free-radicals in reduction of chlorate by iron(II) cation in dilute aqueous acid solution. J. Chem. Soc. Chem. Commun. 1974, 817–818. [Google Scholar] [CrossRef]
- Miki, H.; Nicol, M. The kinetics of the oxidation of iron(II) by chlorate in the leaching of uranium ores. Hydrometallurgy 2009, 100, 47–49. [Google Scholar] [CrossRef]
- Hong, C.C.; Lenzi, F.; Rapson, W.H. Kinetics and mechanism of chloride-chlorate reaction. Can. J. Chem. Eng. 1967, 45, 349–355. [Google Scholar] [CrossRef]
- Shakhashiri, B.Z.; Gordon, G. Oxidation of tris(1, 10-phenanthroline) iron (II) ion by chlorate and chlorite ions and chlorine dioxide. J. Am. Chem. Soc. 1969, 91, 1103–1107. [Google Scholar] [CrossRef]
- Catling, D.C.; Claire, M.W.; Zahnle, K.J.; Quinn, R.C.; Clark, B.C.; Hecht, M.H.; Kounaves, S. Atmospheric origins of perchlorate on Mars and in the Atacama. J. Geophys. Res. Planets 2010, 115, E00E11. [Google Scholar] [CrossRef] [Green Version]
- Wu, Z.C.; Wang, A.; Farrell, W.M.; Yan, Y.C.; Wang, K.; Houghton, J.; Jackson, A.W. Forming perchlorates on Mars through plasma chemistry during dust events. Earth Planet. Sci. Lett. 2018, 504, 94–105. [Google Scholar] [CrossRef]
- Carrier, B.L.; Kounaves, S.P. The origins of perchlorate in the Martian soil. Geophys. Res. Lett. 2015, 42, 3739–3745. [Google Scholar] [CrossRef]
- Turner, A.M.; Abplanalp, M.J.; Kaiser, R.I. Mechanistic studies on the radiolytic decomposition of perchlorates on the Martian surface. Astrophys. J. 2016, 820, 127. [Google Scholar] [CrossRef] [Green Version]
- Wilson, E.H.; Atreya, S.K.; Kaiser, R.I.; Mahaffy, P.R. Perchlorate formation on Mars through surface radiolysis-initiated atmospheric chemistry: A potential mechanism. J. Geophys. Res. Planets 2016, 121, 1472–1487. [Google Scholar] [CrossRef] [Green Version]
- Schuttlefield, J.D.; Sambur, J.B.; Gelwicks, M.; Eggleston, C.M.; Parkinson, B.A. Photooxidation of chloride by oxide minerals: Implications for perchlorate on Mars. J. Am. Chem. Soc. 2011, 133, 17521–17523. [Google Scholar] [CrossRef]
- Squyres, S.W.; Grotzinger, J.P.; Arvidson, R.E.; Bell, J.F.; Calvin, W.; Christensen, P.R.; Clark, B.C.; Crisp, J.A.; Farrand, W.H.; Herkenhoff, K.E.; et al. In situ evidence for an ancient aqueous environment at Meridiani Planum, Mars. Science 2004, 306, 1709–1714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nuding, D.L.; Rivera-Valentin, E.G.; Davis, R.D.; Gough, R.V.; Chevrier, V.F.; Tolbert, M.A. Deliquescence and efflorescence of calcium perchlorate: An investigation of stable aqueous solutions relevant to Mars. Icarus 2014, 243, 420–428. [Google Scholar] [CrossRef]
- Vaniman, D.T.; Bish, D.L.; Chipera, S.J.; Fialips, C.I.; Carey, J.W.; Feldman, W.C. Magnesium sulphate salts and the history of water on Mars. Nature 2004, 431, 663–665. [Google Scholar] [CrossRef]
- Léveille, R.J.; Bridges, J.; Wiens, R.C.; Mangold, N.; Cousin, A.; Lanza, N.; Forni, O.; Ollila, A.; Grotzinger, J.; Clegg, S.; et al. Chemistry of fracture-filling raised ridges in Yellowknife Bay, Gale Crater: Window into past aqueous activity and habitability on Mars. J. Geophys. Res. Planets 2014, 119, 2398–2415. [Google Scholar] [CrossRef]
- Fox-Powell, M.G.; Hallsworth, J.E.; Cousins, C.R.; Cockell, C.S. Ionic strength is a barrier to the habitability of Mars. Astrobiology 2016, 16, 427–442. [Google Scholar] [CrossRef]
- Rapin, W.; Ehlmann, B.L.; Dromart, G.; Schieber, J.; Thomas, N.H.; Fischer, W.W.; Fox, V.K.; Stein, N.T.; Nachon, M.; Clark, B.C.; et al. An interval of high salinity in ancient Gale crater lake on Mars. Nat. Geosci. 2019, 12, 889–895. [Google Scholar] [CrossRef] [Green Version]
- Bibring, J.P.; Langevin, Y.; Mustard, J.; Poulet, F.; Arvidson, R.; Gendrin, A.; Gondet, B.; Mangold, N.; Pinet, P.; Forget, F.; et al. Global mineralogical and aqueous Mars history derived from OMEGA/Mars Express data. Science 2006, 312, 400–404. [Google Scholar] [CrossRef] [Green Version]
- Poulet, F.; Bibring, J.-P.; Mustard, J.; Gendrin, A.; Mangold, N.; Langevin, Y.; Arvidson, R.; Gondet, B.; Gomez, C. Phyllosilicates on Mars and implications for early Martian climate. Nature 2005, 438, 623–627. [Google Scholar] [CrossRef] [PubMed]
- Fairén, A.G.; Schulze-Makuch, D.; Rodríguez, A.P.; Fink, W.; Davila, A.F.; Uceda, E.R.; Furfaro, R.; Amils, R.; McKay, C.P. Evidence for Amazonian acidic liquid water on Mars—A reinterpretation of MER mission results. Planet. Space Sci. 2009, 57, 276–287. [Google Scholar] [CrossRef]
- Viollier, E.; Inglett, P.W.; Hunter, K.; Roychoudhury, A.N.; Van Cappellen, P. The ferrozine method revisited: Fe(II)/Fe(III) determination in natural waters. Appl. Geochem. 2000, 15, 785–790. [Google Scholar] [CrossRef]
- Delany, J.; Lundeen, S.R. The LLNL Thermochemical Database; Report UCRL-21658; Lawrence Livermore National Laboratory: Livermore, CA, USA, 1990; p. 150.
- Catalano, J.G. Thermodynamic and mass balance constraints on iron-bearing phyllosilicate formation and alteration pathways on early Mars. J. Geophys. Res. Planets 2013, 118, 2124–2136. [Google Scholar] [CrossRef]
- Doebelin, N.; Kleeberg, R. Profex: A graphical user interface for the Rietveld refinement program BGMN. J. Appl. Crystallogr. 2015, 48, 1573–1580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bergmann, J.; Friedel, P.; Kleeberg, R. BGMN—A new fundamental parameters based Rietveld program for laboratory X-ray sources, its use in quantitative analysis and structure investigations. CPD Newslett. 1998, 20, 5–8. [Google Scholar]
- Detournay, P.J.; Derie, R.; Ghodsi, M. Etude de l’oxydation par aeration de Fe(OH)2 en milieu chlorure. Z. Anorg. Allg. Chem. 1976, 427, 265–273. [Google Scholar] [CrossRef]
- Taylor, R. Influence of chloride on the formation of iron oxides from Fe (II) chloride. II. Effect of [Cl] on the formation of lepidocrocite and its crystallinity. Clays Clay Miner. 1984, 32, 175–180. [Google Scholar] [CrossRef]
- Bibi, I.; Singh, B.; Silvester, E. Akaganéite (β-FeOOH) precipitation in inland acid sulfate soils of south-western New South Wales (NSW), Australia. Geochim. Cosmochim. Acta 2011, 75, 6429–6438. [Google Scholar] [CrossRef]
- Peretyazhko, T.S.; Pan, M.J.; Ming, D.W.; Rampe, E.B.; Morris, R.V.; Agresti, D.G. Reaction of akaganeite with Mars-relevant anions. ACS Earth Space Chem. 2019, 3, 314–323. [Google Scholar] [CrossRef]
- Rémazeilles, C.; Refait, P. On the formation of β-FeOOH (akaganéite) in chloride-containing environments. Corros. Sci. 2007, 49, 844–857. [Google Scholar] [CrossRef]
- Ståhl, K.; Nielsen, K.; Jiang, J.Z.; Lebech, B.; Hanson, J.C.; Norby, P.; van Lanschot, J. On the akaganeite crystal structure, phase transformations and possible role in post-excavational corrosion of iron artifacts. Corros. Sci. 2003, 45, 2563–2575. [Google Scholar] [CrossRef]
- Tosca, N.J.; McLennan, S.M.; Dyar, M.D.; Sklute, E.C.; Michel, F.M. Fe oxidation processes at Meridiani Planum and implications for secondary Fe mineralogy on Mars. J. Geophys. Res. Planets 2008, 113, E05005. [Google Scholar] [CrossRef]
- Regenspurg, S.; Brand, A.; Peiffer, S. Formation and stability of schwertmannite in acidic mining lakes. Geochim. Cosmochim. Acta 2004, 68, 1185–1197. [Google Scholar] [CrossRef]
- Caraballo, M.A.; Rimstidt, J.D.; Macías, F.; Nieto, J.M.; Hochella, M.F., Jr. Metastability, nanocrystallinity and pseudo-solid solution effects on the understanding of schwertmannite solubility. Chem. Geol. 2013, 360, 22–31. [Google Scholar] [CrossRef]
- Acero, P.; Ayora, C.; Torrentó, C.; Nieto, J.-M. The behavior of trace elements during schwertmannite precipitation and subsequent transformation into goethite and jarosite. Geochim. Cosmochim. Acta 2006, 70, 4130–4139. [Google Scholar] [CrossRef]
- Schwertmann, U.; Carlson, L. The pH-dependent transformation of schwertmannite to goethite at 25 °C. Clay Miner. 2005, 40, 63–66. [Google Scholar] [CrossRef]
- Burton, E.D.; Bush, R.T.; Sullivan, L.A.; Mitchell, D.R. Schwertmannite transformation to goethite via the Fe (II) pathway: Reaction rates and implications for iron–sulfide formation. Geochim. Cosmochim. Acta 2008, 72, 4551–4564. [Google Scholar] [CrossRef]
- Kumpulainen, S.; Räisänen, M.-L.; Von der Kammer, F.; Hofmann, T. Ageing of synthetic and natural schwertmannites at pH 2–8. Clay Miner. 2008, 43, 437–448. [Google Scholar] [CrossRef]
- Bigham, J.; Schwertmann, U.; Traina, S.; Winland, R.; Wolf, M. Schwertmannite and the chemical modeling of iron in acid sulfate waters. Geochim. Cosmochim. Acta 1996, 60, 2111–2121. [Google Scholar] [CrossRef]
- Peretyazhko, T.; Zachara, J.M.; Boily, J.-F.; Xia, Y.; Gassman, P.L.; Arey, B.W.; Burgos, W.D. Mineralogical transformations controlling acid mine drainage chemistry. Chem. Geol. 2009, 262, 169–178. [Google Scholar] [CrossRef] [Green Version]
- Sidhu, P.S.; Gilkes, R.J.; Cornell, R.M.; Posner, A.M.; Quirk, J.P. Dissolution of iron-oxides and oxyhydroxides in hydrochloric and perchloric acids. Clays Clay Miner. 1981, 29, 269–276. [Google Scholar] [CrossRef]
- Sweeton, F.; Baes, C., Jr. The solubility of magnetite and hydrolysis of ferrous ion in aqueous solutions at elevated temperatures. J. Chem. Thermodyn. 1970, 2, 479–500. [Google Scholar] [CrossRef]
- Salmimies, R.; Mannila, M.; Kallas, J.; Häkkinen, A. Acidic dissolution of magnetite: Experimental study on the effects of acid concentration and temperature. Clays Clay Miner. 2011, 59, 136–146. [Google Scholar] [CrossRef]
- Salmimies, R.; Vehmaanperä, P.; Häkkinen, A. Acidic dissolution of magnetite in mixtures of oxalic and sulfuric acid. Hydrometallurgy 2016, 163, 91–98. [Google Scholar] [CrossRef]
- Murad, E.; Cashion, J. Iron oxides. In Mössbauer Spectroscopy of Environmental Materials and Their Industrial Utilization; Springer: Boston, MA, USA, 2004; pp. 159–188. [Google Scholar]
- Jolivet, J.-P.; Tronc, E. Interfacial electron transfer in colloidal spinel iron oxide. Conversion of Fe3O4-γFe2O3 in aqueous medium. J. Colloid Interface Sci. 1988, 125, 688–701. [Google Scholar] [CrossRef]
- Schwertmann, U.; Cornell, R.M. Iron Oxides in the Laboratory: Preparation and Characterization, 2nd ed.; John Wiley & Sons: Weinheim, Germany, 2008. [Google Scholar]
- Burns, R.G.; Fisher, D.S. Rates of oxidative weathering on the surface of Mars. J. Geophys. Res. Planets 1993, 98, 3365–3372. [Google Scholar] [CrossRef]
- Schwertmann, U.; Stanjek, H.; Becher, H.H. Long-term in vitro transformation of 2-line ferrihydrite to goethite/hematite at 4, 10, 15 and 25 °C. Clay Miner. 2004, 39, 433–438. [Google Scholar] [CrossRef]
- Schwertmann, U.; Friedl, J.; Stanjek, H. From Fe(III) ions to ferrihydrite and then to hematite. J. Colloid Interface Sci. 1999, 209, 215–223. [Google Scholar] [CrossRef]
- Schwertmann, U.; Friedl, J.; Stanjek, H.; Schulze, D.G. The effect of Al on Fe oxides. XIX. Formation of Al-substituted hematite from ferrihydrite at 25 °C and pH 4 to 7. Clays Clay Miner. 2000, 48, 159–172. [Google Scholar] [CrossRef]
- Schwertmann, U.; Friedl, J.; Stanjek, H.; Schulze, D.G. The effect of clay minerals on the formation of goethite and hematite from ferrihydrite after 16 years’ ageing at 25 °C and pH 4–7. Clay Miner. 2000, 35, 613–623. [Google Scholar] [CrossRef]
- Fischer, W.R.; Schwertmann, U. Formation of hematite from amorphous iron(III) hydroxide. Clays Clay Miner. 1975, 23, 33–37. [Google Scholar] [CrossRef]
- Schwertmann, U.; Murad, E. Effect of pH on the formation of goethite and hematite from ferrihydrite. Clays Clay Miner. 1983, 31, 277–284. [Google Scholar] [CrossRef]
- Jiang, Z.; Liu, Q.; Roberts, A.P.; Barrón, V.; Torrent, J.; Zhang, Q. A new model for transformation of ferrihydrite to hematite in soils and sediments. Geology 2018, 46, 987–990. [Google Scholar] [CrossRef] [Green Version]
- Tosca, N.J.; Knoll, A.H. Juvenile chemical sediments and the long term persistence of water at the surface of Mars. Earth Planet. Sci. Lett. 2009, 286, 379–386. [Google Scholar] [CrossRef] [Green Version]
- Cornell, R.; Giovanoli, R. Transformation of akaganeite into goethite and hematite in alkaline media. Clays Clay Miner. 1990, 38, 469–476. [Google Scholar] [CrossRef]
- Goñi-Elizalde, S.; Garcia-Clavel, M.E.; Tejedor-Tejedor, M.I. Mechanism of akaganeite-hematite transformation via solution. React. Solids 1987, 3, 139–154. [Google Scholar] [CrossRef]
- Frandsen, C.; Legg, B.A.; Comolli, L.R.; Zhang, H.; Gilbert, B.; Johnson, E.; Banfield, J.F. Aggregation-induced growth and transformation of β-FeOOH nanorods to micron-sized α-Fe2O3 spindles. CrystEngComm 2014, 16, 1451–1458. [Google Scholar] [CrossRef] [Green Version]
- Peterson, K.M.; Heaney, P.J.; Post, J.E. Evolution in the structure of akaganeite and hematite during hydrothermal growth: An in situ synchrotron X-ray diffraction analysis. Powder Diffr. 2018, 33, 287–297. [Google Scholar] [CrossRef]
- Gendrin, A.; Mangold, N.; Bibring, J.P.; Langevin, Y.; Gondet, B.; Poulet, F.; Bonello, G.; Quantin, C.; Mustard, J.; Arvidson, R.; et al. Sulfates in Martian layered terrains: The OMEGA/Mars Express view. Science 2005, 307, 1587–1591. [Google Scholar] [CrossRef] [Green Version]
- Bibring, J.P.; Langevin, Y.; Gendrin, A.; Gondet, B.; Poulet, F.; Berthe, M.; Soufflot, A.; Arvidson, R.; Mangold, N.; Mustard, J.; et al. Mars surface diversity as revealed by the OMEGA/Mars Express observations. Science 2005, 307, 1576–1581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glotch, T.D.; Rogers, A.D. Evidence for aqueous deposition of hematite- and sulfate-rich light-toned layered deposits in Aureum and Iani Chaos, Mars. J. Geophys. Res. Planets 2007, 112, E06001. [Google Scholar] [CrossRef]
- Fernández-Remolar, D.C.; Morris, R.V.; Gruener, J.E.; Amils, R.; Knoll, A.H. The Río Tinto Basin, Spain: Mineralogy, sedimentary geobiology, and implications for interpretation of outcrop rocks at Meridiani Planum, Mars. Earth Planet. Sci. Lett. 2005, 240, 149–167. [Google Scholar] [CrossRef]
- Archer, P.D.; Franz, H.B.; Sutter, B.; Arevalo, R.D.; Coll, P.; Eigenbrode, J.L.; Glavin, D.P.; Jones, J.J.; Leshin, L.A.; Mahaffy, P.R. Abundances and implications of volatile-bearing species from evolved gas analysis of the Rocknest aeolian deposit, Gale Crater, Mars. J. Geophys. Res. Planets 2014, 119, 237–254. [Google Scholar] [CrossRef]
- David, G.; Cousin, A.; Forni, O.; Meslin, P.Y.; Dehouck, E.; Mangold, N.; L’Haridon, J.; Rapin, W.; Gasnault, O.; Johnson, J. Analyses of high-iron sedimentary bedrock and diagenetic features observed with ChemCam at Vera Rubin ridge, Gale crater, Mars: Calibration and characterization. J. Geophys. Res. Planets 2020. [Google Scholar] [CrossRef]
- Grotzinger, J.P.; Sumner, D.Y.; Kah, L.; Stack, K.; Gupta, S.; Edgar, L.; Rubin, D.; Lewis, K.; Schieber, J.; Mangold, N. A habitable fluvio-lacustrine environment at Yellowknife Bay, Gale Crater, Mars. Science 2014, 343, 1242777. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Sample a | Initial [Fe2+] (mmol L−1) | Initial [ClO3−] (mmol L−1) | [Fe2+]/[ClO3−] | Initial pH | Model Final [Fe2+] (mmol L−1) b | Experimental Final [Fe2+] (mmol L−1) b | Final [ClO3−] (mmol L−1) | Model Final pH d | Experimental Final pH d |
|---|---|---|---|---|---|---|---|---|---|
| Cl-6:1-10-100-3 | 10.5 | 1.73 | 6.1 | 3.04 | 0.98 | 0.65 | BDL c | 2.05 | 1.96 |
| Cl-6:1-10-100-5 | 10.4 | 1.73 | 6.0 | 5.34 | 1.03 | 0.84 | BDL | 2.07 | 1.98 |
| Cl-6:1-10-100-7 | 10.2 | 1.70 | 6.0 | 6.58 | 1.01 | 0.49 | BDL | 2.08 | 2.04 |
| Cl-10:1-10-100-3 | 11.1 | 1.08 | 10.3 | 3.02 | 4.68 | 4.99 | BDL | 2.14 | 2.01 |
| Cl-10:1-10-100-5 | 11.2 | 1.10 | 10.2 | 4.34 | 4.67 | 5.09 | BDL | 2.16 | 2.07 |
| Cl-10:1-10-100-7 | 11.3 | 1.13 | 10.0 | 6.31 | 4.57 | 5.36 | BDL | 2.16 | 2.16 |
| S-6:1-10-100-3 | 10.8 | 1.71 | 6.3 | 3.07 | 1.70 | 0.93 | BDL | 2.45 | 2.44 |
| S-6:1-10-100-5 | 10.7 | 1.71 | 6.3 | 4.68 | 1.81 | 0.96 | BDL | 2.45 | 2.44 |
| S-6:1-10-100-7 | 10.9 | 1.73 | 6.3 | 6.77 | 1.82 | 0.95 | BDL | 2.45 | 2.43 |
| S-10:1-10-100-3 | 11.5 | 1.10 | 10.4 | 3.06 | 5.02 | 5.28 | BDL | 2.45 | 2.49 |
| S-10:1-10-100-5 | 11.5 | 1.10 | 10.5 | 4.88 | 5.19 | 5.46 | BDL | 2.48 | 2.53 |
| S-10:1-10-100-7 | 11.5 | 1.10 | 10.4 | 6.54 | 5.18 | 5.44 | BDL | 2.48 | 2.55 |
| Sample a | Initial [Fe2+] (mmol L−1) | Initial [ClO3−] (mmol L−1) | Initial [Fe2+]/[ClO3−] | Initial pH | Model Final [Fe2+] (mmol L−1) | Experimental Final [Fe2+] (mmol L−1) | Final Total [Fe] (mmol L−1) | Final [ClO3−] (mmol L−1) | Final pH | Mineral Products |
|---|---|---|---|---|---|---|---|---|---|---|
| Cl-6:1-10-100-3 | 11.6 | 1.92 | 6.1 | 3.08 | 0.12 | 1.53 | 4.11 | BDL b | 1.95 | Trace c |
| Cl-6:1-10-100-7 | 8.00 | 1.42 | 5.6 | 7.02 | 0.00 | 2.14 | 1.96 | BDL | 2.51 | M,G,L |
| Cl-6:1-100-100-3 | 90.1 | 15.5 | 5.8 | 3.00 | 0.00 | 0.57 | 43.6 | BDL | 1.56 | Trace |
| Cl-6:1-100-100-7 | 90.9 | 15.1 | 6.0 | 7.25 | 0.16 | 0.35 | 0.35 | BDL | 2.54 | L,M,G |
| Cl-10:1-10-100-3 | 12.5 | 1.35 | 9.3 | 3.04 | 4.43 | 3.12 | 6.47 | BDL | 1.92 | Trace |
| Cl-10:1-10-100-7 | 13.2 | 1.32 | 10.0 | 7.02 | 5.28 | 0.00 | 0.13 | BDL | 5.74 | M,G,GR |
| Cl-10:1-100-100-3 | 138 | 15.0 | 9.2 | 2.97 | 48.1 | 39.0 | 89.4 | BDL | 1.46 | Trace |
| Cl-10:1-100-100-7 | 141 | 15.3 | 9.2 | 7.26 | 49.0 | 10.9 | 11.5 | BDL | 4.46 | M,G |
| S-6:1-5-100-3 | 3.85 | 0.63 | 6.1 | 3.01 | 0.07 | 1.57 | 2.24 | BDL | 2.72 | Trace |
| S-6:1-5-100-7 | 3.91 | 0.63 | 6.2 | 6.90 | 0.13 | 0.82 | 0.10 | BDL | 4.60 | M,G |
| S-6:1-10-100-3 | 8.82 | 1.49 | 5.9 | 3.04 | 0.00 | 1.47 | 3.43 | BDL | 2.51 | G |
| S-6:1-10-100-7 | 8.61 | 1.45 | 6.0 | 6.80 | 0.00 | 1.74 | 3.48 | BDL | 2.58 | G |
| S-6:1-100-100-3 | 83.5 | 15.2 | 5.5 | 3.04 | 0.00 | 0.31 | 50.2 | BDL | 2.11 | S |
| S-6:1-100-100-7 | 72.5 | 15.2 | 4.8 | 6.64 | 0.00 | 0.23 | 12.1 | BDL | 2.08 | G |
| S-10:1-10-100-3 | 9.53 | 1.01 | 9.4 | 3.01 | 3.47 | 3.33 | 5.71 | BDL | 2.58 | G |
| S-10:1-10-100-7 | 9.48 | 1.01 | 9.4 | 7.02 | 3.42 | 5.03 | 4.99 | BDL | 3.43 | M,G |
| S-10:1-100-100-3 | 96.4 | 9.65 | 10.0 | 3.03 | 38.5 | 18.6 | 58.9 | BDL | 2.25 | S |
| S-10:1-100-100-7 | 97.1 | 10.2 | 9.5 | 7.04 | 36.0 | 15.6 | 28.6 | BDL | 2.21 | G |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mitra, K.; Moreland, E.L.; Catalano, J.G. Capacity of Chlorate to Oxidize Ferrous Iron: Implications for Iron Oxide Formation on Mars. Minerals 2020, 10, 729. https://doi.org/10.3390/min10090729
Mitra K, Moreland EL, Catalano JG. Capacity of Chlorate to Oxidize Ferrous Iron: Implications for Iron Oxide Formation on Mars. Minerals. 2020; 10(9):729. https://doi.org/10.3390/min10090729
Chicago/Turabian StyleMitra, Kaushik, Eleanor L. Moreland, and Jeffrey G. Catalano. 2020. "Capacity of Chlorate to Oxidize Ferrous Iron: Implications for Iron Oxide Formation on Mars" Minerals 10, no. 9: 729. https://doi.org/10.3390/min10090729
APA StyleMitra, K., Moreland, E. L., & Catalano, J. G. (2020). Capacity of Chlorate to Oxidize Ferrous Iron: Implications for Iron Oxide Formation on Mars. Minerals, 10(9), 729. https://doi.org/10.3390/min10090729